Comprehensive Genomic Profiling of Circulating Tumor DNA in Patients with Previously Treated Metastatic Colorectal Cancer: Analysis of a Real-World Healthcare Claims Database


Abstract
:1. Introduction
2. Methods
2.1. Ethics
2.2. Data Source and Extraction
2.3. Study Population
2.4. Lines of Therapy
2.5. Genomic Profiling
2.6. Patient Outcomes
2.7. Statistical Analysis
3. Results
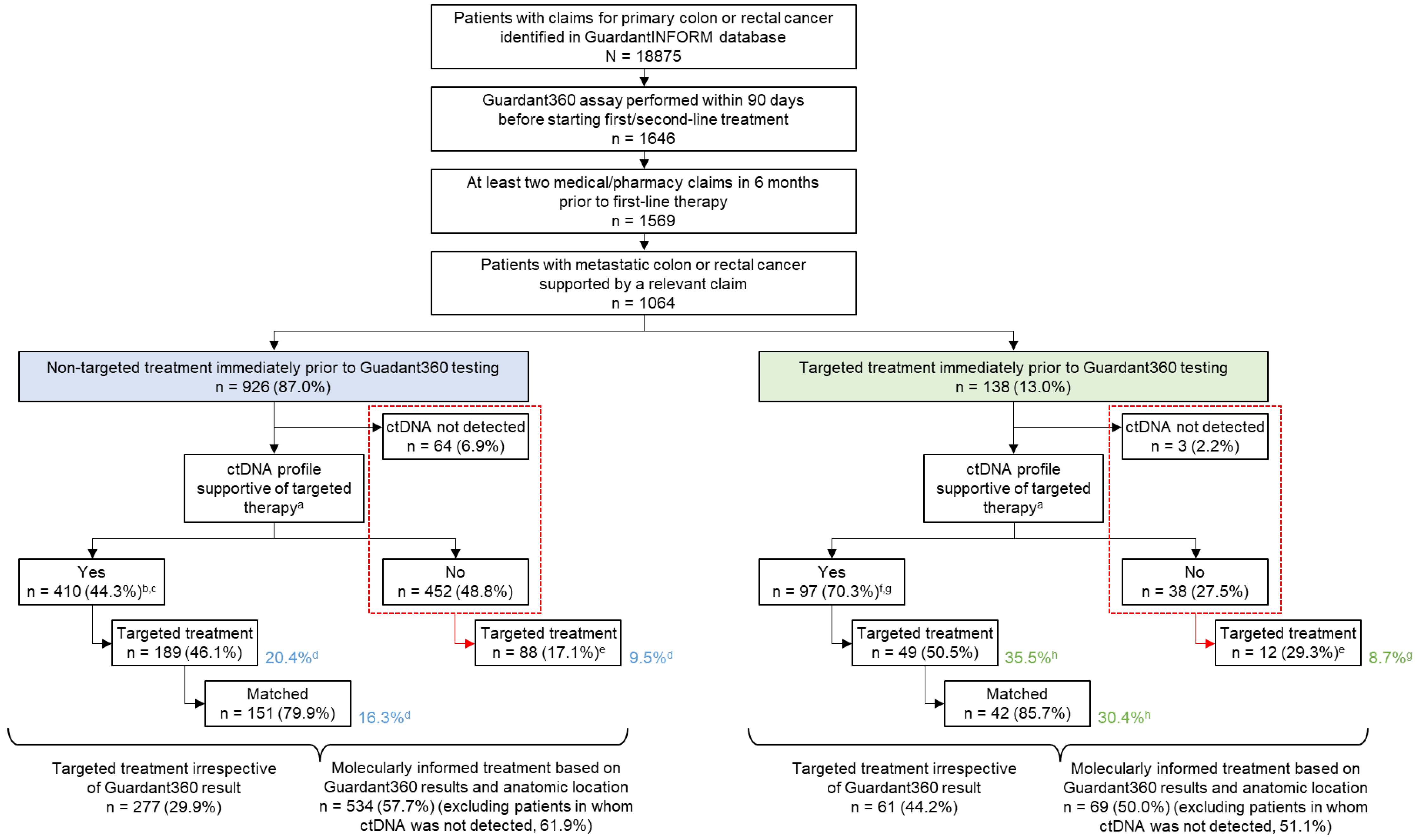
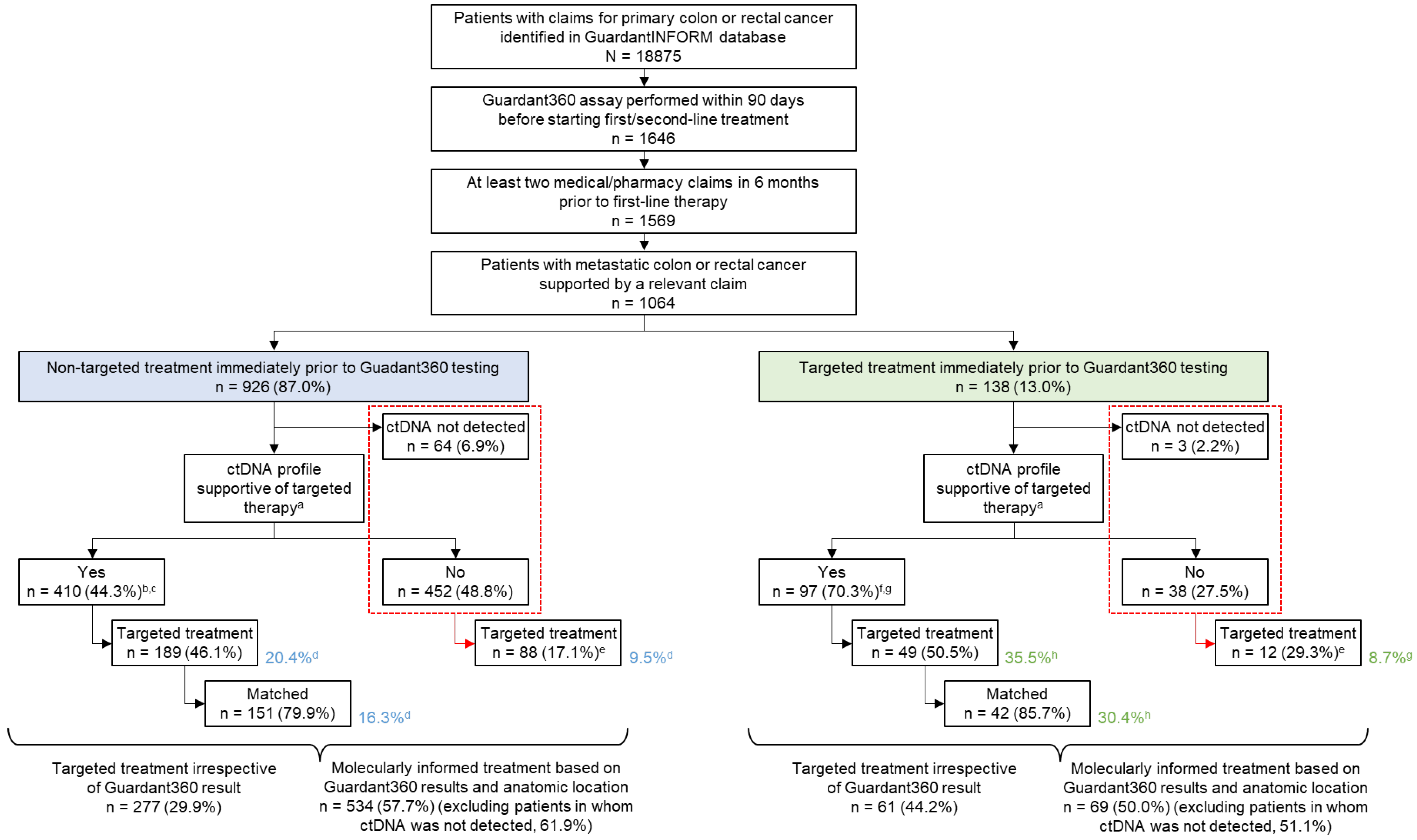
3.1. Patients
3.2. Overview of Alterations Detected
3.3. Evaluation of Molecularly Informed Therapy
3.4. Types of Treatments Administered after ctDNA Testing
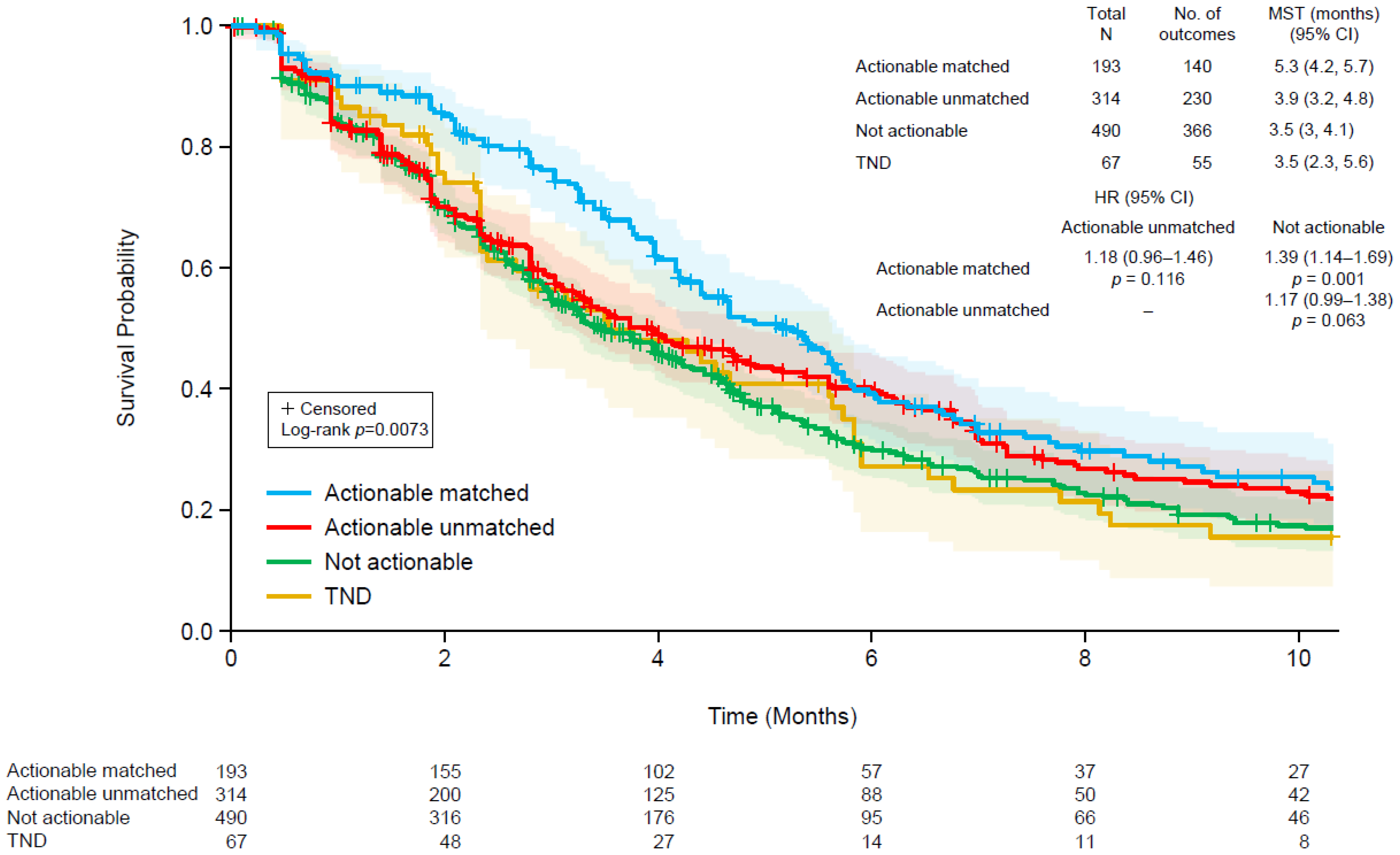
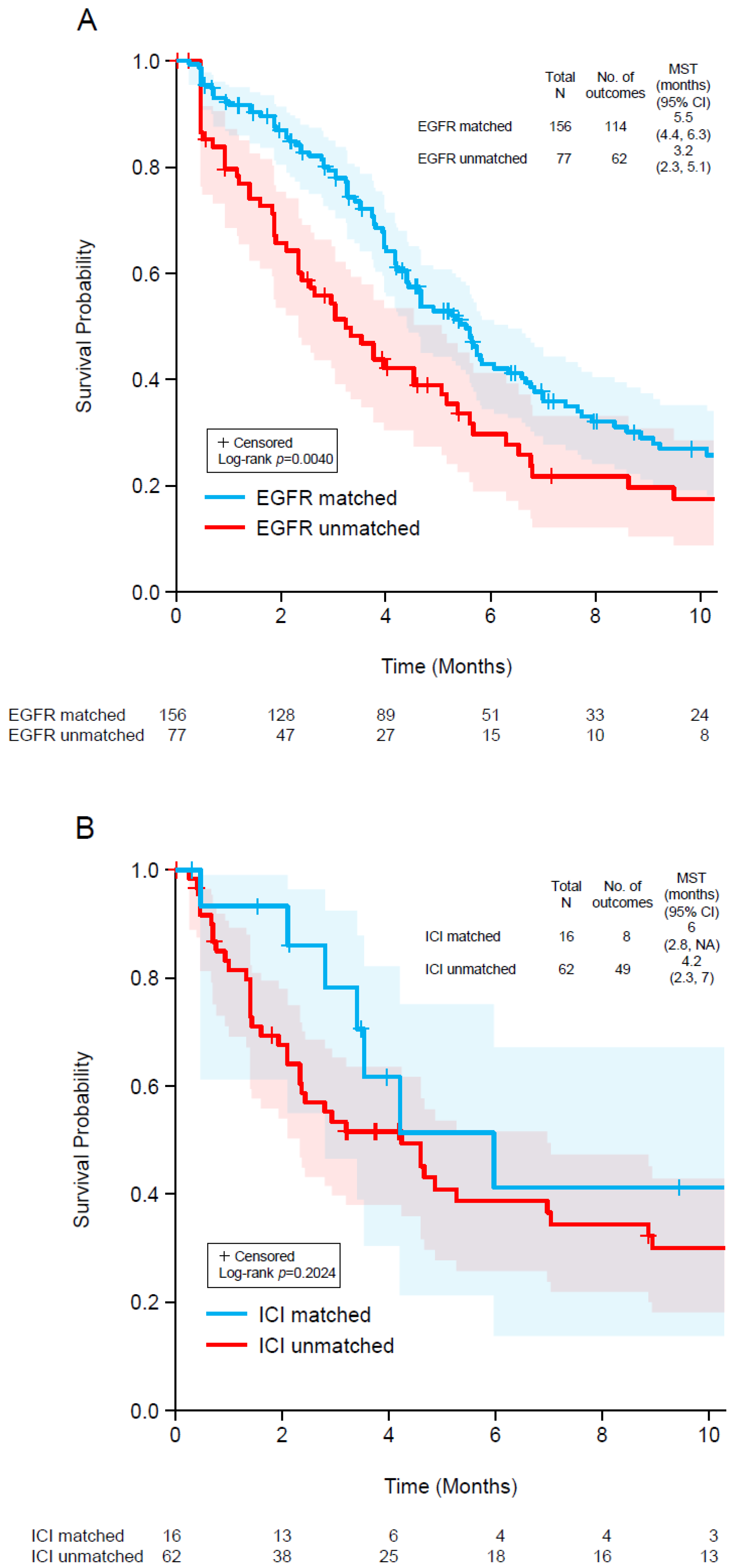
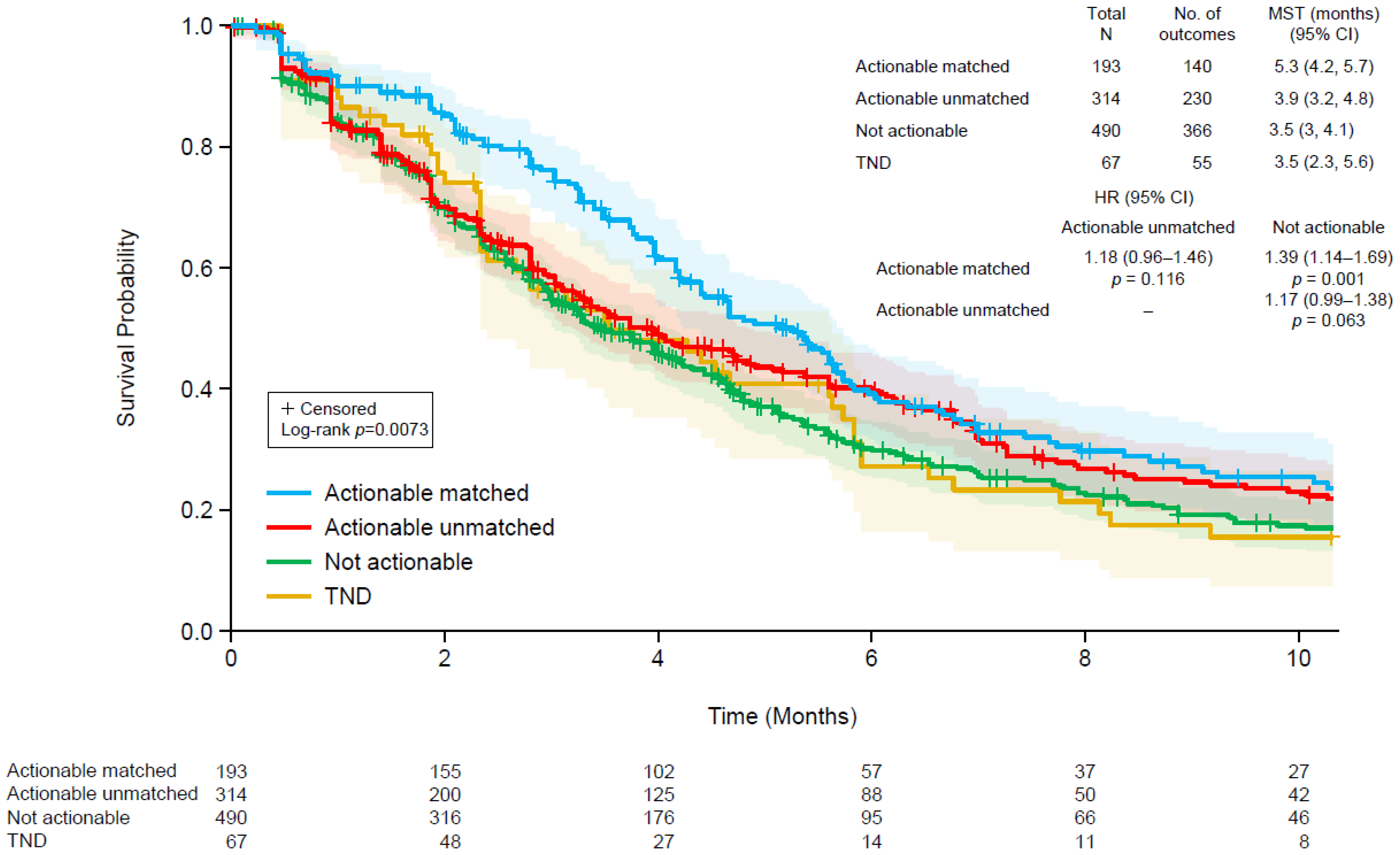
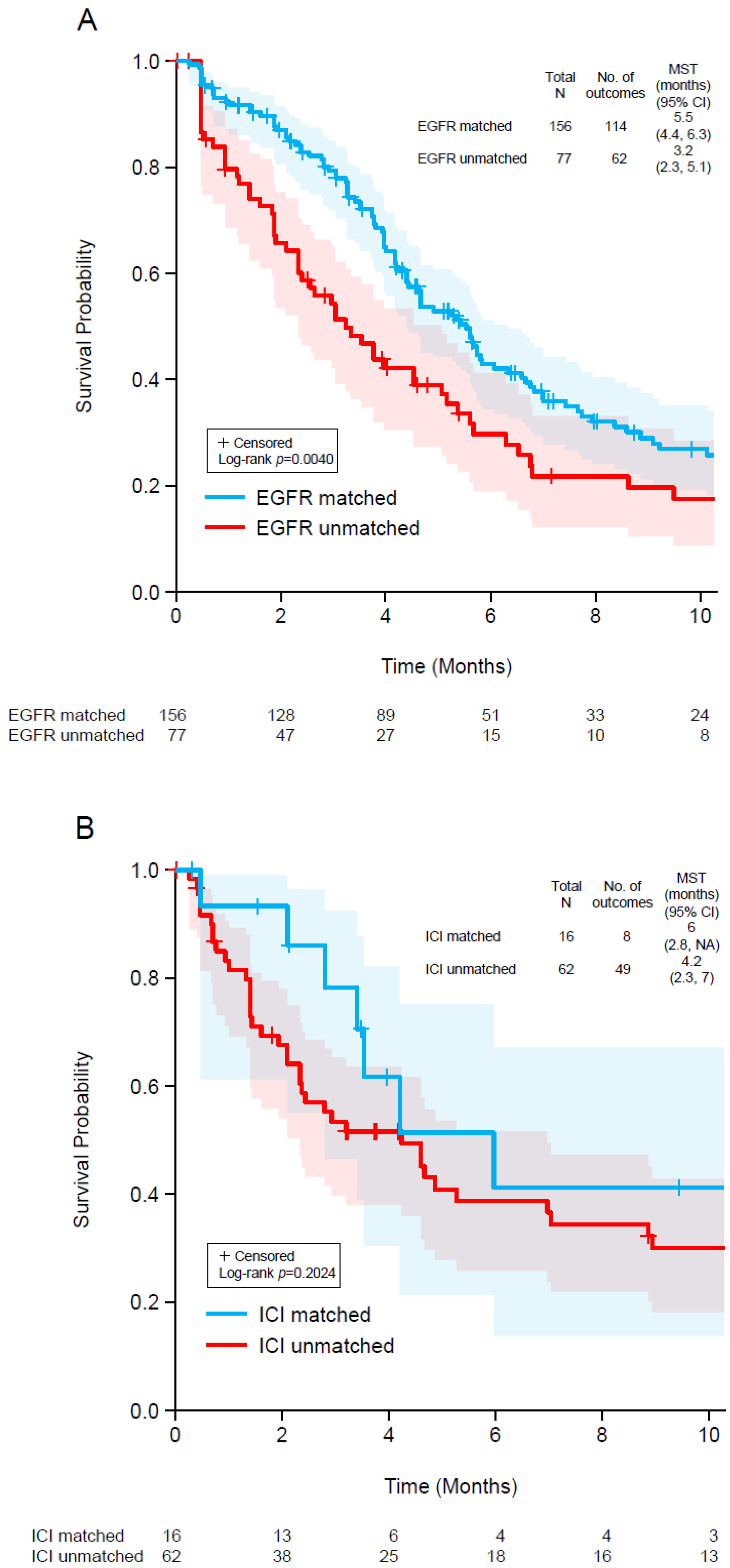
3.5. Patient Outcomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alexovič, M.; Urban, P.L.; Tabani, H.; Sabo, J. Recent advances in robotic protein sample preparation for clinical analysis and other biomedical applications. Clin. Chim. Acta 2020, 507, 104–116. [Google Scholar] [CrossRef]
- Ou, F.S.; Michiels, S.; Shyr, Y.; Adjei, A.A.; Oberg, A.L. Biomarker discovery and validation: Statistical considerations. J. Thorac. Oncol. 2021, 16, 537–545. [Google Scholar] [CrossRef]
- Rogers, J.E.; Dasari, A. Pharmacotherapy for unresectable metastatic colorectal cancer. Expert Opin. Pharm. 2022, 23, 211–220. [Google Scholar] [CrossRef]
- Yu, I.S.; Kopetz, S. The emergence of targetable pathways in colorectal cancer. Clin. Adv. Hematol. Oncol. 2021, 19, 774–783. [Google Scholar]
- De Roock, W.; De Vriendt, V.; Normanno, N.; Ciardiello, F.; Tejpar, S. KRAS, BRAF, PIK3CA, and PTEN mutations: Implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011, 12, 594–603. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Köhne, C.H.; Ciardiello, F.; Lenz, H.J.; Heinemann, V.; Klinkhardt, U.; Beier, F.; Duecker, K.; van Krieken, J.H.; Tejpar, S. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur. J. Cancer 2015, 51, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douillard, J.Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Final results from PRIME: Randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann. Oncol. 2014, 25, 1346–1355. [Google Scholar] [CrossRef]
- Kawazoe, A.; Shitara, K.; Fukuoka, S.; Kuboki, Y.; Bando, H.; Okamoto, W.; Kojima, T.; Fuse, N.; Yamanaka, T.; Doi, T.; et al. A retrospective observational study of clinicopathological features of KRAS, NRAS, BRAF and PIK3CA mutations in Japanese patients with metastatic colorectal cancer. BMC Cancer 2015, 15, 258. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Köhne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Lenz, H.J.; Köhne, C.H.; Heinemann, V.; Tejpar, S.; Melezínek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zheng, J.; Yang, Y.; Lu, J.; Gao, J.; Lu, T.; Sun, J.; Jiang, H.; Zhu, Y.; Zheng, Y.; et al. Molecular spectrum of KRAS, NRAS, BRAF and PIK3CA mutations in Chinese colorectal cancer patients: Analysis of 1110 cases. Sci. Rep. 2015, 5, 18678. [Google Scholar] [CrossRef]
- Lièvre, A.; Laurent-Puig, P. Toward an individualizing therapy for colorectal cancer: The example of the anti-EGFR monoclonal antibodies. Per. Med. 2009, 6, 145–157. [Google Scholar] [CrossRef]
- Stein, A.; Bokemeyer, C. How to select the optimal treatment for first line metastatic colorectal cancer. World J. Gastroenterol. 2014, 20, 899–907. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Siravegna, G.; Sartore-Bianchi, A.; Nagy, R.J.; Raghav, K.; Odegaard, J.I.; Lanman, R.B.; Trusolino, L.; Marsoni, S.; Siena, S.; Bardelli, A. Plasma HER2 (ERBB2) Copy Number Predicts Response to HER2-targeted Therapy in Metastatic Colorectal Cancer. Clin. Cancer Res. 2019, 25, 3046–3053. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Okamoto, W.; Kato, T.; Esaki, T.; Kato, K.; Komatsu, Y.; Yuki, S.; Masuishi, T.; Nishina, T.; Ebi, H.; et al. Circulating tumor DNA-guided treatment with pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer: A phase 2 trial. Nat. Med. 2021, 27, 1899–1903. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Colon Cancer Version 3.2021—10 September 2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf. (accessed on 17 February 2022).
- Parseghian, C.M.; Napolitano, S.; Loree, J.M.; Kopetz, S. Mechanisms of Innate and Acquired Resistance to Anti-EGFR Therapy: A Review of Current Knowledge with a Focus on Rechallenge Therapies. Clin. Cancer Res. 2019, 25, 6899–6908. [Google Scholar] [CrossRef]
- Piawah, S.; Venook, A.P. Targeted therapy for colorectal cancer metastases: A review of current methods of molecularly targeted therapy and the use of tumor biomarkers in the treatment of metastatic colorectal cancer. Cancer 2019, 125, 4139–4147. [Google Scholar] [CrossRef]
- Shuford, R.A.; Cairns, A.L.; Moaven, O. Precision Approaches in the Management of Colorectal Cancer: Current Evidence and Latest Advancements Towards Individualizing the Treatment. Cancers 2020, 12, 3481. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hurwitz, H.; Raghav, K.P.S.; McWilliams, R.R.; Fakih, M.; VanderWalde, A.; Swanton, C.; Kurzrock, R.; Burris, H.; Sweeney, C.; et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): An updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019, 20, 518–530. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Lonardi, S.; Martino, C.; Fenocchio, E.; Tosi, F.; Ghezzi, S.; Leone, F.; Bergamo, F.; Zagonel, V.; Ciardiello, F.; et al. Pertuzumab and trastuzumab emtansine in patients with HER2-amplified metastatic colorectal cancer: The phase II HERACLES-B trial. ESMO Open 2020, 5, e000911. [Google Scholar] [CrossRef]
- Siena, S.; Di Bartolomeo, M.; Raghav, K.; Masuishi, T.; Loupakis, F.; Kawakami, H.; Yamaguchi, K.; Nishina, T.; Fakih, M.; Elez, E.; et al. Trastuzumab deruxtecan (DS-8201) in patients with HER2-expressing metastatic colorectal cancer (DESTINY-CRC01): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2021, 22, 779–789. [Google Scholar] [CrossRef]
- Strickler, J.H.; Zemla, T.; Ou, F.S.; Cercek, A.; Wu, C.; Sanchez, F.A.; Hubbard, J.; Jaszewski, B.; Bandel, L.; Schweitzer, B.; et al. Trastuzumab and tucatinib for the treatment of HER2 amplified metastatic colorectal cancer (mCRC): Initial results from the MOUNTAINEER trial. Ann. Oncol. 2019, 30, v200. [Google Scholar] [CrossRef]
- Tosi, F.; Sartore-Bianchi, A.; Lonardi, S.; Amatu, A.; Leone, F.; Ghezzi, S.; Martino, C.; Bencardino, K.; Bonazzina, E.; Bergamo, F.; et al. Long-term Clinical Outcome of Trastuzumab and Lapatinib for HER2-positive Metastatic Colorectal Cancer. Clin. Colorectal. Cancer 2020, 19, 256–262.e252. [Google Scholar] [CrossRef] [PubMed]
- Dasari, A.; Morris, V.K.; Allegra, C.J.; Atreya, C.; Benson, A.B., 3rd; Boland, P.; Chung, K.; Copur, M.S.; Corcoran, R.B.; Deming, D.A.; et al. ctDNA applications and integration in colorectal cancer: An NCI Colon and Rectal-Anal Task Forces whitepaper. Nat. Rev. Clin. Oncol. 2020, 17, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Taniguchi, H.; Ikeda, M.; Bando, H.; Kato, K.; Morizane, C.; Esaki, T.; Komatsu, Y.; Kawamoto, Y.; Takahashi, N.; et al. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat. Med. 2020, 26, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Vincent, J.J.; Mortimer, S.; Vowles, J.V.; Ulrich, B.C.; Banks, K.C.; Fairclough, S.R.; Zill, O.A.; Sikora, M.; Mokhtari, R.; et al. Validation of a Plasma-Based Comprehensive Cancer Genotyping Assay Utilizing Orthogonal Tissue- and Plasma-Based Methodologies. Clin. Cancer Res. 2018, 24, 3539–3549. [Google Scholar] [CrossRef] [Green Version]
- Guardant Health Inc. Guardant360 CDx Technical Information, D-001590 R3, July 2021. Available online: https://guardant360cdx.com/technicalinfo/ (accessed on 11 April 2022).
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, 1–16. [Google Scholar] [CrossRef]
- Brulé, S.Y.; Jonker, D.J.; Karapetis, C.S.; O’Callaghan, C.J.; Moore, M.J.; Wong, R.; Tebbutt, N.C.; Underhill, C.; Yip, D.; Zalcberg, J.R.; et al. Location of colon cancer (right-sided versus left-sided) as a prognostic factor and a predictor of benefit from cetuximab in NCIC CO.17. Eur. J. Cancer 2015, 51, 1405–1414. [Google Scholar] [CrossRef]
- Zill, O.A.; Banks, K.C.; Fairclough, S.R.; Mortimer, S.A.; Vowles, J.V.; Mokhtari, R.; Gandara, D.R.; Mack, P.C.; Odegaard, J.I.; Nagy, R.J.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer Res. 2018, 24, 3528–3538. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.S.; Fang, H.Y.; Hung, Y.C.; Ke, T.W.; Chang, C.M.; Liu, T.Y.; Chen, Y.C.; Chao, D.S.; Huang, H.Y.; Chang, J.G. Correlation of genomic alterations between tumor tissue and circulating tumor DNA by next-generation sequencing. J. Cancer Res. Clin. Oncol. 2018, 144, 2167–2175. [Google Scholar] [CrossRef]
- Choi, I.S.; Kato, S.; Fanta, P.T.; Leichman, L.; Okamura, R.; Raymond, V.M.; Lanman, R.B.; Lippman, S.M.; Kurzrock, R. Genomic Profiling of Blood-Derived Circulating Tumor DNA from Patients with Colorectal Cancer: Implications for Response and Resistance to Targeted Therapeutics. Mol. Cancer 2019, 18, 1852–1862. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Schwaederlé, M.C.; Fanta, P.T.; Okamura, R.; Leichman, L.; Lippman, S.M.; Lanman, R.B.; Raymond, V.M.; Talasaz, A.; Kurzrock, R. Genomic Assessment of Blood-Derived Circulating Tumor DNA in Patients With Colorectal Cancers: Correlation With Tissue Sequencing, Therapeutic Response, and Survival. JCO Precis. Oncol. 2019, 3, 1–16. [Google Scholar] [CrossRef]
- Oellerich, M.; Schütz, E.; Beck, J.; Kanzow, P.; Plowman, P.N.; Weiss, G.J.; Walson, P.D. Using circulating cell-free DNA to monitor personalized cancer therapy. Crit. Rev. Clin. Lab. Sci. 2017, 54, 205–218. [Google Scholar] [CrossRef]
- Innocenti, F.; Ou, F.S.; Qu, X.; Zemla, T.J.; Niedzwiecki, D.; Tam, R.; Mahajan, S.; Goldberg, R.M.; Bertagnolli, M.M.; Blanke, C.D.; et al. Mutational Analysis of Patients With Colorectal Cancer in CALGB/SWOG 80405 Identifies New Roles of Microsatellite Instability and Tumor Mutational Burden for Patient Outcome. J. Clin. Oncol. 2019, 37, 1217–1227. [Google Scholar] [CrossRef]
- Fabrizio, D.A.; George, T.J., Jr.; Dunne, R.F.; Frampton, G.; Sun, J.; Gowen, K.; Kennedy, M.; Greenbowe, J.; Schrock, A.B.; Hezel, A.F.; et al. Beyond microsatellite testing: Assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J. Gastrointest. Oncol. 2018, 9, 610–617. [Google Scholar] [CrossRef]
- Pereira, A.A.L.; Morelli, M.P.; Overman, M.; Kee, B.; Fogelman, D.; Vilar, E.; Shureiqi, I.; Raghav, K.; Eng, C.; Manuel, S.; et al. Clinical utility of circulating cell-free DNA in advanced colorectal cancer. PLoS ONE 2017, 12, e0183949. [Google Scholar] [CrossRef] [Green Version]
- Massard, C.; Michiels, S.; Ferté, C.; Le Deley, M.C.; Lacroix, L.; Hollebecque, A.; Verlingue, L.; Ileana, E.; Rosellini, S.; Ammari, S.; et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017, 7, 586–595. [Google Scholar] [CrossRef] [Green Version]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Lyons, E.; DeArbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sahai, V.; Sohal, D.P.S.; et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: A retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020, 21, 508–518. [Google Scholar] [CrossRef]
- Schwaederle, M.; Parker, B.A.; Schwab, R.B.; Daniels, G.A.; Piccioni, D.E.; Kesari, S.; Helsten, T.L.; Bazhenova, L.A.; Romero, J.; Fanta, P.T.; et al. Precision Oncology: The UC San Diego Moores Cancer Center PREDICT Experience. Mol. Cancer 2016, 15, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Eggermont, A.M.; Schilsky, R.L.; Mendelsohn, J.; Lazar, V.; Kurzrock, R. Impact of Precision Medicine in Diverse Cancers: A Meta-Analysis of Phase II Clinical Trials. J. Clin. Oncol. 2015, 33, 3817–3825. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Iskander, N.G.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Luthra, R.; et al. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin. Cancer Res. 2012, 18, 6373–6383. [Google Scholar] [CrossRef] [Green Version]
- Pairawan, S.; Hess, K.R.; Janku, F.; Sanchez, N.S.; Mills Shaw, K.R.; Eng, C.; Damodaran, S.; Javle, M.; Kaseb, A.O.; Hong, D.S.; et al. Cell-free Circulating Tumor DNA Variant Allele Frequency Associates with Survival in Metastatic Cancer. Clin. Cancer Res. 2020, 26, 1924–1931. [Google Scholar] [CrossRef]
- Wang, F.; Zhao, Q.; Wang, Y.N.; Jin, Y.; He, M.M.; Liu, Z.X.; Xu, R.H. Evaluation of POLE and POLD1 Mutations as Biomarkers for Immunotherapy Outcomes Across Multiple Cancer Types. JAMA Oncol. 2019, 5, 1504–1506. [Google Scholar] [CrossRef] [Green Version]
- Inc, M.C. KEYTRUDA® (Pembrolizumab) Injection, for Intravenous Use. Highlights of Prescribing Information, Revised February 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/125514s125lbl.pdf (accessed on 17 February 2022).
- Rousseau, B.; Foote, M.B.; Maron, S.B.; Diplas, B.H.; Lu, S.; Argilés, G.; Cercek, A.; Diaz, L.A., Jr. The Spectrum of Benefit from Checkpoint Blockade in Hypermutated Tumors. N. Engl. J. Med. 2021, 384, 1168–1170. [Google Scholar] [CrossRef]
- Strickler, J.H.; Loree, J.M.; Ahronian, L.G.; Parikh, A.R.; Niedzwiecki, D.; Pereira, A.A.L.; McKinney, M.; Korn, W.M.; Atreya, C.E.; Banks, K.C.; et al. Genomic Landscape of Cell-Free DNA in Patients with Colorectal Cancer. Cancer Discov. 2018, 8, 164–173. [Google Scholar] [CrossRef] [Green Version]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e123. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Class of Treatment | Second-Line Cohort (n = 642) | Third-Line Cohort (n = 422) | Total (n = 1064) | |||
|---|---|---|---|---|---|---|
| n | % | n | % | n | % | |
| Any non-targeted * | 580 | 90.3% | 346 | 82.0% | 926 | 87.0% |
| Chemotherapy only | 262 | 40.8% | 103 | 24.4% | 365 | 34.3% |
| VEGF inhibitor (with or without chemotherapy, no targeted treatment) | 318 | 49.5% | 243 | 57.6% | 561 | 52.7% |
| Any targeted | 62 | 9.7% | 76 | 18.0% | 138 | 13.0% |
| EGFR-targeted | 56 | 8.7% | 69 | 16.4% | 125 | 11.7% |
| BRAF-targeted | 0 | 0.0% | 3 | 0.7% | 3 | 0.3% |
| ERBB2-targeted | 3 | 0.5% | 1 | 0.2% | 4 | 0.4% |
| Other targeted | 1 | 0.2% | 0 | 0.0% | 1 | 0.1% |
| Immune checkpoint inhibitor | 3 | 0.5% | 5 | 1.2% | 8 | 0.8% |
| Mutation | Prior Therapy: Targeted | Prior Therapy: Non-Targeted | p-Value * | Total | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | All Tested (%) | With ctDNA (%) | n | All Tested (%) | With ctDNA (%) | All Tested | With ctDNA | n | All Tested (%) | With ctDNA (%) | |
| n | 138 | 135 | 926 | 862 | 1064 | 997 | |||||
| KRAS mutation † | 29 ‡ | 21.0% | 21.5% | 395 § | 42.7% | 45.8% | <0.001 | <0.001 | 424 | 39.8% | 42.5% |
| NRAS mutation † | 12 | 8.7% | 8.9% | 40 | 4.3% | 4.6% | 0.044 | 0.063 | 52 | 4.9% | 5.2% |
| Any RAS mutation † | 33 | 23.9% | 24.4% | 422 ‡ | 45.6% | 49.0% | <0.001 | <0.001 | 455 | 42.8% | 45.6% |
| BRAF V600E † | 17 | 12.3% | 12.6% | 56 | 6.0% | 6.5% | 0.011 | 0.019 | 73 | 6.9% | 7.3% |
| ERBB2 amplification † | 8 | 5.8% | 5.9% | 20 | 2.2% | 2.3% | 0.021 | 0.043 | 28 | 2.6% | 2.8% |
| MSI-H † | 3 | 2.2% | 2.2% | 17 | 1.8% | 2.0% | 0.736 | 0.745 | 20 | 1.9% | 2.0% |
| NTRK1 fusion † | 1 | 0.7% | 0.7% | 0 | 0 | 0 | 0 | 0 | 1 | 0.1% | 0.1% |
| RET fusion | 1 | 0.7% | 0.7% | 1 | 0.1% | 0.1% | 0 | 0 | 2 | 0.2% | 0.2% |
| ALK fusion | 1 | 0.7% | 0.7% | 0 | 0 | 0 | 0 | 1 | 0.1% | 0.1% | |
| ROS1 fusion | 0 | 0 | 0 | 1 | 0.1% | 0.1% | 0 | 0 | 1 | 0.1% | 0.1% |
| FGFR3 fusion | 1 | 0.7% | 0.7% | 1 | 0.1% | 0.1% | 0 | 0 | 2 | 0.2% | 0.2% |
| MET exon 14 skipping | 0 | 0 | 0 | 1 | 0.1% | 0.1% | 0 | 0 | 1 | 0.1% | 0.1% |
| Second-Line Cohort (n = 642) | Third-Line Cohort (n = 422) | Total (n = 1064) | ||||
|---|---|---|---|---|---|---|
| Class of Treatment | n | Informed, n (%) | n | Informed, n (%) | n | Informed, n (%) |
| Chemotherapy only | 184 | 96 (52.2%) | 105 | 68 (64.8%) | 289 | 164 (56.7%) |
| VEGF inhibitor (with or without chemotherapy, no targeted treatment) | 275 | 148 (53.8%) | 162 | 96 (59.3%) | 437 | 244 (55.8%) |
| Any non-targeted * | 459 | 244 (53.2%) | 267 | 164 (61.4%) | 726 | 408 (56.2%) |
| EGFR-targeted | 132 | 77 (58.3%) | 101 | 79 (78.2%) | 233 | 156 (67.0%) |
| BRAF-targeted | 9 | 8 (88.9%) | 7 | 5 (71.4%) | 16 | 13 (81.3%) |
| ERBB2-targeted | 11 | 11 (100%) | 10 | 8 (80.0%) | 21 | 19 (90.5%) |
| Other targeted | 1 | 1 (100%) | 0 | 0 | 1 | 1 (100%) |
| Immune checkpoint inhibitor | 37 | 11 (32.4%) | 41 † | 5 (12.2%) | 78 | 16 (20.5%) |
| Any treatment | 642 | 346 (53.9%) | 422 | 257 (60.9%) | 1064 | 603 (56.7%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, Y.; Olsen, S.; Zhang, N.; Liao, J.; Yoshino, T. Comprehensive Genomic Profiling of Circulating Tumor DNA in Patients with Previously Treated Metastatic Colorectal Cancer: Analysis of a Real-World Healthcare Claims Database. Curr. Oncol. 2022, 29, 3433-3448. https://doi.org/10.3390/curroncol29050277
Nakamura Y, Olsen S, Zhang N, Liao J, Yoshino T. Comprehensive Genomic Profiling of Circulating Tumor DNA in Patients with Previously Treated Metastatic Colorectal Cancer: Analysis of a Real-World Healthcare Claims Database. Current Oncology. 2022; 29(5):3433-3448. https://doi.org/10.3390/curroncol29050277
Chicago/Turabian StyleNakamura, Yoshiaki, Steven Olsen, Nicole Zhang, Jiemin Liao, and Takayuki Yoshino. 2022. "Comprehensive Genomic Profiling of Circulating Tumor DNA in Patients with Previously Treated Metastatic Colorectal Cancer: Analysis of a Real-World Healthcare Claims Database" Current Oncology 29, no. 5: 3433-3448. https://doi.org/10.3390/curroncol29050277
APA StyleNakamura, Y., Olsen, S., Zhang, N., Liao, J., & Yoshino, T. (2022). Comprehensive Genomic Profiling of Circulating Tumor DNA in Patients with Previously Treated Metastatic Colorectal Cancer: Analysis of a Real-World Healthcare Claims Database. Current Oncology, 29(5), 3433-3448. https://doi.org/10.3390/curroncol29050277