Clinical Impact of High Throughput Sequencing on Liquid Biopsy in Advanced Solid Cancer

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design, Endpoints, and Patients
2.2. Genomic Analyses and Targeted Therapies
2.3. Statistical Analysis
3. Results
3.1. Patients
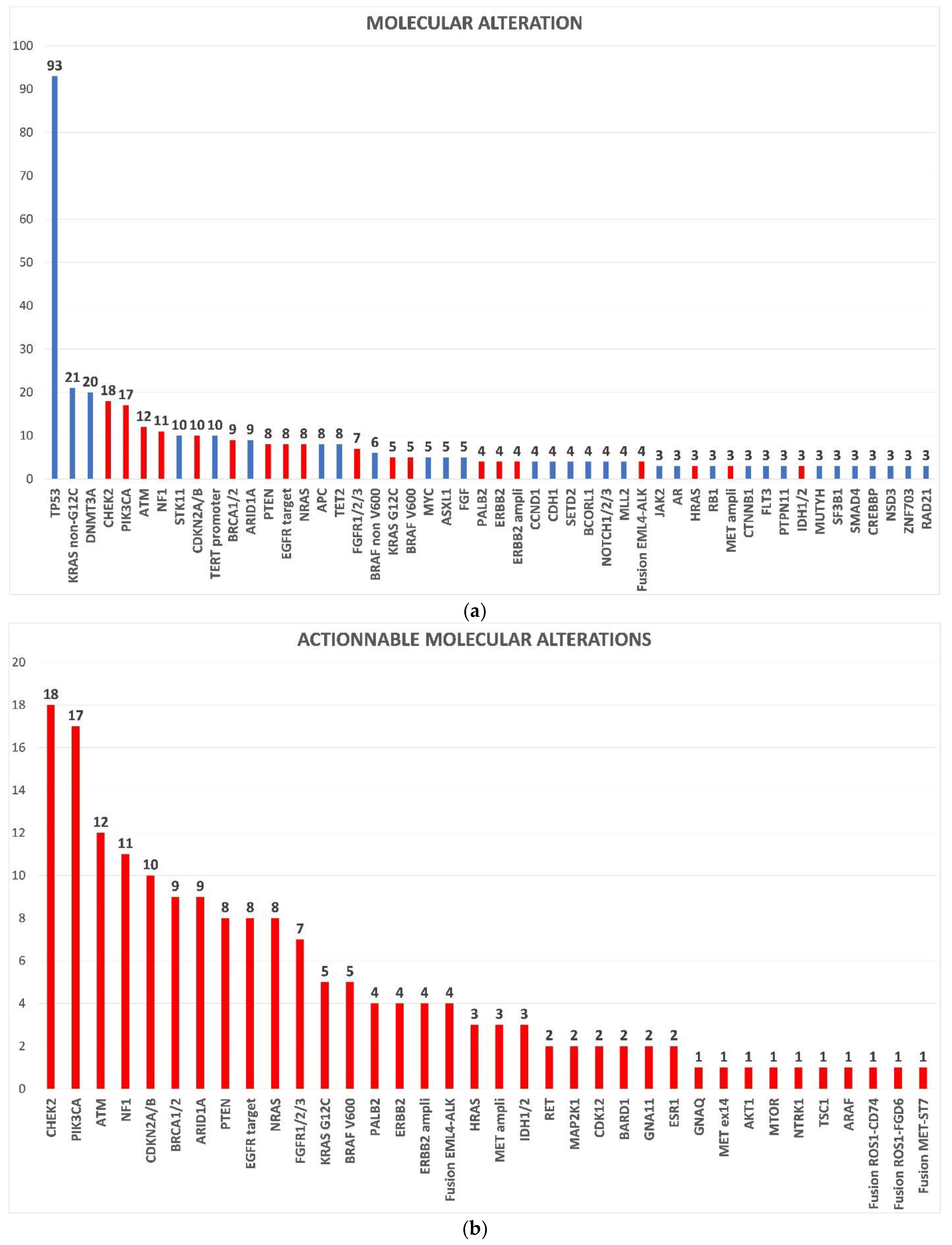
3.2. Molecular Profiling and Treatments
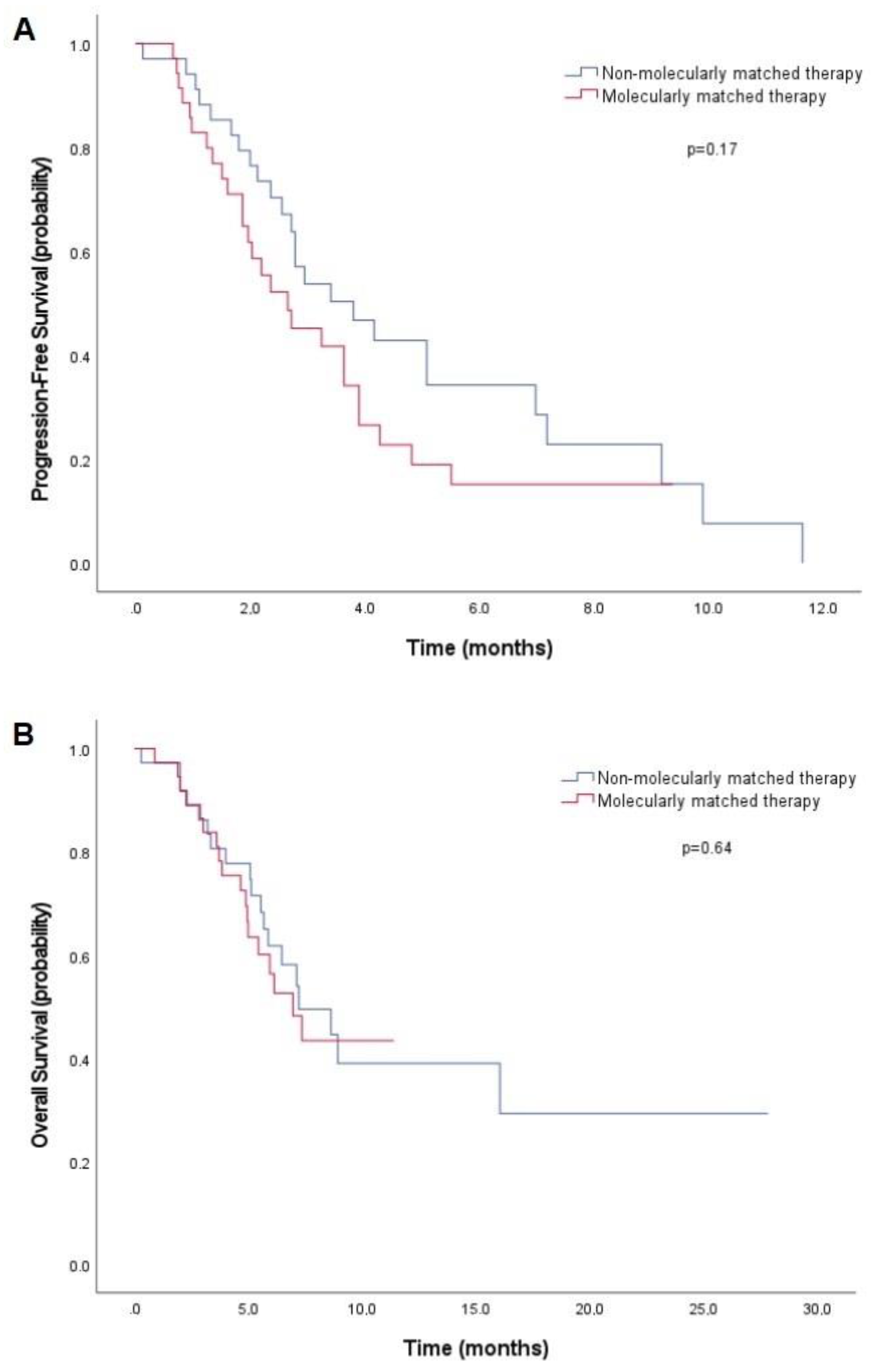
3.3. Efficacy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, M.F.; Mardis, E.R. The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 2018, 15, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Wakai, T.; Prasoon, P.; Hirose, Y.; Shimada, Y.; Ichikawa, H.; Nagahashi, M. Next-generation sequencing-based clinical sequencing: Toward precision medicine in solid tumors. Int. J. Clin. Oncol. 2019, 24, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or Chemotherapy for Non–Small-Cell Lung Cancer with Mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.T.; Kim, D.-W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus Chemotherapy in AdvancedALK-Positive Lung Cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.T.; Ou, S.-H.I.; Bang, Y.-J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in ROS1-Rearranged Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planchard, D.; Besse, B.; Groen, H.J.; Souquet, P.-J.; Quoix, E.; Baik, C.S.; Barlesi, F.; Kim, T.M.; Mazieres, J.; Novello, S.; et al. Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non-small cell lung cancer: An open-label, multicentre phase 2 trial. Lancet Oncol. 2016, 17, 984–993. [Google Scholar] [CrossRef] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib Compared with Interferon and Low-Dose Cytarabine for Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef] [Green Version]
- Wirth, L.J.; Robinson, B.; Boni, V.; Tan, D.S.; McCoach, C.; Massarelli, E.; Hess, L.M.; Jen, M.-H.; Kherani, J.; Olek, E.; et al. Patient-Reported Outcomes with Selpercatinib Treatment Among Patients with RET-Mutant Medullary Thyroid Cancer in the Phase I/II LIBRETTO-001 Trial. Oncologist 2021, 27, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282, Erratum in Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.-Y.; Wolf, J.L.; Raje, N.S.; Diamond, E.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Fountzilas, E.; Nikanjam, M.; Kurzrock, R. Review of precision cancer medicine: Evolution of the treatment paradigm. Cancer Treat. Rev. 2020, 86, 102019. [Google Scholar] [CrossRef]
- Schwaederle, M.; Parker, B.A.; Schwab, R.B.; Daniels, G.A.; Piccioni, D.; Kesari, S.; Helsten, T.L.; Bazhenova, L.A.; Romero, J.; Fanta, P.T.; et al. Precision Oncology: The UC San Diego Moores Cancer Center PREDICT Experience. Mol. Cancer Ther. 2016, 15, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Stockley, T.; Oza, A.; Berman, H.K.; Leighl, N.B.; Knox, J.J.; Shepherd, F.A.; Chen, E.X.; Krzyzanowska, M.; Dhani, N.; Joshua, A.; et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: The Princess Margaret IMPACT/COMPACT trial. Genome Med. 2016, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wheler, J.J.; Janku, F.; Naing, A.; Filip, J.; Stephen, B.; Zinner, R.; Subbiah, V.; Fu, S.; Karp, D.; Falchook, G.S.; et al. Cancer Therapy Directed by Comprehensive Genomic Profiling: A Single Center Study. Cancer Res. 2016, 76, 3690–3701. [Google Scholar] [CrossRef] [Green Version]
- Massard, C.; Michiels, S.; Ferté, C.; Le Deley, M.-C.; Lacroix, L.; Hollebecque, A.; Verlingue, L.; Ileana, E.; Rosellini, S.; Ammari, S.; et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017, 7, 586–595. [Google Scholar] [CrossRef] [Green Version]
- Tsimberidou, A.-M.; Wen, S.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Aldape, K.; et al. Personalized Medicine for Patients with Advanced Cancer in the Phase I Program at MD Anderson: Validation and Landmark Analyses. Clin. Cancer Res. 2014, 20, 4827–4836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tredan, O.; Corset, V.; Wang, Q.; Varnier, R.; Pacaud, C.; Torroja, A.; Luppi, N.; Ezzalfani, M.; Myard, M.; Jiang, X.; et al. Routine molecular screening of advanced refractory cancer patients: An analysis of the first 2490 patients of the ProfiLER study. J. Clin. Oncol. 2017, 35, LBA100. [Google Scholar] [CrossRef]
- Seeber, A.; Gastl, G.; Ensinger, C.; Spizzo, G.; Willenbacher, W.; Kocher, F.; Leitner, C.; Willenbacher, E.; Amann, A.; Steiner, N.; et al. Treatment of patients with refractory metastatic cancer according to molecular profiling on tumor tissue in the clinical routine: An interim-analysis of the ONCO-T-PROFILE project. Genes Cancer 2016, 7, 301–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodon, J.; Soria, J.-C.; Berger, R.; Miller, W.H.; Lazar, V.; Rubin, E.; Tsimberidou, A.M.; Saintigny, P.; Ackerstein, A.; Brana, I.; et al. WINTHER: An international WIN Consortium precision medicine trial using genomic and transcriptomic analysis in patients with advanced malignancies. J. Clin. Oncol. 2018, 36, 12011. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Delord, J.-P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Tredan, O.; Massiani, M.-A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Hiley, C.; De Bruin, E.C.; McGranahan, N.; Swanton, C. Deciphering intratumor heterogeneity and temporal acquisition of driver events to refine precision medicine. Genome Biol. 2014, 15, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasini, P.; Barlesi, F.; Gilles, S.; Nanni-Metellus, I.; Soffietti, R.; Denicolai, E.; Pellegrino, E.; Bialecki, E.; Ouafik, L.; Metellus, P. Comparative genomic analysis of primary tumors and paired brain metastases in lung cancer patients by whole exome sequencing: A pilot study. Oncotarget 2020, 11, 4648–4654. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Lázaro, D.; Hernández, J.L.G.; Garcia, J.; Del Castillo, A.C.; Hueso, M.V.; Cruz-Hernández, J.J. Clinical Perspective and Translational Oncology of Liquid Biopsy. Diagnostics 2020, 10, 443. [Google Scholar] [CrossRef] [PubMed]
- Sabari, J.K.; Offin, M.; Stephens, D.; Ni, A.; Lee, A.; Pavlakis, N.; Clarke, S.; Diakos, C.I.; Datta, S.; Tandon, N.; et al. A Prospective Study of Circulating Tumor DNA to Guide Matched Targeted Therapy in Lung Cancers. JNCI J. Natl. Cancer Inst. 2019, 111, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Lin, D.; Wu, C.; Li, X.; Zhao, C.; Zheng, L.; Chuai, S.; Fei, K.; Zhou, C.; Hirsch, F.R. Intratumoral Heterogeneity of ALK-Rearranged and ALK/EGFR Coaltered Lung Adenocarcinoma. J. Clin. Oncol. 2015, 33, 3701–3709. [Google Scholar] [CrossRef]
- Rolfo, C.; Mack, P.C.; Scagliotti, G.V.; Baas, P.; Barlesi, F.; Bivona, T.G.; Herbst, R.S.; Mok, T.; Peled, N.; Pirker, R.; et al. Liquid Biopsy for Advanced Non-Small Cell Lung Cancer (NSCLC): A Statement Paper from the IASLC. J. Thorac. Oncol. 2018, 13, 1248–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hoff, D.D.; Stephenson, J.J.S., Jr.; Rosen, P.; Loesch, D.M.; Borad, M.J.; Anthony, S.; Jameson, G.S.; Brown, S.; Cantafio, N.; Richards, D.A.; et al. Pilot Study Using Molecular Profiling of Patients’ Tumors to Find Potential Targets and Select Treatments for Their Refractory Cancers. J. Clin. Oncol. 2010, 28, 4877–4883. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. FoundationOne® Liquid CDx. Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf19/P190032C.pdf (accessed on 8 February 2022).
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Macdonald, D.R.; Reardon, D.A.; Cloughesy, T.F.; Sorensen, A.G.; Galanis, E.; DeGroot, J.; Wick, W.; Gilbert, M.R.; Lassman, A.B.; et al. Updated Response Assessment Criteria for High-Grade Gliomas: Response Assessment in Neuro-Oncology Working Group. J. Clin. Oncol. 2010, 28, 1963–1972. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Gonçalves, A.; Guille, A.; Adelaïde, J.; Garnier, S.; Carbuccia, N.; Billon, E.; Finetti, P.; Sfumato, P.; Monneur, A.; et al. Prospective high-throughput genome profiling of advanced cancers: Results of the PERMED-01 clinical trial. Genome Med. 2021, 13, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Réda, M.; Richard, C.; Bertaut, A.; Niogret, J.; Collot, T.; Fumet, J.D.; Blanc, J.; Truntzer, C.; Desmoulins, I.; Ladoire, S.; et al. Implementation and use of whole exome sequencing for metastatic solid cancer. EBioMedicine 2020, 51, 102624. [Google Scholar] [CrossRef] [PubMed]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S. Poly (ADP-Ribose) Polymerase Inhibitors: Recent Advances and Future Development. J. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquard, A.M.; Eklund, A.C.; Joshi, T.; Krzystanek, M.; Favero, F.; Wang, Z.C.; Richardson, A.L.; Silver, D.P.; Szallasi, Z.; Birkbak, N.J. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark. Res. 2015, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.; Martens, J.W.M.; Van Hoeck, A.; Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination–Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis From the Phase II TRITON2 Study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29 (Suppl. 4), iv192–iv237. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.; Chirieac, L.R.; D’Amico, T.A.; DeCamp, M.M.; Dilling, T.J.; Dobelbower, M.; et al. Non–Small Cell Lung Cancer, Version 5.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 504–535. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Janku, F.; Jung, B.; Hou, C.; Madwani, K.; Alden, R.; Razavi, P.; Reis-Filho, J.; Shen, R.; Isbell, J.; et al. Ultra-deep next-generation sequencing of plasma cell-free DNA in patients with advanced lung cancers: Results from the Actionable Genome Consortium. Ann. Oncol. 2019, 30, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Rijavec, E.; Coco, S.; Genova, C.; Rossi, G.; Longo, L.; Grossi, F. Liquid Biopsy in Non-Small Cell Lung Cancer: Highlights and Challenges. Cancers 2019, 12, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonanno, L.; Pavan, A.; Ferro, A.; Calvetti, L.; Frega, S.; Pasello, G.; Aprile, G.; Guarneri, V.; Conte, P.; Rete Oncologica Veneta (ROV). Clinical Impact of Plasma and Tissue Next-Generation Sequencing in Advanced Non-Small Cell Lung Cancer: A Real-World Experience. Oncologist 2020, 25, e1996–e2005. [Google Scholar] [CrossRef]
- Woodhouse, R.; Li, M.; Hughes, J.; Delfosse, D.; Skoletsky, J.; Ma, P.; Meng, W.; Dewal, N.; Milbury, C.; Clark, T.; et al. Clinical and Analytical Validation of FoundationOne Liquid CDx, a Novel 324-Gene CfDNA-Based Comprehensive Genomic Profiling Assay for Cancers of Solid Tumor Origin. PLoS ONE 2020, 18, e0237802. [Google Scholar] [CrossRef] [PubMed]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.; Barlesi, F.; Lolkema, M.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Kim, K.H.; Lim, H.J.; Boichard, A.; Nikanjam, M.; Weihe, E.; Kuo, D.J.; Eskander, R.N.; Goodman, A.; Galanina, N.; et al. Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hoefflin, R.; Geißler, A.-L.; Fritsch, R.; Claus, R.; Wehrle, J.; Metzger, P.; Reiser, M.; Mehmed, L.; Fauth, L.; Heiland, D.H.; et al. Personalized Clinical Decision Making Through Implementation of a Molecular Tumor Board: A German Single-Center Experience. JCO Precis. Oncol. 2018, 2, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, C.; Manca, P.; Salgado, R.; Van Dam, P.; Dendooven, A.; Gandia, J.F.; Rutten, A.; Lybaert, W.; Vermeij, J.; Gevaert, T.; et al. Multidisciplinary molecular tumour board: A tool to improve clinical practice and selection accrual for clinical trials in patients with cancer. ESMO Open 2018, 3, e000398. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient’s Characteristics | Overall Population n = 191 | AMA + MMT n = 37 (19%) | AMA + Non-MMT n = 37 (19%) | p-Value * |
|---|---|---|---|---|
| Age | 0.1 | |||
| Median, years (range) | 61 (15–88) | 57 (24–88) | 61 (45–78) | |
| Sex | 0.35 | |||
| Female | 107 (56%) | 19 (51%) | 13 (35%) | |
| Male | 84 (44%) | 18 (49%) | 24 (65%) | |
| ECOG PS | 0.63 | |||
| 0 | 67 (35%) | 10 (27%) | 11 (30%) | |
| 1 | 93 (49%) | 21 (57%) | 23 (62%) | |
| ≥2 | 31 (16%) | 6 (16%) | 3 (8%) | |
| Smoking Status | 0.81 | |||
| Smoker (active or former) | 94 (49%) | 18 (49%) | 19 (51%) | |
| Missing | 17 (9%) | 2 (5%) | 3 (8%) | |
| Tumor Type | 0.84 | |||
| Lung | 88 (46%) | 18 (49%) | 17 (46%) | |
| Melanoma | 21 (11%) | 6 (16%) | 5 (14%) | |
| Breast | 19 (10%) | 7 (19%) | 4 (10%) | |
| Pancreas | 12 (6%) | 1 (3%) | 3 (8%) | |
| Sarcoma | 7 (4%) | 0 | 1 (3%) | |
| Colorectal | 7 (4%) | 1 (3%) | 2 (5%) | |
| Other | 37 (19%) | 4 (10%) | 5 (14%) | |
| Extension Stage | 1 | |||
| Metastatic | 180 (94%) | 36 (97%) | 35 (94%) | |
| Other | 11 (6%) | 1 (3%) | 2 (5%) | |
| Previous Systemic Treatment | 0.15 | |||
| Median (range) | 3 (0–10) | 3 (0–9) | 3 (0–7) | |
| Missing | 1 | 0 | 0 | |
| Subsequent Systemic Treatment | 0.73 | |||
| Median (range) | 1 (0–4) | 1 (1–3) | 1 (1–3) | |
| Missing | 32 | 0 | 0 | |
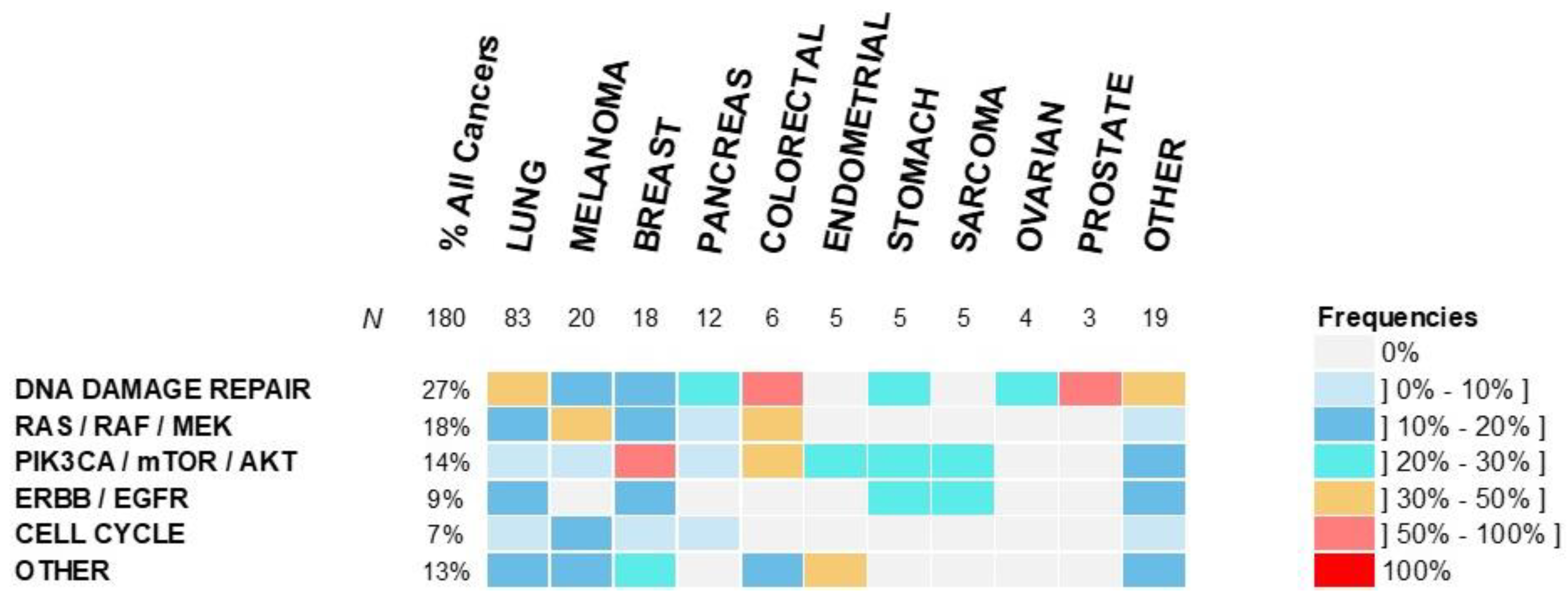
| Molecular Pathways Altered | ||||
| DNA damage repair system | - | 15 (41%) | 18 (49%) | 0.64 |
| RAS/RAF/MEK | - | 14 (38%) | 10 (27%) | 0.46 |
| PIK3CA/mTOR/AKT | - | 13 (35%) | 5 (14%) | 0.06 |
| ERBB/EGFR | - | 10 (27%) | 5 (14%) | 0.25 |
| Cell cycle | - | 3 (8%) | 5 (14%) | 0.71 |
| Other | - | 11 (30%) | 5 (14%) | 0.16 |
| F1LCDx’s Characteristics | Overall Population n = 180 | AMA n = 100 (52%) | Non-AMA n = 80 (42%) | p-Value * |
|---|---|---|---|---|
| Result Delay | 0.56 | |||
| Median, days (range) | 13 (5–38) | 13 (5–28) | 13 (7–27) | |
| Timing of F1LCDx testing | 0.0004 | |||
| Progression on non-TT | 109 (61%) | 74 (74%) | 35 (44%) | |
| During treatment | 44 (24%) | 16 (16%) | 28 (35%) | |
| Progression on TT | 11 (6%) | 6 (6%) | 5 (6%) | |
| Diagnosis | 7 (4%) | 1 (1%) | 6 (7%) | |
| Metastatic relapse | 6 (3%) | 2 (2%) | 4 (5%) | |
| Other | 3 (2%) | 1 (1%) | 2 (3%) | |
| Previous Molecular Profile | ||||
| n (%) | 147 (82%) | 83 (83%) | 64 (80%) | - |
| with previous MA | 99/147 (67%) | 60/83 (72%) | 39/64 (61%) | 0.7 |
| with previous AMA | 53/147 (36%) | 38/83 (46%) | 15/64 (23%) | 0.16 |
| Tumor Fraction (%) | 0.91 | |||
| Median (range) | 21 (10–72) | 21 (10–72) | 19 (11–71) | |
| Tumor Mutational Burden | 0.01 | |||
| <10 mut/Mb | 84 (47%) | 43 (43%) | 41 (51%) | |
| ≥10 mut/Mb | 12 (6%) | 11 (11%) | 1 (1%) | |
| Missing | 84 (47%) | 46 (46%) | 38 (48%) | |
| MSI status | 0.35 | |||
| MSI high | 1 (1%) | 1 (1%) | 0 | |
| MSI high not detected | 28 (15%) | 13 (13%) | 15 (19%) | |
| Undetermined | 151 (84%) | 86 (86%) | 65 (81%) |
| Efficacy | AMA + MMT n = 37 | AMA + Non-MMT n = 37 | p-Value * |
|---|---|---|---|
| PFS2/PFS1 | |||
| Median (range) | 0.63 (0–11.9) | 0.84 (0–7.2) | - |
| Ratio ≥ 1.3 | 7/35 (20%) | 8/34 (24%) | 0.72 |
| Missing | 2 | 3 | - |
| PFS2 | 0.17 | ||
| Median, months (95%CI) | 2.7 (1.3–3.9) | 3.8 (2.1–5.5) | |
| OS | 0.64 | ||
| Median, months (95%CI) | 6.9 (5.0–8.9) | 7.2 (4.3–10.1) | |
| Tumor Response | |||
| Complete response | 1 (3%) | 0 | - |
| Partial response | 6 (16%) | 6 (16%) | - |
| Stable disease | 5 (14%) | 12 (32%) | - |
| Progressive disease | 17 (46%) | 8 (22%) | - |
| ORR | 7 (19%) | 6 (16%) | 0.87 |
| Disease control | 12 (32%) | 18 (49%) | 0.05 |
| Not available | 8 (22%) | 11 (30%) | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gouton, E.; Malissen, N.; André, N.; Jeanson, A.; Pelletier, A.; Testot-Ferry, A.; Gaudy-Marqueste, C.; Dahan, L.; Tabouret, E.; Chevalier, T.; et al. Clinical Impact of High Throughput Sequencing on Liquid Biopsy in Advanced Solid Cancer. Curr. Oncol. 2022, 29, 1902-1918. https://doi.org/10.3390/curroncol29030155
Gouton E, Malissen N, André N, Jeanson A, Pelletier A, Testot-Ferry A, Gaudy-Marqueste C, Dahan L, Tabouret E, Chevalier T, et al. Clinical Impact of High Throughput Sequencing on Liquid Biopsy in Advanced Solid Cancer. Current Oncology. 2022; 29(3):1902-1918. https://doi.org/10.3390/curroncol29030155
Chicago/Turabian StyleGouton, Etienne, Nausicaa Malissen, Nicolas André, Arnaud Jeanson, Annick Pelletier, Albane Testot-Ferry, Caroline Gaudy-Marqueste, Laetitia Dahan, Emeline Tabouret, Thomas Chevalier, and et al. 2022. "Clinical Impact of High Throughput Sequencing on Liquid Biopsy in Advanced Solid Cancer" Current Oncology 29, no. 3: 1902-1918. https://doi.org/10.3390/curroncol29030155
APA StyleGouton, E., Malissen, N., André, N., Jeanson, A., Pelletier, A., Testot-Ferry, A., Gaudy-Marqueste, C., Dahan, L., Tabouret, E., Chevalier, T., Greillier, L., & Tomasini, P. (2022). Clinical Impact of High Throughput Sequencing on Liquid Biopsy in Advanced Solid Cancer. Current Oncology, 29(3), 1902-1918. https://doi.org/10.3390/curroncol29030155