A 3-miRNA Signature Enables Risk Stratification in Glioblastoma Multiforme Patients with Different Clinical Outcomes

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mixed-Methods Content Analysis
2.2. Glioblastoma Multiforme Patient Cohort and Clinical Samples
2.3. Immunohistochemical Analysis
2.4. MiRNA Extraction
2.5. cDNA Synthesis
2.6. Quantitative Real-Time PCR (qRT-PCR)
2.7. Statistical Analysis
2.7.1. MiRNA Signature Analysis
2.7.2. Survival Analysis
2.8. Discriminant Analysis
2.8.1. MiRNA Raw Data
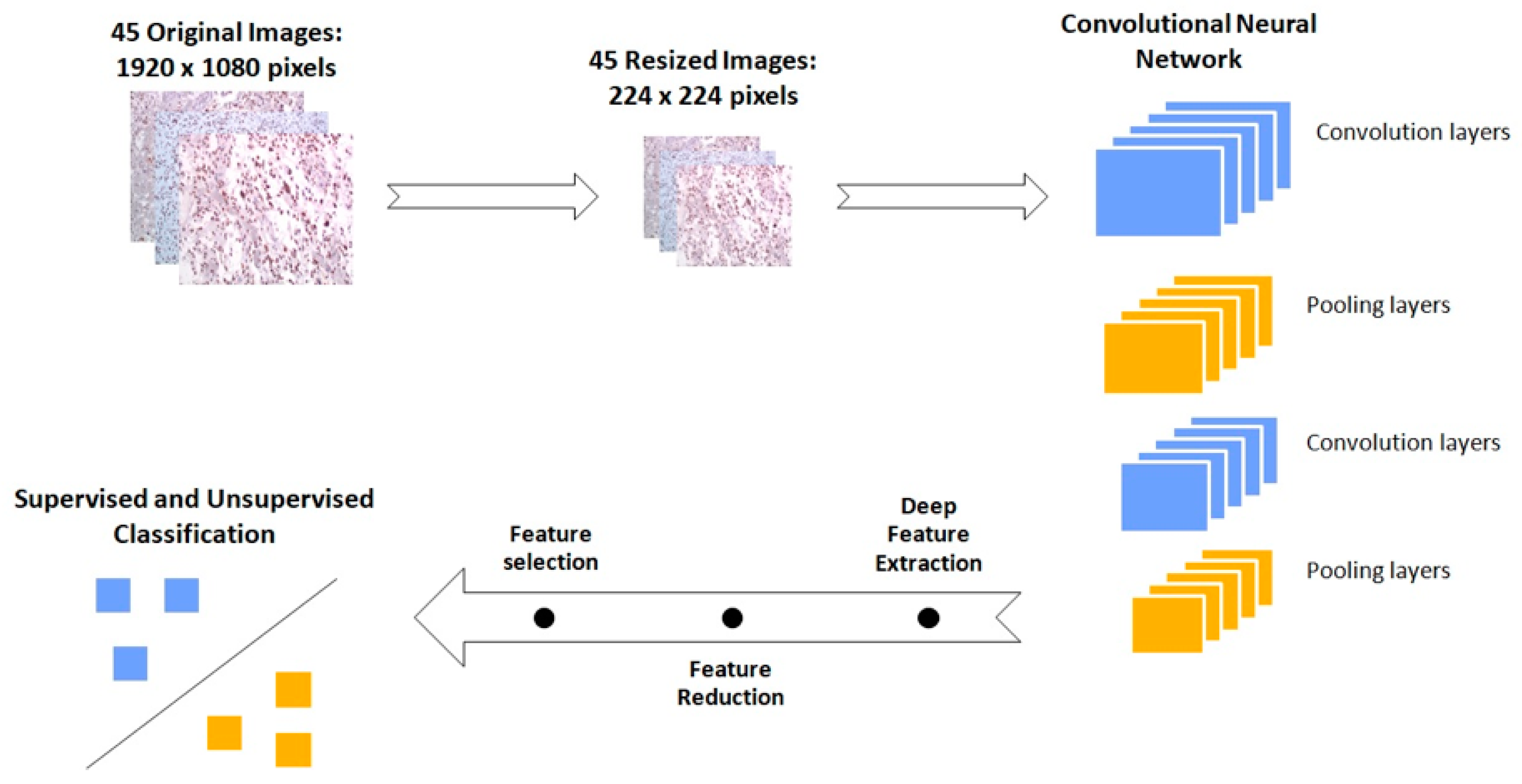
2.8.2. IHC Images
3. Results
3.1. A Mechanistic View of Our 3-miRNA Signature in GBM
3.2. Deciphering Age and Sex Dependence in Age- and Sex-Matched Standard-of-Care Treated Patient Cohort
3.3. The 3-miRNA Signature Expression May Stratify the Standard-of-Care Treated Patients to Lower (OS > 12 Months) vs. Higher (OS < 12 Months) Risk Groups
3.4. In Silico Validation of the 3-miRNA Signature
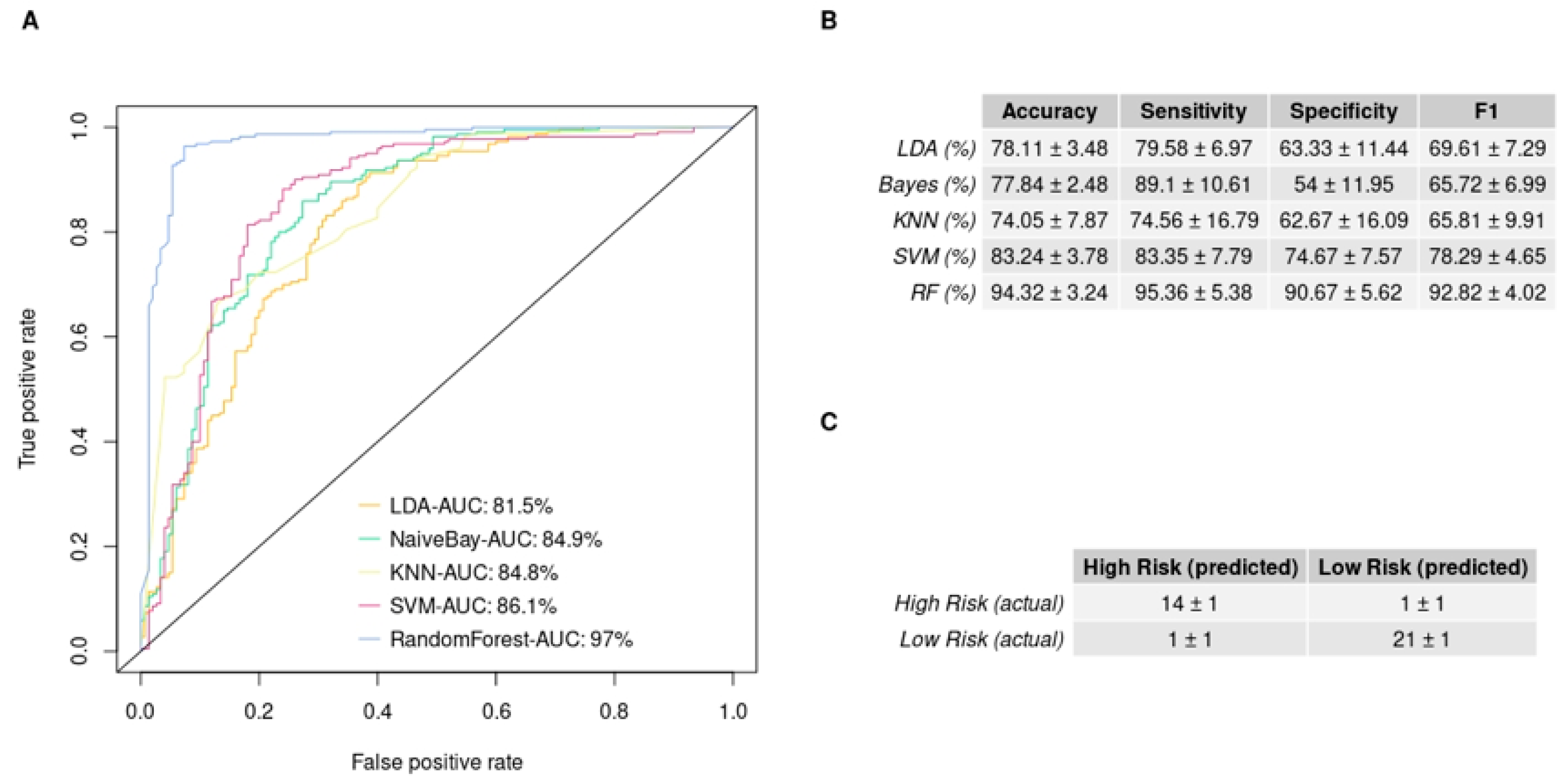
3.5. GBM IHC Images Can Be Discriminated into Two Groups, Depending on Overall Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reni, M.; Mazza, E.; Zanon, S.; Gatta, G.; Vecht, C.J. Central Nervous System Gliomas. Crit. Rev. Oncol. Hematol. 2017, 113, 213–234. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.T.; Hochheimer, C.J.; Rock, A.K.; Dincer, A.; Ravindra, L.; Zhang, F.L.; Opalak, C.F.; Poulos, N.; Sima, A.P.; Broaddus, W.C. Comorbid Medical Conditions as Predictors of Overall Survival in Glioblastoma Patients. Sci. Rep. 2019, 9, 20018. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Brada, M.; van den Bent, M.J.; Tonn, J.C.; Pentheroudakis, G. High-Grade Glioma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2014, 25, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; le Rhun, E.; Preusser, M.; Tonn, J.C.; Roth, P. How We Treat Glioblastoma. ESMO Open 2019, 4, e000520. [Google Scholar] [CrossRef]
- Seyhan, A.A.; Carini, C. Are Innovation and New Technologies in Precision Medicine Paving a New Era in Patients Centric Care? J. Transl. Med. 2019, 17, 114. [Google Scholar] [CrossRef]
- Anjum, K.; Shagufta, B.I.; Abbas, S.Q.; Patel, S.; Khan, I.; Shah, S.A.A.; Akhter, N.; ul Hassan, S.S. Current Status and Future Therapeutic Perspectives of Glioblastoma Multiforme (GBM) Therapy: A Review. Biomed. Pharmacother. 2017, 92, 681–689. [Google Scholar] [CrossRef]
- Toraih, E.A.; Aly, N.M.; Abdallah, H.Y.; Al-Qahtani, S.A.; Shaalan, A.A.M.; Hussein, M.H.; Fawzy, M.S. MicroRNA–Target Cross-Talks: Key Players in Glioblastoma Multiforme. Tumor Biol. 2017, 39, 1010428317726842. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhang, H.; Liu, X.; Lu, Z.; Li, G.; Lu, M.; Tao, X. MicroRNA Signatures Predict Prognosis of Patients with Glioblastoma Multiforme through the Cancer Genome Atlas. Oncotarget 2017, 8, 58386. [Google Scholar] [CrossRef]
- Unger, K.; Fleischmann, D.F.; Ruf, V.; Felsberg, J.; Piehlmaier, D.; Samaga, D.; Hess, J.; Suresh, M.P.; Mittelbronn, M.; Lauber, K.; et al. Improved Risk Stratification in Younger IDH Wild-Type Glioblastoma Patients by Combining a 4-MiRNA Signature with MGMT Promoter Methylation Status. Neurooncol. Adv. 2020, 2, vdaa137. [Google Scholar] [CrossRef]
- Theodosi, A.; Ouzounis, S.; Kostopoulos, S.; Glotsos, D.; Kalatzis, I.; Tzelepi, V.; Ravazoula, P.; Asvestas, P.; Cavouras, D.; Sakellaropoulos, G. Design of a Hybrid Deep Learning System for Discriminating between Low- and High-Grade Colorectal Cancer Lesions, Using Microscopy Images of IHC Stained for AIB1 Expression Biopsy Material. Mach. Vis. Appl. 2021, 32, 58. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Therneau, T.M. Survival Analysis [R Package Survival Version 3.2-11]. Available online: https://cran.r-project.org/web/packages/survival/index.html (accessed on 20 June 2021).
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2000. [Google Scholar]
- Package “survminer” Type Package Title Drawing Survival Curves Using “Ggplot2”. Available online: https://cran.r-project.org/web/packages/survminer/index.html (accessed on 20 June 2021).
- Næs, T.; Mevik, B.-H. Understanding the Collinearity Problem in Regression and Discriminant Analysis. J. Chemom. 2001, 15, 413–426. [Google Scholar] [CrossRef]
- Ambroise, C.; McLachlan, G.J. Selection Bias in Gene Extraction on the Basis of Microarray Gene-Expression Data. Proc. Natl. Acad. Sci. USA 2002, 99, 6562–6566. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. Building Predictive Models in R Using the Caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Xu, Y.; Jia, Z.; Wang, L.-B.; Ai, Y.; Zhang, F.; Lai, M.; Chang, E.I.-C. Large Scale Tissue Histopathology Image Classification, Segmentation, and Visualization via Deep Convolutional Activation Features. BMC Bioinform. 2017, 18, 281. [Google Scholar] [CrossRef]
- Yonekura, A.; Kawanaka, H.; Prasath, V.B.S.; Aronow, B.J.; Takase, H. Automatic Disease Stage Classification of Glioblastoma Multiforme Histopathological Images Using Deep Convolutional Neural Network. Biomed. Eng. Lett. 2018, 8, 321. [Google Scholar] [CrossRef]
- Simonyan, K.; Zisserman, A. Very Deep Convolutional Networks for Large-Scale Image Recognition. In Proceedings of the 3rd International Conference on Learning Representations, ICLR 2015—Conference Track Proceedings 2014, San Diego, CA, USA, 7–9 May 2015. [Google Scholar]
- GitHub—Keras-Team/Keras: Deep Learning for Humans. Available online: https://github.com/keras-team/keras (accessed on 20 June 2021).
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. TensorFlow: A System for Large-Scale Machine Learning. In Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI ’16), Savannah, GA, USA, 2–4 November 2016. [Google Scholar]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene Selection for Cancer Classification Using Support Vector Machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Müller, A.; Nothman, J.; Louppe, G.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2012, 12, 2825–2830. [Google Scholar]
- Wang, Z.; Wang, B.; Shi, Y.; Xu, C.; Xiao, H.L.; Ma, L.N.; Xu, S.L.; Yang, L.; Wang, Q.L.; Dang, W.Q.; et al. Oncogenic MiR-20a and MiR-106a Enhance the Invasiveness of Human Glioma Stem Cells by Directly Targeting TIMP-2. Oncogene 2015, 34, 1407–1419. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, N.; Liu, B.; Huang, Y.; Zeng, H.; Yang, Z.; He, Z.; Guo, H. Long Noncoding RNA RP11-838N2.4 Enhances the Cytotoxic Effects of Temozolomide by Inhibiting the Functions of MiR-10a in Glioblastoma Cell Lines. Oncotarget 2016, 7, 43835. [Google Scholar] [CrossRef] [PubMed]
- Maimon, O.; Rokach, L. Introduction to Knowledge Discovery and Data Mining. In Data Mining and Knowledge Discovery Handbook; Springer: Boston, MA, USA, 2009; pp. 1–15. [Google Scholar]
- Henriksen, M.; Johnsen, K.B.; Olesen, P.; Pilgaard, L.; Duroux, M. MicroRNA Expression Signatures and Their Correlation with Clinicopathological Features in Glioblastoma Multiforme. Neuromol. Med. 2014, 16, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Delen, E.; Doganlar, O.; Doganlar, Z.B.; Delen, O. Inhibition of the Invasion of Human Glioblastoma U87 Cell Line by Ruxolitinib: A Molecular Player of MiR-17 and MiR-20a Regulating JAK/STAT Pathway. Turk. Neurosurg. 2020, 30, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.; Tang, W.; Pan, C.; Hu, X.; Hong, Y. Long Non-Coding RNA TUSC7 Inhibits Temozolomide Resistance by Targeting MiR-10a in Glioblastoma. Cancer Chemother. Pharmacol. 2018, 81, 671–678. [Google Scholar] [CrossRef]
- Visani, M.; de Biase, D.; Marucci, G.; Cerasoli, S.; Nigrisoli, E.; Letizia, M.; Reggiani, B.; Albani, F.; Baruzzi, A.; Pession, A. Expression of 19 MicroRNAs in Glioblastoma and Comparison with Other Brain Neoplasia of Grades IeIII. Mol. Oncol. 2013, 8, 417–430. [Google Scholar] [CrossRef]
- Hermansen, S.K.; Sørensen, M.D.; Hansen, A.; Knudsen, S.; Alvarado, A.G.; Lathia, J.D.; Kristensen, B.W. A 4-MiRNA Signature to Predict Survival in Glioblastomas. PLoS ONE 2017, 12, e0188090. [Google Scholar] [CrossRef]
- Marziali, G.; Buccarelli, M.; Giuliani, A.; Ilari, R.; Grande, S.; Palma, A.; D’Alessandris, Q.G.; Martini, M.; Biffoni, M.; Pallini, R.; et al. A Three-MicroRNA Signature Identifies Two Subtypes of Glioblastoma Patients with Different Clinical Outcomes. Mol. Oncol. 2017, 11, 1115–1129. [Google Scholar] [CrossRef][Green Version]
- Labib, E.M.; Ezz El Arab, L.R.; Ghanem, H.M.; Hassan, R.E.; Swellam, M. Relevance of Circulating MiRNA-21 and MiRNA-181 in Prediction of Glioblastoma Multiforme Prognosis. Arch. Physiol. Biochem. 2020, 1–6. [Google Scholar] [CrossRef]
- Palumbo, R.; Pasculli, O.; Galasso, B.; Volinia, M. A MiRNA Signature for Defining Aggressive Phenotype and Prognosis in Gliomas. PLoS ONE 2014, 9, e108950. [Google Scholar] [CrossRef]
- Niyazi, M.; Zehentmayr, F.; Niemöller, O.M.; Eigenbrod, S.; Kretzschmar, H.; Schulze-Osthoff, K.; Tonn, J.-C.; Atkinson, M.; Mörtl, S.; Belka, C. MiRNA Expression Patterns Predict Survival in Glioblastoma. Radiat. Oncol. 2011, 6, 153. [Google Scholar] [CrossRef]
- Li, R.; Gao, K.; Luo, H.; Wang, X.; Shi, Y.; Dong, Q.; Luan, W.; You, Y. Identification of Intrinsic Subtype-Specific Prognostic MicroRNAs in Primary Glioblastoma. J. Exp. Clin. Cancer Res. 2014, 33, 9. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Machado Da Silva, A.; Calin, G.; Pantel, K. Data Normalization Strategies for MicroRNA Quantification. Clin. Chem. 2015, 61, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Ballman, K.V.; Buckner, J.C.; Brown, P.D.; Giannini, C.; Flynn, P.J.; Laplant, B.R.; Jaeckle, K.A.; Ballman, K.; Clinic, M. The Relationship between Six-Month Progression-Free Survival and 12-Month Overall Survival End Points for Phase II Trials in Patients with Glioblastoma Multiforme. J. Neurooncol. 2007, 9, 29–38. [Google Scholar] [CrossRef]
- Kudulaiti, N.; Zhou, Z.; Luo, C.; Zhang, J.; Zhu, F.; Wu, J. A nomogram for individualized prediction of overall survival in patients with newly diagnosed glioblastoma: A real-world retrospective cohort study. BMC Surg. 2021, 21, 238. [Google Scholar] [CrossRef] [PubMed]
- Chahal, M.; Thiessen, B.; Mariano, C. Treatment of Older Adult Patients with Glioblastoma: Moving towards the Inclusion of a Comprehensive Geriatric Assessment for Guiding Management. Curr. Oncol. 2022, 29, 360–376. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Shen, J.; Hodges, T.R.; Song, R.; Fuller, G.N.; Heimberger, A.B. Serum MicroRNA Profiling in Patients with Glioblastoma: A Survival Analysis. Mol. Cancer 2017, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Patric, I.; Somasundaram, K. A Ten-MicroRNA Expression Signature Predicts Survival in Glioblastoma. PLoS ONE 2011, 6, e17438. [Google Scholar] [CrossRef]
- Conroy, S.; Kruyt, F.A.E.; Joseph, J.V.; Balasubramaniyan, V.; Bhat, K.P.; Wagemakers, M.; Enting, R.H.; Walenkamp, A.M.E.; den Dunnen, W.F.A. Subclassification of Newly Diagnosed Glioblastomas through an Immunohistochemical Approach. PLoS ONE 2014, 9, e115687. [Google Scholar] [CrossRef]
- Tiwari, X.P.; Prasanna, X.P.; Wolansky, X.L.; Pinho, X.M.; Cohen, X.M.; Nayate, X.A.P.; Gupta, X.A.; Singh, X.G.; Hatanpaa, X.K.J.; Sloan, X.A.; et al. Computer-Extracted Texture Features to Distinguish Cerebral Radionecrosis from Recurrent Brain Tumors on Multiparametric MRI: A Feasibility Study. AJNR Am. J. Neuroradiol. 2016, 37, 2231–2236. [Google Scholar] [CrossRef]
- Korfiatis, P.; Kline, T.L.; Coufalova, L.; Lachance, D.H.; Parney, I.F.; Carter, R.E.; Buckner, J.C.; Erickson, B.J. MRI Texture Features as Biomarkers to Predict MGMT Methylation Status in Glioblastomas. J. Med. Phys. 2016, 43, 2835–2844. [Google Scholar] [CrossRef]
- Jolliffe, I.T. A Note on the Use of Principal Components in Regression. J. Appl. Stat. 1982, 31, 300. [Google Scholar] [CrossRef]
- Rønning, P.A.; Helseth, E.; Meling, T.R.; Johannesen, T.B. A population-based study on the effect of temozolomide in the treatment of glioblastoma multiforme. Neuro. Oncol. 2012, 14, 1178–1184. [Google Scholar] [CrossRef]
- Gorlia, T.; van den Bent, M.J.; Hegi, M.E.; Mirimanoff, R.O.; Weller, M.; Cairncross, J.G.; Eisenhauer, E.; Belanger, K.; Brandes, A.A.; Allgeier, A.; et al. Nomograms for predicting survival of patients with newly diagnosed glioblastoma: Prognostic factor analysis of EORTC and NCIC trial 26981–22981/CE.3. Lancet Oncol. 2008, 9, 29–38. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; Van Den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Helseth, R.; Helseth, E.; Johannesen, T.B.; Langberg, C.W.; Lote, K.; Rønning, P.; Scheie, D.; Vik, A.; Meling, T.R. Overall survival, prognostic factors, and repeated surgery in a consecutive series of 516 patients with glioblastoma multiforme. Acta Neurol. Scand. 2010, 122, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, P.R. Observational Studies, 2nd ed.; Springer: New York, NY, USA, 2002. [Google Scholar]
- Katsila, T.; Matsoukas, M.T.; Patrinos, G.P.; Kardamakis, D. Pharmacometabolomics Informs Quantitative Radiomics for Glioblastoma Diagnostic Innovation. OMICS 2017, 21, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Katsila, T.; Kardamakis, D. The Role of MicroRNAs in Gliomas–Therapeutic Implications. Curr. Mol. Pharmacol. 2021, 14, 1004–1012. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bafiti, V.; Ouzounis, S.; Chalikiopoulou, C.; Grigorakou, E.; Grypari, I.M.; Gregoriou, G.; Theofanopoulos, A.; Panagiotopoulos, V.; Prodromidi, E.; Cavouras, D.; et al. A 3-miRNA Signature Enables Risk Stratification in Glioblastoma Multiforme Patients with Different Clinical Outcomes. Curr. Oncol. 2022, 29, 4315-4331. https://doi.org/10.3390/curroncol29060345
Bafiti V, Ouzounis S, Chalikiopoulou C, Grigorakou E, Grypari IM, Gregoriou G, Theofanopoulos A, Panagiotopoulos V, Prodromidi E, Cavouras D, et al. A 3-miRNA Signature Enables Risk Stratification in Glioblastoma Multiforme Patients with Different Clinical Outcomes. Current Oncology. 2022; 29(6):4315-4331. https://doi.org/10.3390/curroncol29060345
Chicago/Turabian StyleBafiti, Vivi, Sotiris Ouzounis, Constantina Chalikiopoulou, Eftychia Grigorakou, Ioanna Maria Grypari, Gregory Gregoriou, Andreas Theofanopoulos, Vasilios Panagiotopoulos, Evangelia Prodromidi, Dionisis Cavouras, and et al. 2022. "A 3-miRNA Signature Enables Risk Stratification in Glioblastoma Multiforme Patients with Different Clinical Outcomes" Current Oncology 29, no. 6: 4315-4331. https://doi.org/10.3390/curroncol29060345
APA StyleBafiti, V., Ouzounis, S., Chalikiopoulou, C., Grigorakou, E., Grypari, I. M., Gregoriou, G., Theofanopoulos, A., Panagiotopoulos, V., Prodromidi, E., Cavouras, D., Zolota, V., Kardamakis, D., & Katsila, T. (2022). A 3-miRNA Signature Enables Risk Stratification in Glioblastoma Multiforme Patients with Different Clinical Outcomes. Current Oncology, 29(6), 4315-4331. https://doi.org/10.3390/curroncol29060345