Updates in Pathology for Retroperitoneal Soft Tissue Sarcoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Liposarcoma (LPS)
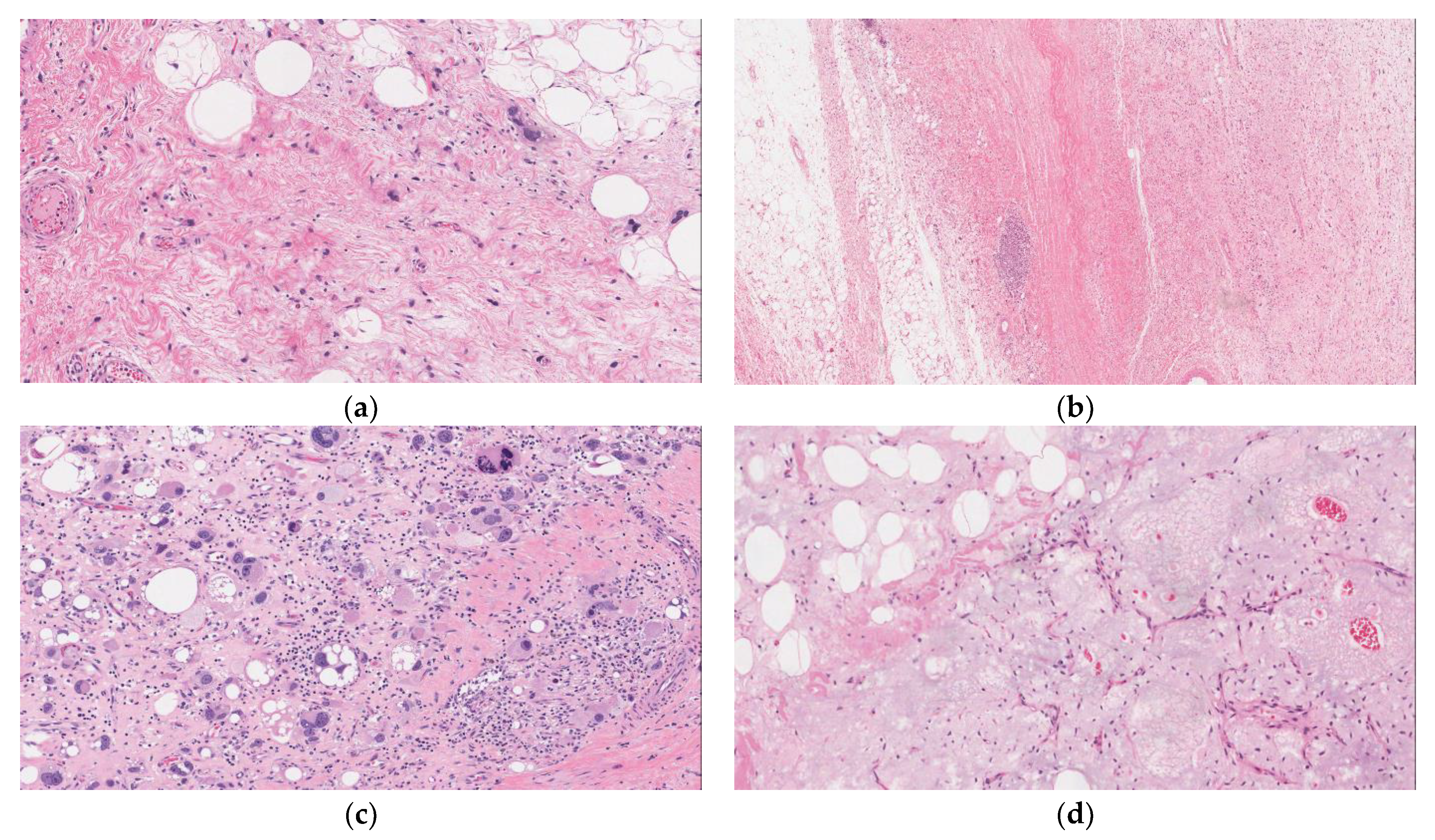
2.1. Well-Differentiated Liposarcoma (WDLPS)
2.2. De-Differentiated Liposarcoma (DDLPS)
2.3. Pleomorphic Liposarcoma (PLPS)
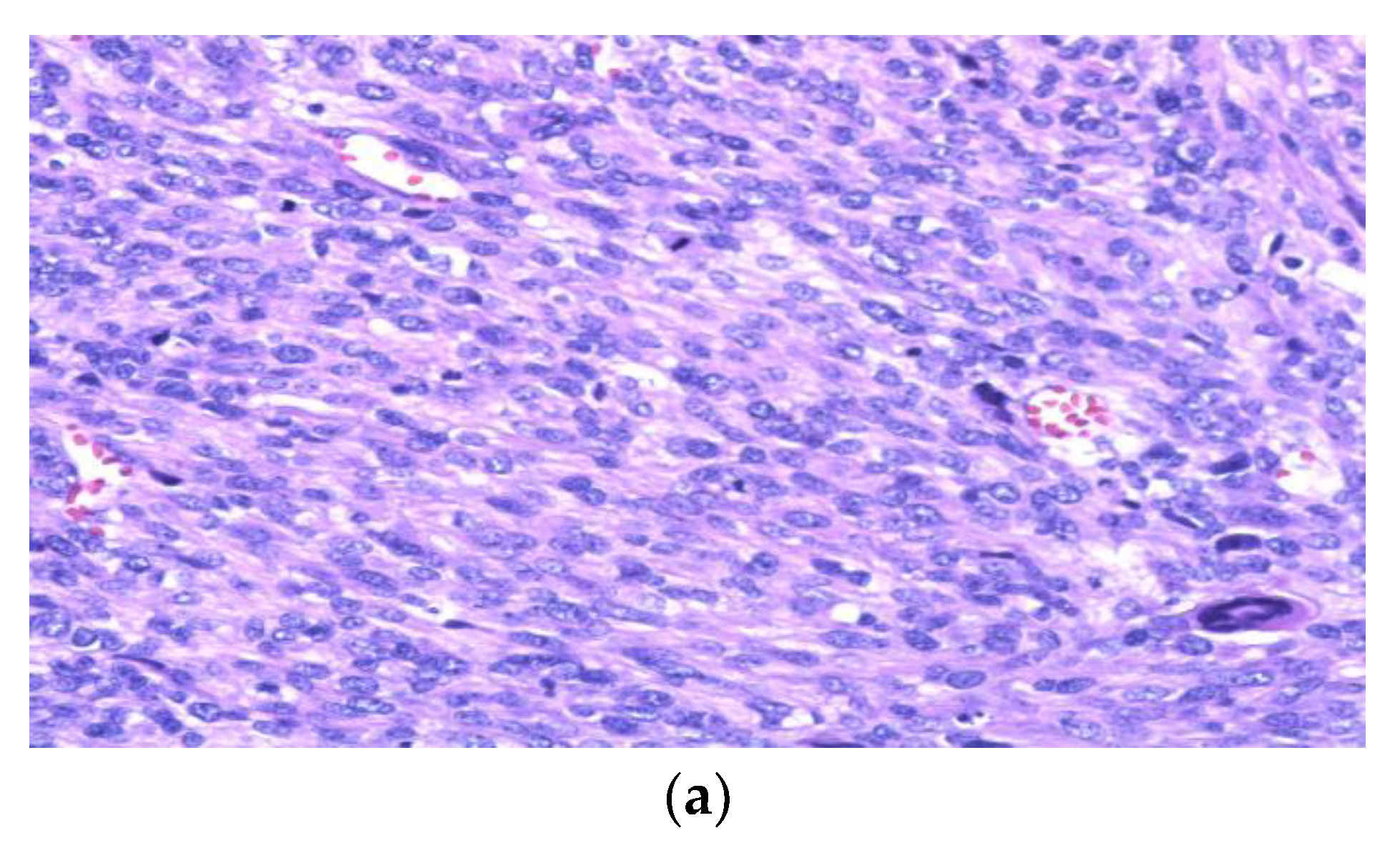
3. Leiomyosarcoma (LMS)
4. Undifferentiated Pleomorphic Sarcoma (UPS)
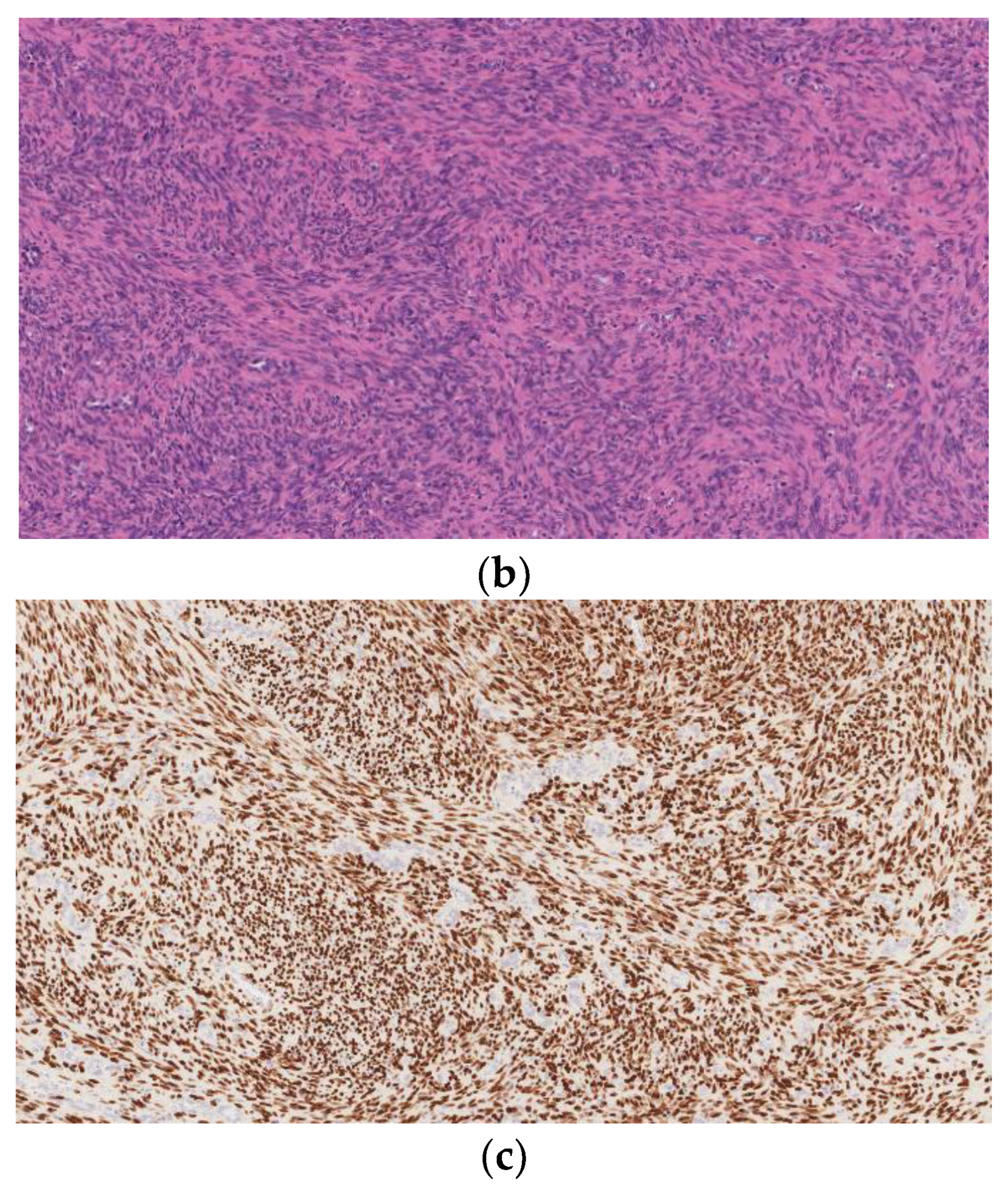
5. Malignant Peripheral Nerve Sheath Tumor (MPNST)
6. Small Round Blue Cell Tumors
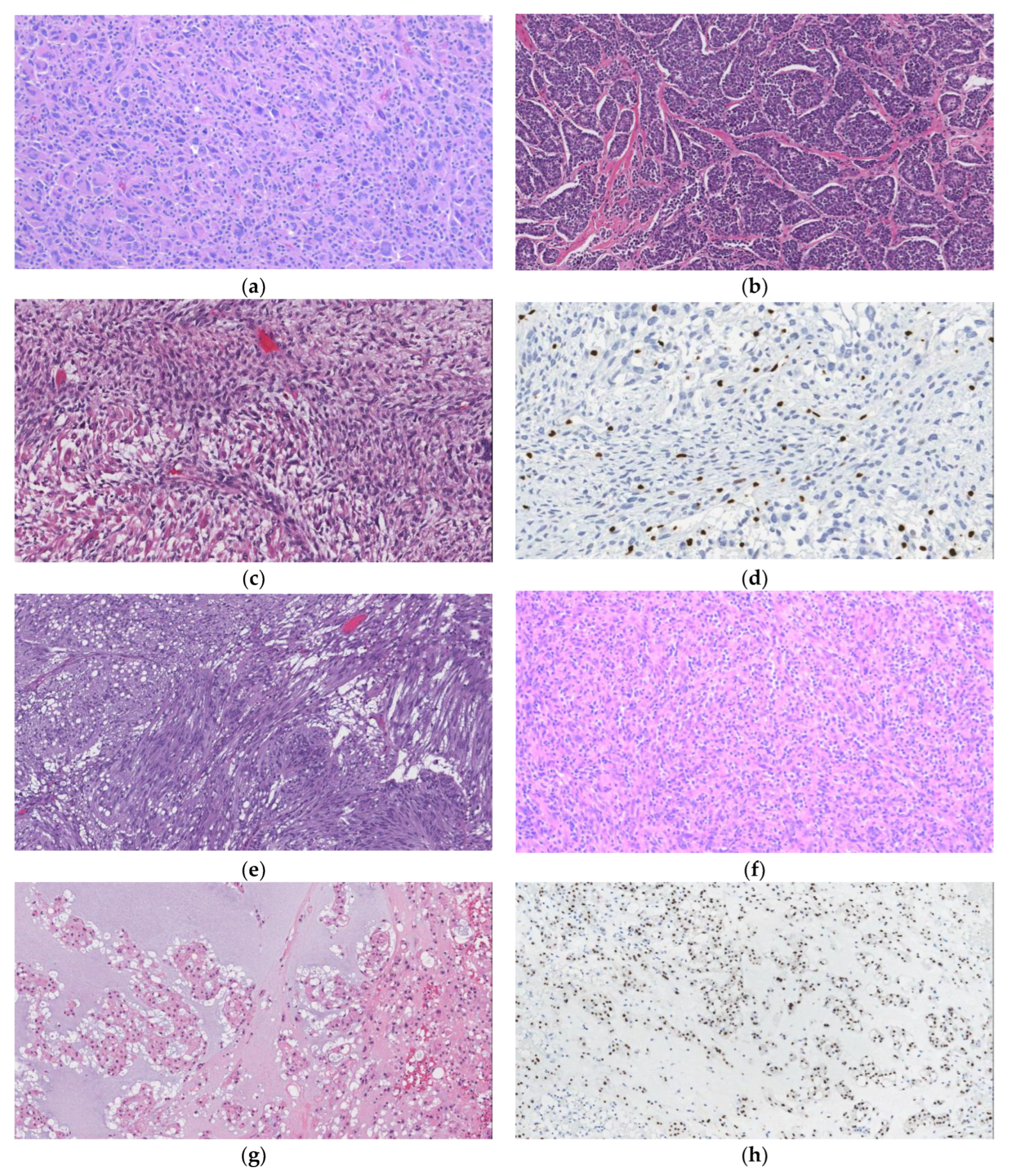
6.1. Desmoplastic Small Round Cell Tumor (DSRCT)
6.2. Ewing Sarcoma
7. Solitary Fibrous Tumor (SFT)
8. Gastrointestinal Stromal Tumor (GIST)
9. Inflammatory Myofibroblastic Tumor (IMT)
10. Angiomyolipoma (AML)
11. Myelolipoma
12. Chordoma
13. Angiosarcoma
14. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Antonescu, C.R.; Blay, J.-Y.; Bovée, J.V.M.G.; Bridge, J.A.; Cunha, I.W.; Dei Tos, A.P.; Flanagan, A.M.; Fletcher, C.D.M.; Folpe, A.L.; Gronchi, A.; et al. WHO Classification of Tumours: Soft Tissue and Bone Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2020; Volume 3. [Google Scholar]
- Goldblum, J. Enzinger and Weiss’s Soft Tissue Tumors; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Goldblum, J.R.; Lamps, L.W.; McKenney, J.K.; Myers, J.L.; Ackerman, L.V. Rosai and Ackerman′s Surgical Pathology, 7th ed.; Elsevier: Philadelphia, PA, USA, 2018. [Google Scholar]
- Surveillance Epidemiology and End Results (SEER) “SEER*Explorer Application”. Available online: https://seer.cancer.gov/statistics-network/ (accessed on 8 June 2022).
- Carbone, F.; Pizzolorusso, A.; Di Lorenzo, G.; Di Marzo, M.; Cannella, L.; Barretta, M.L.; Delrio, P.; Tafuto, S. Multidisciplinary Management of Retroperitoneal Sarcoma: Diagnosis, Prognostic Factors and Treatment. Cancers 2021, 13, 4016. [Google Scholar] [CrossRef] [PubMed]
- Tyler, R.; Lee, M.; Ierodiakonou, V.; Hodson, J.; Taniere, P.; Almond, M.; Ford, S.; Desai, A. Prognostic implications of histological organ involvement in retroperitoneal sarcoma. BJS Open 2021, 5, zrab080. [Google Scholar] [CrossRef] [PubMed]
- Gamboa, A.C.; Gronchi, A.; Cardona, K. Soft-tissue sarcoma in adults: An update on the current state of histiotype-specific management in an era of personalized medicine. CA Cancer J. Clin. 2020, 70, 200–229. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Lock, I.; Ozenberger, B.B.; Jones, K.B. Genetic drivers and cells of origin in sarcomagenesis. J. Pathol. 2021, 254, 474–493. [Google Scholar] [CrossRef] [PubMed]
- Renne, S.L.; Iwenofu, O.H. Pathology of retroperitoneal sarcomas: A brief review. J. Surg. Oncol. 2018, 117, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Ro, J.Y. Retroperitoneal Sarcomas: An Update on the Diagnostic Pathology Approach. Diagnostics 2020, 10, 642. [Google Scholar] [CrossRef]
- Sun, P.; Ma, R.; Liu, G.; Wang, L.; Chang, H.; Li, Y. Pathological prognostic factors of retroperitoneal liposarcoma: Comprehensive clinicopathological analysis of 124 cases. Ann. Transl. Med. 2021, 9, 574. [Google Scholar] [CrossRef]
- Xiao, J.; Liu, J.; Chen, M.; Liu, W.; He, X. Diagnosis and Prognosis of Retroperitoneal Liposarcoma: A Single Asian Center Cohort of 57 Cases. J. Oncol. 2021, 2021, 7594027. [Google Scholar] [CrossRef]
- Swallow, C.J.; Strauss, D.C.; Bonvalot, S.; Rutkowski, P.; Desai, A.; Gladdy, R.A.; Gonzalez, R.; Gyorki, D.E.; Fairweather, M.; van Houdt, W.J.; et al. Management of Primary Retroperitoneal Sarcoma (RPS) in the Adult: An Updated Consensus Approach from the Transatlantic Australasian RPS Working Group. Ann. Surg. Oncol. 2021, 28, 7873–7888. [Google Scholar] [CrossRef]
- Bonvalot, S.; Gronchi, A.; Le Pechoux, C.; Swallow, C.J.; Strauss, D.; Meeus, P.; van Coevorden, F.; Stoldt, S.; Stoeckle, E.; Rutkowski, P.; et al. Preoperative radiotherapy plus surgery versus surgery alone for patients with primary retroperitoneal sarcoma (EORTC-62092: STRASS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2020, 21, 1366–1377. [Google Scholar] [CrossRef]
- Xu, C.; Ma, Z.; Zhang, H.; Yu, J.; Chen, S. Giant retroperitoneal liposarcoma with a maximum diameter of 37 cm: A case report and review of literature. Ann. Transl. Med. 2020, 8, 1248. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.L. Atypical lipomatous tumor, its variants, and its combined forms: A study of 61 cases, with a minimum follow-up of 10 years. Am. J. Surg. Pathol. 2007, 31, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kraus, M.D.; Guillou, L.; Fletcher, C.D. Well-differentiated inflammatory liposarcoma: An uncommon and easily overlooked variant of a common sarcoma. Am. J. Surg. Pathol. 1997, 21, 518–527. [Google Scholar] [CrossRef]
- Bestic, J.M.; Kransdorf, M.J.; White, L.M.; Bridges, M.D.; Murphey, M.D.; Peterson, J.J.; Garner, H.W. Sclerosing variant of well-differentiated liposarcoma: Relative prevalence and spectrum of CT and MRI features. AJR Am. J. Roentgenol. 2013, 201, 154–161. [Google Scholar] [CrossRef]
- Tran, T.A.N.; de La Fuente, S. Retroperitoneal Well-Differentiated Liposarcoma with Uterine-Type Leiomyomatous Differentiation: A First Case Report with Literature Analysis of Soft Tissue Sarcomas with Dual Lipomatous and Low-Grade Smooth Muscle Differentiation. Int. J. Surg. Pathol. 2019, 27, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.L. Smooth muscle in atypical lipomatous tumors. A report of three cases. Am. J. Surg. Pathol. 1990, 14, 714–718. [Google Scholar] [CrossRef]
- Yoshida, A.; Ushiku, T.; Motoi, T.; Shibata, T.; Fukayama, M.; Tsuda, H. Well-differentiated liposarcoma with low-grade osteosarcomatous component: An underrecognized variant. Am. J. Surg. Pathol. 2010, 34, 1361–1366. [Google Scholar] [CrossRef]
- Lu, J.; Wood, D.; Ingley, E.; Koks, S.; Wong, D. Update on genomic and molecular landscapes of well-differentiated liposarcoma and dedifferentiated liposarcoma. Mol. Biol. Rep. 2021, 48, 3637–3647. [Google Scholar] [CrossRef]
- Tyler, R.; Wanigasooriya, K.; Taniere, P.; Almond, M.; Ford, S.; Desai, A.; Beggs, A. A review of retroperitoneal liposarcoma genomics. Cancer Treat. Rev. 2020, 86, 102013. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.; Gruel, N.; Le Loarer, F. New developments in the pathology and molecular biology of retroperitoneal sarcomas. Eur. J. Surg. Oncol. 2022; In Press . [Google Scholar] [CrossRef]
- Dal Cin, P.; Kools, P.; Sciot, R.; De Wever, I.; Van Damme, B.; Van de Ven, W.; Van den Berghe, H. Cytogenetic and fluorescence in situ hybridization investigation of ring chromosomes characterizing a specific pathologic subgroup of adipose tissue tumors. Cancer Genet. Cytogenet. 1993, 68, 85–90. [Google Scholar] [CrossRef]
- Thway, K.; Flora, R.; Shah, C.; Olmos, D.; Fisher, C. Diagnostic utility of p16, CDK4, and MDM2 as an immunohistochemical panel in distinguishing well-differentiated and dedifferentiated liposarcomas from other adipocytic tumors. Am. J. Surg. Pathol. 2012, 36, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Clay, M.R.; Martinez, A.P.; Weiss, S.W.; Edgar, M.A. MDM2 and CDK4 Immunohistochemistry: Should It Be Used in Problematic Differentiated Lipomatous Tumors?: A New Perspective. Am. J. Surg. Pathol. 2016, 40, 1647–1652. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Chen, X.L.; Yu, Q.; Fan, Q.B. Giant retroperitoneal lipoma presenting with abdominal distention: A case report and review of the literature. World J. Clin. Cases 2022, 10, 1675–1683. [Google Scholar] [CrossRef]
- Clay, M.R.; Martinez, A.P.; Weiss, S.W.; Edgar, M.A. MDM2 Amplification in Problematic Lipomatous Tumors: Analysis of FISH Testing Criteria. Am. J. Surg. Pathol. 2015, 39, 1433–1439. [Google Scholar] [CrossRef]
- Setsu, N.; Miyake, M.; Wakai, S.; Nakatani, F.; Kobayashi, E.; Chuman, H.; Hiraoka, N.; Kawai, A.; Yoshida, A. Primary Retroperitoneal Myxoid Liposarcomas. Am. J. Surg. Pathol. 2016, 40, 1286–1290. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.W.; Chen, J.; Patel, D.; Miao, C.; Ching, A.; Yang, S.; Feng, Y.; Matcuk, G.; Lara, K.; Hu, J.; et al. Multidisciplinary sarcoma tumor board: Retroperitoneal liposarcoma. Chin. Clin. Oncol. 2020, 9, 20. [Google Scholar] [CrossRef]
- Young, R.; Snow, H.; Hendry, S.; Mitchell, C.; Slavin, J.; Schlicht, S.; Na, L.; Hofman, M.S.; Gyorki, D.E. Correlation between percutaneous biopsy and final histopathology for retroperitoneal sarcoma: A single-centre study. ANZ J. Surg. 2020, 90, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.L.; Khurana, K.K.; Kemp, B.L.; Ayala, A.G. Heterologous elements in the dedifferentiated component of dedifferentiated liposarcoma. Am. J. Surg. Pathol. 1994, 18, 1150–1157. [Google Scholar] [CrossRef]
- Gronchi, A.; Collini, P.; Miceli, R.; Valeri, B.; Renne, S.L.; Dagrada, G.; Fiore, M.; Sanfilippo, R.; Barisella, M.; Colombo, C.; et al. Myogenic differentiation and histologic grading are major prognostic determinants in retroperitoneal liposarcoma. Am. J. Surg. Pathol. 2015, 39, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Casadei, L.; de Faria, F.C.C.; Lopez-Aguiar, A.; Pollock, R.E.; Grignol, V. Targetable Pathways in the Treatment of Retroperitoneal Liposarcoma. Cancers 2022, 14, 1362. [Google Scholar] [CrossRef]
- WELLIVER, M.X.; Tine, B.A.V.; Houghton, P.; Rudek, M.A.; Pollock, R.E.; Kane, J.M.; Schwartz, G.K.; Zhang, P.; Kirsch, D.G.; Wakely, P.; et al. MDM2 inhibitor AMG-232 and radiation therapy in treating patients with soft tissue sarcoma with wild-type TP53: A phase IB study (NRG-DT001). J. Clin. Oncol. 2019, 37, TPS11076. [Google Scholar] [CrossRef]
- Dehner, C.A.; Hagemann, I.S.; Chrisinger, J.S.A. Retroperitoneal Dedifferentiated Liposarcoma. Am. J. Clin. Pathol. 2021, 156, 920–925. [Google Scholar] [CrossRef]
- Bonvalot, S.; Miceli, R.; Berselli, M.; Causeret, S.; Colombo, C.; Mariani, L.; Bouzaiene, H.; Le Pechoux, C.; Casali, P.G.; Le Cesne, A.; et al. Aggressive surgery in retroperitoneal soft tissue sarcoma carried out at high-volume centers is safe and is associated with improved local control. Ann. Surg Oncol. 2010, 17, 1507–1514. [Google Scholar] [CrossRef]
- Kelley, T.W.; Borden, E.C.; Goldblum, J.R. Estrogen and progesterone receptor expression in uterine and extrauterine leiomyosarcomas: An immunohistochemical study. Appl. Immunohistochem. Mol. Morphol. 2004, 12, 338–341. [Google Scholar] [CrossRef]
- Hussein, K.; Rath, B.; Ludewig, B.; Kreipe, H.; Jonigk, D. Clinico-pathological characteristics of different types of immunodeficiency-associated smooth muscle tumours. Eur. J. Cancer 2014, 50, 2417–2424. [Google Scholar] [CrossRef]
- Tan, C.S.; Loh, H.L.; Foo, M.W.; Choong, L.H.; Wong, K.S.; Kee, T.Y. Epstein-Barr virus-associated smooth muscle tumors after kidney transplantation: Treatment and outcomes in a single center. Clin. Transpl. 2013, 27, E462–E468. [Google Scholar] [CrossRef]
- Moore Dalal, K.; Antonescu, C.R.; Dematteo, R.P.; Maki, R.G. EBV-Associated Smooth Muscle Neoplasms: Solid Tumors Arising in the Presence of Immunosuppression and Autoimmune Diseases. Sarcoma 2008, 2008, 859407. [Google Scholar] [CrossRef] [PubMed]
- Magg, T.; Schober, T.; Walz, C.; Ley-Zaporozhan, J.; Facchetti, F.; Klein, C.; Hauck, F. Epstein-Barr Virus(+) Smooth Muscle Tumors as Manifestation of Primary Immunodeficiency Disorders. Front. Immunol. 2018, 9, 368. [Google Scholar] [CrossRef]
- Schneider, N.; Strauss, D.C.; Smith, M.J.; Miah, A.B.; Zaidi, S.; Benson, C.; van Houdt, W.J.; Jones, R.L.; Hayes, A.J.; Fisher, C.; et al. The Adequacy of Core Biopsy in the Assessment of Smooth Muscle Neoplasms of Soft Tissues: Implications for Treatment and Prognosis. Am. J. Surg. Pathol. 2017, 41, 923–931. [Google Scholar] [CrossRef]
- Wang, W.L.; Bones-Valentin, R.A.; Prieto, V.G.; Pollock, R.E.; Lev, D.C.; Lazar, A.J. Sarcoma metastases to the skin: A clinicopathologic study of 65 patients. Cancer 2012, 118, 2900–2904. [Google Scholar] [CrossRef]
- Billings, S.D.; Folpe, A.L.; Weiss, S.W. Do leiomyomas of deep soft tissue exist? An analysis of highly differentiated smooth muscle tumors of deep soft tissue supporting two distinct subtypes. Am. J. Surg. Pathol. 2001, 25, 1134–1142. [Google Scholar] [CrossRef]
- Widemann, B.C.; Italiano, A. Biology and Management of Undifferentiated Pleomorphic Sarcoma, Myxofibrosarcoma, and Malignant Peripheral Nerve Sheath Tumors: State of the Art and Perspectives. J. Clin. Oncol. 2018, 36, 160–167. [Google Scholar] [CrossRef]
- Fletcher, C.D.; Gustafson, P.; Rydholm, A.; Willen, H.; Akerman, M. Clinicopathologic re-evaluation of 100 malignant fibrous histiocytomas: Prognostic relevance of subclassification. J. Clin. Oncol. 2001, 19, 3045–3050. [Google Scholar] [CrossRef] [PubMed]
- Savina, M.; Le Cesne, A.; Blay, J.Y.; Ray-Coquard, I.; Mir, O.; Toulmonde, M.; Cousin, S.; Terrier, P.; Ranchere-Vince, D.; Meeus, P.; et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: The METASARC observational study. BMC Med. 2017, 15, 78. [Google Scholar] [CrossRef]
- Mustapar, N.; Zawawi, M.S.F.; Tuan Sharif, S.E. The Value of H3K27me3 Immunohistochemistry in Differentiating Malignant Peripheral Nerve Sheath Tumour with Its Histologic Mimickers. Asian Pac. J. Cancer Prev. 2020, 21, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, I.M.; Fletcher, C.D.; Hornick, J.L. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod. Pathol. 2016, 29, 4–13. [Google Scholar] [CrossRef]
- Otsuka, H.; Kohashi, K.; Yoshimoto, M.; Ishihara, S.; Toda, Y.; Yamada, Y.; Yamamoto, H.; Nakashima, Y.; Oda, Y. Immunohistochemical evaluation of H3K27 trimethylation in malignant peripheral nerve sheath tumors. Pathol. Res. Pract. 2018, 214, 417–425. [Google Scholar] [CrossRef]
- Schaefer, I.M.; Dong, F.; Garcia, E.P.; Fletcher, C.D.M.; Jo, V.Y. Recurrent SMARCB1 Inactivation in Epithelioid Malignant Peripheral Nerve Sheath Tumors. Am. J. Surg. Pathol. 2019, 43, 835–843. [Google Scholar] [CrossRef]
- Le Guellec, S.; Decouvelaere, A.V.; Filleron, T.; Valo, I.; Charon-Barra, C.; Robin, Y.M.; Terrier, P.; Chevreau, C.; Coindre, J.M. Malignant Peripheral Nerve Sheath Tumor Is a Challenging Diagnosis: A Systematic Pathology Review, Immunohistochemistry, and Molecular Analysis in 160 Patients From the French Sarcoma Group Database. Am. J. Surg. Pathol. 2016, 40, 896–908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.J.; Goldblum, J.R.; Pawel, B.R.; Fisher, C.; Pasha, T.L.; Barr, F.G. Immunophenotype of desmoplastic small round cell tumors as detected in cases with EWS-WT1 gene fusion product. Mod. Pathol. 2003, 16, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Lamhamedi-Cherradi, S.E.; Cuglievan, B.; Menegaz, B.A.; Camacho, P.; Huh, W.; Ramamoorthy, V.; Anderson, P.M.; Pollock, R.E.; Lev, D.C.; et al. Multimodality Treatment of Desmoplastic Small Round Cell Tumor: Chemotherapy and Complete Cytoreductive Surgery Improve Patient Survival. Clin. Cancer Res. 2018, 24, 4865–4873. [Google Scholar] [CrossRef]
- Machado, I.; Yoshida, A.; Morales, M.G.N.; Abrahao-Machado, L.F.; Navarro, S.; Cruz, J.; Lavernia, J.; Parafioriti, A.; Picci, P.; Llombart-Bosch, A. Review with novel markers facilitates precise categorization of 41 cases of diagnostically challenging, "undifferentiated small round cell tumors". A clinicopathologic, immunophenotypic and molecular analysis. Ann. Diagn. Pathol. 2018, 34, 1–12. [Google Scholar] [CrossRef]
- Folpe, A.L.; Goldblum, J.R.; Rubin, B.P.; Shehata, B.M.; Liu, W.; Dei Tos, A.P.; Weiss, S.W. Morphologic and immunophenotypic diversity in Ewing family tumors: A study of 66 genetically confirmed cases. Am. J. Surg. Pathol. 2005, 29, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Albergo, J.I.; Gaston, C.L.; Laitinen, M.; Darbyshire, A.; Jeys, L.M.; Sumathi, V.; Parry, M.; Peake, D.; Carter, S.R.; Tillman, R.; et al. Ewing′s sarcoma: Only patients with 100% of necrosis after chemotherapy should be classified as having a good response. Bone Jt. J. 2016, 98-B, 1138–1144. [Google Scholar] [CrossRef]
- Demicco, E.G.; Wagner, M.J.; Maki, R.G.; Gupta, V.; Iofin, I.; Lazar, A.J.; Wang, W.L. Risk assessment in solitary fibrous tumors: Validation and refinement of a risk stratification model. Mod. Pathol. 2017, 30, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Salas, S.; Resseguier, N.; Blay, J.Y.; Le Cesne, A.; Italiano, A.; Chevreau, C.; Rosset, P.; Isambert, N.; Soulie, P.; Cupissol, D.; et al. Prediction of local and metastatic recurrence in solitary fibrous tumor: Construction of a risk calculator in a multicenter cohort from the French Sarcoma Group (FSG) database. Ann. Oncol. 2017, 28, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.A.; Tao, D.; Marino-Enriquez, A. STAT6 is amplified in a subset of dedifferentiated liposarcoma. Mod. Pathol. 2014, 27, 1231–1237. [Google Scholar] [CrossRef]
- Creytens, D.; Libbrecht, L.; Ferdinande, L. Nuclear expression of STAT6 in dedifferentiated liposarcomas with a solitary fibrous tumor-like morphology: A diagnostic pitfall. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 462–463. [Google Scholar] [CrossRef]
- Pasini, B.; McWhinney, S.R.; Bei, T.; Matyakhina, L.; Stergiopoulos, S.; Muchow, M.; Boikos, S.A.; Ferrando, B.; Pacak, K.; Assie, G.; et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur. J. Hum. Genet. 2008, 16, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.A.; Carney, J.A. The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): Molecular genetics and clinical implications. J. Intern. Med. 2009, 266, 43–52. [Google Scholar] [CrossRef]
- Joensuu, H.; Wardelmann, E.; Sihto, H.; Eriksson, M.; Sundby Hall, K.; Reichardt, A.; Hartmann, J.T.; Pink, D.; Cameron, S.; Hohenberger, P.; et al. Effect of KIT and PDGFRA Mutations on Survival in Patients With Gastrointestinal Stromal Tumors Treated With Adjuvant Imatinib: An Exploratory Analysis of a Randomized Clinical Trial. JAMA Oncol. 2017, 3, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Debiec-Rychter, M.; Sciot, R.; Le Cesne, A.; Schlemmer, M.; Hohenberger, P.; van Oosterom, A.T.; Blay, J.Y.; Leyvraz, S.; Stul, M.; Casali, P.G.; et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur. J. Cancer 2006, 42, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Martignoni, G.; Pea, M.; Reghellin, D.; Zamboni, G.; Bonetti, F. PEComas: The past, the present and the future. Virchows Arch. 2008, 452, 119–132. [Google Scholar] [CrossRef]
- Folpe, A.L.; Kwiatkowski, D.J. Perivascular epithelioid cell neoplasms: Pathology and pathogenesis. Hum. Pathol. 2010, 41, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.B.; Berney, D.M.; Compérat, E.M.; Hartmann, A.; Menon, S.; Netto, G.J.; Raspollini, M.R.; Rubin, M.A.; Tickoo, S.K.; Turajlic, S.; et al. WHO Classification of Tumours: Urinary and Male Genital Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2022; Volume 8. [Google Scholar]
- Cho, J.; Kinsey, D.; Kimchi, E.T.; O’Carroll, K.S.; Nguyen, V.; Alsabbagh, M.; Gaballah, A. Retroperitoneal extra-adrenal myelolipoma misdiagnosed as liposarcoma: A case report. Radiol. Case Rep. 2021, 16, 364–368. [Google Scholar] [CrossRef]
- Katsimantas, A.; Filippou, D.; Melloy, A.; Paparidis, S.; Ferakis, N. Macroscopic Appearance of Giant Adrenal Myelolipoma During Laparoscopy: An Adjunct in Differential Diagnosis. Cureus 2020, 12, e6582. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.J.; Shi, J.; Ballew, B.; Hyland, P.L.; Li, W.Q.; Rotunno, M.; Alcorta, D.A.; Liebsch, N.J.; Mitchell, J.; Bass, S.; et al. Characterization of T gene sequence variants and germline duplications in familial and sporadic chordoma. Hum. Genet. 2014, 133, 1289–1297. [Google Scholar] [CrossRef]
- Lee-Jones, L.; Aligianis, I.; Davies, P.A.; Puga, A.; Farndon, P.A.; Stemmer-Rachamimov, A.; Ramesh, V.; Sampson, J.R. Sacrococcygeal chordomas in patients with tuberous sclerosis complex show somatic loss of TSC1 or TSC2. Genes Chromosomes Cancer 2004, 41, 80–85. [Google Scholar] [CrossRef]
- Presneau, N.; Shalaby, A.; Ye, H.; Pillay, N.; Halai, D.; Idowu, B.; Tirabosco, R.; Whitwell, D.; Jacques, T.S.; Kindblom, L.G.; et al. Role of the transcription factor T (brachyury) in the pathogenesis of sporadic chordoma: A genetic and functional-based study. J. Pathol. 2011, 223, 327–335. [Google Scholar] [CrossRef]
- Tarpey, P.S.; Behjati, S.; Young, M.D.; Martincorena, I.; Alexandrov, L.B.; Farndon, S.J.; Guzzo, C.; Hardy, C.; Latimer, C.; Butler, A.P.; et al. The driver landscape of sporadic chordoma. Nat. Commun. 2017, 8, 890. [Google Scholar] [CrossRef]
- Ross, J.A.; Severson, R.K.; Davis, S.; Brooks, J.J. Trends in the incidence of soft tissue sarcomas in the United States from 1973 through 1987. Cancer 1993, 72, 486–490. [Google Scholar] [CrossRef]
- Toro, J.R.; Travis, L.B.; Wu, H.J.; Zhu, K.; Fletcher, C.D.; Devesa, S.S. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int. J. Cancer 2006, 119, 2922–2930. [Google Scholar] [CrossRef]
- Meis-Kindblom, J.M.; Kindblom, L.G. Angiosarcoma of soft tissue: A study of 80 cases. Am. J. Surg. Pathol. 1998, 22, 683–697. [Google Scholar] [CrossRef]
- Vuletin, J.C.; Wajsbort, R.R.; Ghali, V. Primary retroperitoneal angiosarcoma with eosinophilic globules. A combined light-microscopic, immunohistochemical, and ultrastructural study. Arch. Pathol. Lab. Med. 1990, 114, 618–622. [Google Scholar] [PubMed]
- Rossi, S.; Fletcher, C.D. Angiosarcoma arising in hemangioma/vascular malformation: Report of four cases and review of the literature. Am. J. Surg. Pathol. 2002, 26, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, W.K.; Acs, G.; Simon, M.C.; Vaughn, D.J. HIF transcription factor expression and induction of hypoxic response genes in a retroperitoneal angiosarcoma. Anticancer Res. 2004, 24, 167–169. [Google Scholar]
- Lang, E.K.; Rudman, E.; Colon, I.; Macchia, R.J. Hematuria: The presenting symptom of an angiosarcoma of the inferior vena cava. J. Urol. 2009, 182, 2470. [Google Scholar] [CrossRef]
- Chiang, W.; Tynski, Z. Primary Peritoneal Angiosarcoma Metastatic to Liver and Bone without History of Radiation Therapy. Case Rep. Pathol. 2018, 2018, 1257284. [Google Scholar] [CrossRef]
- Jennings, T.A.; Peterson, L.; Axiotis, C.A.; Friedlaender, G.E.; Cooke, R.A.; Rosai, J. Angiosarcoma associated with foreign body material. A report of three cases. Cancer 1988, 62, 2436–2444. [Google Scholar] [CrossRef]
- Mentzel, T.; Katenkamp, D. Intraneural angiosarcoma and angiosarcoma arising in benign and malignant peripheral nerve sheath tumours: Clinicopathological and immunohistochemical analysis of four cases. Histopathology 1999, 35, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Malagon, H.D.; Valdez, A.M.; Moran, C.A.; Suster, S. Germ cell tumors with sarcomatous components: A clinicopathologic and immunohistochemical study of 46 cases. Am. J. Surg. Pathol. 2007, 31, 1356–1362. [Google Scholar] [CrossRef]
- Manner, J.; Radlwimmer, B.; Hohenberger, P.; Mossinger, K.; Kuffer, S.; Sauer, C.; Belharazem, D.; Zettl, A.; Coindre, J.M.; Hallermann, C.; et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am. J. Pathol. 2010, 176, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Zhang, L.; Chang, N.E.; Singer, S.; Maki, R.G.; Antonescu, C.R. Consistent MYC and FLT4 gene amplification in radiation-induced angiosarcoma but not in other radiation-associated atypical vascular lesions. Genes Chromosomes Cancer 2011, 50, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Chen, C.L.; Thomas, R.; Breen, M.; Bonnet, F.; Sevenet, N.; Longy, M.; Maki, R.G.; Coindre, J.M.; Antonescu, C.R. Alterations of the p53 and PIK3CA/AKT/mTOR pathways in angiosarcomas: A pattern distinct from other sarcomas with complex genomics. Cancer 2012, 118, 5878–5887. [Google Scholar] [CrossRef] [PubMed]
- Guillou, L.; Aurias, A. Soft tissue sarcomas with complex genomic profiles. Virchows Arch. 2010, 456, 201–217. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mack, T.; Purgina, B. Updates in Pathology for Retroperitoneal Soft Tissue Sarcoma. Curr. Oncol. 2022, 29, 6400-6418. https://doi.org/10.3390/curroncol29090504
Mack T, Purgina B. Updates in Pathology for Retroperitoneal Soft Tissue Sarcoma. Current Oncology. 2022; 29(9):6400-6418. https://doi.org/10.3390/curroncol29090504
Chicago/Turabian StyleMack, Tanner, and Bibianna Purgina. 2022. "Updates in Pathology for Retroperitoneal Soft Tissue Sarcoma" Current Oncology 29, no. 9: 6400-6418. https://doi.org/10.3390/curroncol29090504
APA StyleMack, T., & Purgina, B. (2022). Updates in Pathology for Retroperitoneal Soft Tissue Sarcoma. Current Oncology, 29(9), 6400-6418. https://doi.org/10.3390/curroncol29090504