NUTM1-Rearranged Neoplasms—A Heterogeneous Group of Primitive Tumors with Expanding Spectrum of Histology and Molecular Alterations—An Updated Review



Abstract
:1. Introduction
2. Clinical Aspects
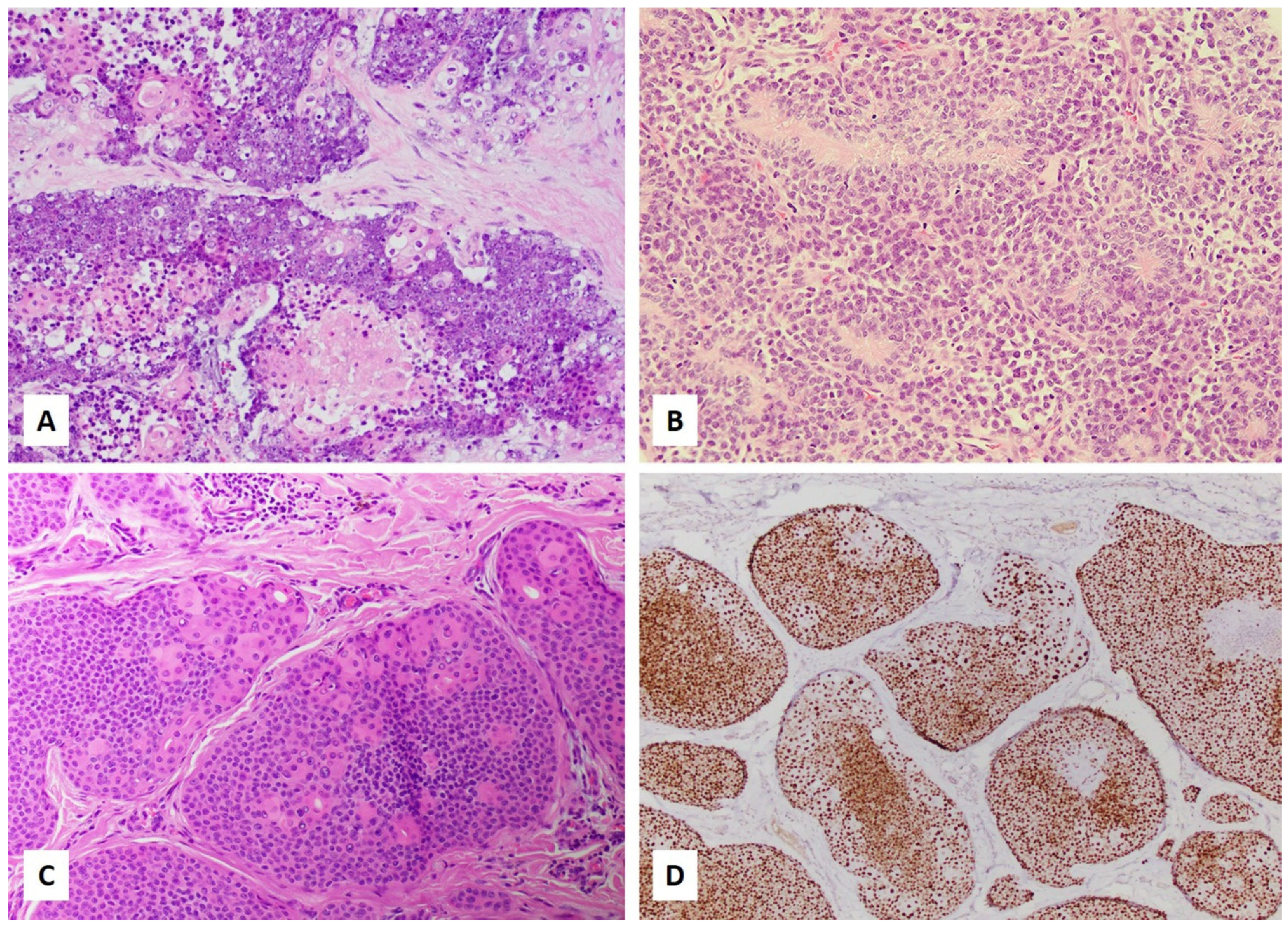
3. Histologic and Immunohistochemical Diagnosis
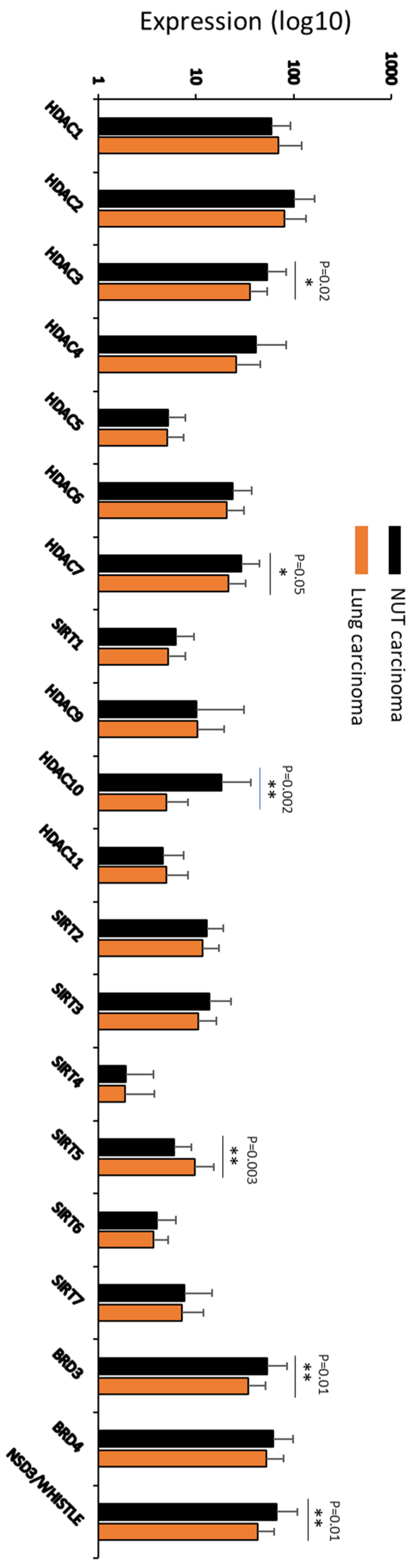
4. Pathogenic Mechanism
5. Targeted Therapy
6. Other NUTM1-Rearranged Tumors
6.1. Skin Tumors
6.2. Sarcomas
6.3. Brain Tumors
6.4. Hematologic Neoplasms
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shiota, H.; Barral, S.; Buchou, T.; Tan, M.; Coute, Y.; Charbonnier, G.; Reynoird, N.; Boussouar, F.; Gerard, M.; Zhu, M.; et al. Nut Directs p300-Dependent, Genome-Wide H4 Hyperacetylation in Male Germ Cells. Cell Rep. 2018, 24, 3477–3487.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEvoy, C.R.; Fox, S.B.; Prall, O.W.J. Emerging entities in NUTM1-rearranged neoplasms. Genes Chromosomes Cancer 2020, 59, 375–385. [Google Scholar] [CrossRef]
- Kees, U.R.; Mulcahy, M.T.; Willoughby, M.L. Intrathoracic carcinoma in an 11-year-old girl showing a translocation t(15;19). Am. J. Pediatric Hematol. Oncol. 1991, 13, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Kubonishi, I.; Takehara, N.; Iwata, J.; Sonobe, H.; Ohtsuki, Y.; Abe, T.; Miyoshi, I. Novel t(15;19)(q15;p13) chromosome abnormality in a thymic carcinoma. Cancer Res. 1991, 51, 3327–3328. [Google Scholar]
- Lee, A.C.; Kwong, Y.I.; Fu, K.H.; Chan, G.C.; Ma, L.; Lau, Y.L. Disseminated mediastinal carcinoma with chromosomal translocation (15;19). A distinctive clinicopathologic syndrome. Cancer 1993, 72, 2273–2276. [Google Scholar] [CrossRef]
- Dang, T.P.; Gazdar, A.F.; Virmani, A.K.; Sepetavec, T.; Hande, K.R.; Minna, J.D.; Roberts, J.R.; Carbone, D.P. Chromosome 19 Translocation, Overexpression of Notch3, and Human Lung Cancer. J. Natl. Cancer Inst. 2000, 92, 1355–1357. [Google Scholar] [CrossRef] [Green Version]
- Vargas, S.O.; French, C.A.; Faul, P.N.; Fletcher, J.A.; Davis, I.J.; Dal Cin, P.; Perez-Atayde, A.R. Upper respiratory tract carcinoma with chromosomal translocation 15;19: Evidence for a distinct disease entity of young patients with a rapidly fatal course. Cancer 2001, 92, 1195–1203. [Google Scholar] [CrossRef]
- French, C.A.; Miyoshi, I.; Kubonishi, I.; Grier, H.E.; Perez-Atayde, A.R.; Fletcher, J.A. BRD4-NUT fusion oncogene: A novel mechanism in aggressive carcinoma. Cancer Res. 2003, 63, 304–307. [Google Scholar] [PubMed]
- French, C.A.; Ramirez, C.L.; Kolmakova, J.; Hickman, T.T.; Cameron, M.J.; Thyne, M.E.; Kutok, J.L.; Toretsky, J.A.; Tadavarthy, A.K.; Kees, U.R.; et al. BRD-NUT oncoproteins: A family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene 2008, 27, 2237–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, C.A.; Rahman, S.; Walsh, E.M.; Kuhnle, S.; Grayson, A.R.; Lemieux, M.E.; Grunfeld, N.; Rubin, B.P.; Antonescu, C.R.; Zhang, S.; et al. NSD3-NUT fusion oncoprotein in NUT midline carcinoma: Implications for a novel oncogenic mechanism. Cancer Discov. 2014, 4, 928–941. [Google Scholar] [CrossRef] [Green Version]
- Chau, N.G.; Ma, C.; Danga, K.; Al-Sayegh, H.; Nardi, V.; Barrette, R.; Lathan, C.S.; DuBois, S.G.; Haddad, R.I.; Shapiro, G.I.; et al. An Anatomical Site and Genetic-Based Prognostic Model for Patients With Nuclear Protein in Testis (NUT) Midline Carcinoma: Analysis of 124 Patients. JNCI Cancer Spectr. 2020, 4, pkz094. [Google Scholar] [CrossRef]
- Stelow, E.B.; Bellizzi, A.M.; Taneja, K.; Mills, S.E.; Legallo, R.D.; Kutok, J.L.; Aster, J.C.; French, C.A. NUT rearrangement in undifferentiated carcinomas of the upper aerodigestive tract. Am. J. Surg. Pathol. 2008, 32, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Dickson, B.C.; Sung, Y.S.; Rosenblum, M.K.; Reuter, V.E.; Harb, M.; Wunder, J.S.; Swanson, D.; Antonescu, C.R. NUTM1 Gene Fusions Characterize a Subset of Undifferentiated Soft Tissue and Visceral Tumors. Am. J. Surg. Pathol. 2018, 42, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Kiyono, T.; Ryo, E.; Ogawa, R.; Wakai, S.; Ichikawa, H.; Suzuki, K.; Arai, S.; Tsuta, K.; Ishida, M. Recurrent YAP1-MAML2 and YAP1-NUTM1 fusions in poroma and porocarcinoma. J. Clin. Investig. 2019, 129, 3827–3832. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.E.; Mitchell, C.M.; Strait, K.M.; Lathan, C.S.; Stelow, E.B.; Luer, S.C.; Muhammed, S.; Evans, A.G.; Sholl, L.M.; Rosai, J.; et al. Clinicopathologic features and long-term outcomes of NUT midline carcinoma. Clin. Cancer Res. 2012, 18, 5773–5779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, B.E.; Hofer, M.D.; Lemieux, M.E.; Bauer, D.E.; Cameron, M.J.; West, N.H.; Agoston, E.S.; Reynoird, N.; Khochbin, S.; Ince, T.A.; et al. Differentiation of NUT midline carcinoma by epigenomic reprogramming. Cancer Res. 2011, 71, 2686–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boer, J.M.; Valsecchi, M.G.; Hormann, F.M.; Antic, Z.; Zaliova, M.; Schwab, C.; Cazzaniga, G.; Arfeuille, C.; Cave, H.; Attarbaschi, A.; et al. Favorable outcome of NUTM1-rearranged infant and pediatric B cell precursor acute lymphoblastic leukemia in a collaborative international study. Leukemia 2021, 35, 2978–2982. [Google Scholar] [CrossRef]
- Underwood, C.I.M.; Cardona, D.M.; Bentley, R.C.; Shen, G.; Feng, X.; Jour, G.; Al-Rohil, R.N. Epithelioid Hyalinizing Sarcoma With MGA-NUTM1 Fusion. Am. J. Clin. Pathol. 2020, 154, 859–866. [Google Scholar] [CrossRef]
- Diolaiti, D.; Cruz, F.S.D.; Gundem, G.; Bouvier, N.; Boulad, M.; Zhang, Y.; Chou, A.J.; Dunkel, I.J.; Sanghvi, R.; Shah, M. A recurrent novel MGA–NUTM1 fusion identifies a new subtype of high-grade spindle cell sarcoma. Mol. Case Stud. 2018, 4, a003194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Loarer, F.; Pissaloux, D.; Watson, S.; Godfraind, C.; Galmiche-Rolland, L.; Silva, K.; Mayeur, L.; Italiano, A.; Michot, A.; Pierron, G. Clinicopathologic features of CIC-NUTM1 sarcomas, a new molecular variant of the family of CIC-fused sarcomas. Am. J. Surg. Pathol. 2019, 43, 268–276. [Google Scholar] [CrossRef]
- Van Treeck, B.J.; Thangaiah, J.J.; Torres-Mora, J.; Stevens, T.M.; Rothermundt, C.; Fassan, M.; Loupakis, F.; Diebold, J.; Hornick, J.L.; Halling, K.C.; et al. NUTM1-rearranged colorectal sarcoma: A clinicopathologically and genetically distinctive malignant neoplasm with a poor prognosis. Mod. Pathol. 2021, 34, 1547–1557. [Google Scholar] [CrossRef]
- Giridhar, P.; Mallick, S.; Kashyap, L.; Rath, G.K. Patterns of care and impact of prognostic factors in the outcome of NUT midline carcinoma: A systematic review and individual patient data analysis of 119 cases. Eur. Arch. Otorhinolaryngol. 2018, 275, 815–821. [Google Scholar] [CrossRef]
- Shehata, B.M.; Steelman, C.K.; Abramowsky, C.R.; Olson, T.A.; French, C.A.; Saxe, D.F.; Ricketts, R.R.; Katzenstein, H.M. NUT midline carcinoma in a newborn with multiorgan disseminated tumor and a 2-year-old with a pancreatic/hepatic primary. Pediatr. Dev. Pathol. 2010, 13, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Chau, N.G.; Hurwitz, S.; Mitchell, C.M.; Aserlind, A.; Grunfeld, N.; Kaplan, L.; Hsi, P.; Bauer, D.E.; Lathan, C.S.; Rodriguez-Galindo, C.; et al. Intensive treatment and survival outcomes in NUT midline carcinoma of the head and neck. Cancer 2016, 122, 3632–3640. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Kim, S.; Lee, J.K.; Yoon, S.O.; Park, H.S.; Hong, S.W.; Park, W.S.; Kim, J.E.; Kim, J.; Keam, B.; et al. Clinicopathological and Preclinical Findings of NUT Carcinoma: A Multicenter Study. Oncologist 2019, 24, e740–e748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, W.; French, C.A.; Cameron, M.J.; Han, Y.; Liu, H. Clinicopathological significance of NUT rearrangements in poorly differentiated malignant tumors of the upper respiratory tract. Int. J. Surg. Pathol. 2013, 21, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Salles, P.G.; Moura Rde, D.; Menezes, L.M.; Bacchi, C.E. Expression of P16 in NUT carcinomas with no association with human papillomavirus (HPV). Appl. Immunohistochem. Mol. Morphol. 2014, 22, 262–265. [Google Scholar] [CrossRef]
- Cho, Y.A.; Choi, Y.L.; Hwang, I.; Lee, K.; Cho, J.H.; Han, J. Clinicopathological characteristics of primary lung nuclear protein in testis carcinoma: A single-institute experience of 10 cases. Thorac. Cancer 2020, 11, 3205–3212. [Google Scholar] [CrossRef]
- French, C.A.; Kutok, J.L.; Faquin, W.C.; Toretsky, J.A.; Antonescu, C.R.; Griffin, C.A.; Nose, V.; Vargas, S.O.; Moschovi, M.; Tzortzatou-Stathopoulou, F.; et al. Midline carcinoma of children and young adults with NUT rearrangement. J. Clin. Oncol. 2004, 22, 4135–4139. [Google Scholar] [CrossRef]
- Den Bakker, M.A.; Beverloo, B.H.; van den Heuvel-Eibrink, M.M.; Meeuwis, C.A.; Tan, L.M.; Johnson, L.A.; French, C.A.; van Leenders, G.J. NUT midline carcinoma of the parotid gland with mesenchymal differentiation. Am. J. Surg. Pathol. 2009, 33, 1253–1258. [Google Scholar] [CrossRef]
- Agaimy, A.; Fonseca, I.; Martins, C.; Thway, K.; Barrette, R.; Harrington, K.J.; Hartmann, A.; French, C.A.; Fisher, C. NUT Carcinoma of the Salivary Glands. Am. J. Surg. Pathol. 2018, 42, 877–884. [Google Scholar] [CrossRef]
- Kakkar, A.; Antony, V.M.; Irugu, D.V.K.; Adhikari, N.; Jain, D. NUT Midline Carcinoma: A Series of Five Cases, Including One with Unusual Clinical Course. Head Neck Pathol. 2018, 12, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Haack, A.H.; Johnson, J.L.; Fry, D.C.; Crosby, B.K.; Polakiewicz, E.R.; Stelow, J.E.; Hong, A.S.-M.; Schwartz, C.B.; Cameron, C.M.; Rubin, A.M.; et al. Diagnosis of NUT Midline Carcinoma Using a NUT-specific Monoclonal Antibody. Am. J. Surg. Pathol. 2009, 33, 984–991. [Google Scholar] [CrossRef] [Green Version]
- Hung, Y.P.; Chen, A.L.; Taylor, M.S.; Huynh, T.G.; Kem, M.; Selig, M.K.; Nielsen, G.P.; Lennerz, J.K.; Azzoli, C.G.; Dagogo-Jack, I.; et al. Thoracic nuclear protein in testis (NUT) carcinoma: Expanded pathological spectrum with expression of thyroid transcription factor-1 and neuroendocrine markers. Histopathology 2021, 78, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Cho, J.; Baek, C.H.; Son, Y.I.; Jeong, H.S.; Chung, M.K.; Hong, S.D.; Ahn, Y.C.; Oh, D.R.; Noh, J.M.; et al. Prevalence of NUT carcinoma in head and neck: Analysis of 362 cases with literature review. Head Neck 2020, 42, 924–938. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.A.; Westra, W.H. NUT midline carcinomas of the sinonasal tract. Am. J. Surg. Pathol. 2012, 36, 1216–1221. [Google Scholar] [CrossRef] [Green Version]
- Evans, A.G.; French, C.A.; Cameron, M.J.; Fletcher, C.D.; Jackman, D.M.; Lathan, C.S.; Sholl, L.M. Pathologic characteristics of NUT midline carcinoma arising in the mediastinum. Am. J. Surg. Pathol. 2012, 36, 1222–1227. [Google Scholar] [CrossRef] [Green Version]
- Petrini, P.; French, C.A.; Rajan, A.; Cameron, M.J.; Jaffe, E.S.; Zucali, P.A.; Xie, J.; Wang, Y.; Giaccone, G. NUT rearrangement is uncommon in human thymic epithelial tumors. J. Thorac. Oncol. 2012, 7, 744–750. [Google Scholar] [CrossRef] [Green Version]
- Virarkar, M.; Saleh, M.; Ramani, N.S.; Morani, A.C.; Bhosale, P. Imaging spectrum of NUT carcinomas. Clin. Imaging 2020, 67, 198–206. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, L.; Palesandro, E.; Moretti, M.; Pelosi, G.; Fabbri, A.; Carnevale Schianca, F.; Aglietta, M.; Grignani, G. Alpha-fetoprotein elevation in NUT midline carcinoma: A case report. BMC Cancer 2017, 17, 266. [Google Scholar] [CrossRef] [Green Version]
- Arimizu, K.; Hirano, G.; Makiyama, C.; Matsuo, M.; Sasaguri, T.; Makiyama, A. NUT carcinoma of the nasal cavity that responded to a chemotherapy regimen for Ewing’s sarcoma family of tumors: A case report. BMC Cancer 2018, 18, 1134. [Google Scholar] [CrossRef]
- Watanabe, S.; Hirano, S.; Mine, S.; Yoshida, A.; Motoi, T.; Ishii, S.; Naka, G.; Takeda, Y.; Igari, T.; Sugiyama, H.; et al. A case of endobronchial NUT midline carcinoma with intraluminal growth. Anticancer Res. 2015, 35, 1607–1612. [Google Scholar]
- Mertens, F.; Wiebe, T.; Adlercreutz, C.; Mandahl, N.; French, C.A. Successful treatment of a child with t(15;19)-positive tumor. Pediatr. Blood Cancer 2007, 49, 1015–1017. [Google Scholar] [CrossRef] [PubMed]
- Engleson, J.; Soller, M.; Panagopoulos, I.; Dahlen, A.; Dictor, M.; Jerkeman, M. Midline carcinoma with t(15;19) and BRD4-NUT fusion oncogene in a 30-year-old female with response to docetaxel and radiotherapy. BMC Cancer 2006, 6, 69. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.; Chang, J.S. Durable Complete Remission of PD-L1 Positive NUT Carcinoma Treated With Concurrent Chemotherapy and Radiation. Perm. J. 2020, 25, 1–3. [Google Scholar] [CrossRef]
- Fekkar, A.; Emprou, C.; Lefebvre, C.; Ferretti, G.; Stephanov, O.; Pissaloux, D.; Mc Leer, A.; Toffart, A.C.; Rousseaux, S.; Khochbin, S.; et al. Thoracic NUT carcinoma: Common pathological features despite diversity of clinical presentations. Lung Cancer 2021, 158, 55–59. [Google Scholar] [CrossRef]
- Stevens, T.M.; Morlote, D.; Xiu, J.; Swensen, J.; Brandwein-Weber, M.; Miettinen, M.M.; Gatalica, Z.; Bridge, J.A. NUTM1-rearranged neoplasia: A multi-institution experience yields novel fusion partners and expands the histologic spectrum. Mod. Pathol. 2019, 32, 764. [Google Scholar] [CrossRef]
- Wang, R.; Liu, W.; Helfer, C.M.; Bradner, J.E.; Hornick, J.L.; Janicki, S.M.; French, C.A.; You, J. Activation of SOX2 expression by BRD4-NUT oncogenic fusion drives neoplastic transformation in NUT midline carcinoma. Cancer Res. 2014, 74, 3332–3343. [Google Scholar] [CrossRef] [Green Version]
- Grayson, A.R.; Walsh, E.M.; Cameron, M.J.; Godec, J.; Ashworth, T.; Ambrose, J.M.; Aserlind, A.B.; Wang, H.; Evan, G.; Kluk, M.J.; et al. MYC, a downstream target of BRD-NUT, is necessary and sufficient for the blockade of differentiation in NUT midline carcinoma. Oncogene 2014, 33, 1736–1742. [Google Scholar] [CrossRef] [Green Version]
- Sholl, L.M.; Barletta, J.A.; Yeap, B.Y.; Chirieac, L.R.; Hornick, J.L. Sox2 protein expression is an independent poor prognostic indicator in stage I lung adenocarcinoma. Am. J. Surg. Pathol. 2010, 34, 1193–1198. [Google Scholar] [CrossRef]
- Santagata, S.; Ligon, K.L.; Hornick, J.L. Embryonic stem cell transcription factor signatures in the diagnosis of primary and metastatic germ cell tumors. Am. J. Surg. Pathol. 2007, 31, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Kwak, Y.; Nam, K.H.; Kim, D.W.; Kang, S.B.; Choe, G.; Kim, W.H.; Lee, H.S. Favorable prognosis in colorectal cancer patients with co-expression of c-MYC and ss-catenin. BMC Cancer 2016, 16, 730. [Google Scholar] [CrossRef] [Green Version]
- Chisholm, K.M.; Krishnan, C.; Heerema-McKenney, A.; Natkunam, Y. Immunohistochemical Profile of MYC Protein in Pediatric Small Round Blue Cell Tumors. Pediatr. Dev. Pathol. 2017, 20, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Numakura, S.; Saito, K.; Motoi, N.; Mori, T.; Saito, Y.; Yokote, F.; Kanamoto, Y.; Asami, M.; Sakai, T.; Yamauchi, Y.; et al. P63-negative pulmonary NUT carcinoma arising in the elderly: A case report. Diagn. Pathol. 2020, 15, 134. [Google Scholar] [CrossRef]
- Prall, O.W.J.; Thio, N.; Yerneni, S.; Kumar, B.; McEvoy, C.R. A NUT carcinoma lacking squamous differentiation and expressing TTF1. Pathology 2021, 53, 663–666. [Google Scholar] [CrossRef]
- Kuo, L.E.; Barletta, J.; Schoenfeld, J.D.; White, A.; French, C.A.; Wong, K.S.; Swanson, J.R.; Lowe, C.A.; Danga, K.; Moore, F.D., Jr.; et al. NUT Carcinoma of the Thyroid: An Unusual Case with a Complete Response to Treatment. Clin. Thyroidol. 2021, 33, 38–47. [Google Scholar] [CrossRef]
- Agaimy, A.; Haller, F.; Renner, A.; Niedermeyer, J.; Hartmann, A.; French, C.A. Misleading Germ Cell Phenotype in Pulmonary NUT Carcinoma Harboring the ZNF532-NUTM1 Fusion. Am. J. Surg. Pathol. 2021. [Google Scholar] [CrossRef]
- Ko, K.; Kitani, T.; Harris, B.T.; Anaizi, A.N.; Solomon, D.; Perry, A.; Toretsky, J.; Ozdemirli, M. A novel PARD3B-NUTM1 fusion in an aggressive primary CNS embryonal tumor in a young adult. Acta. Neuropathol. Commun. 2020, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Chen, X.; Cao, X.; Wang, F.; Zhang, Y.; Ma, X.; Cao, P.; Fang, J.; Chen, J.; Zhou, X.; et al. Identification of a novel AVEN-NUTM1 fusion gene in acute myeloid leukemia. Int. J. Lab. Hematol. 2021, 43, O207–O210. [Google Scholar] [CrossRef]
- Pincez, T.; Landry, J.R.; Roussy, M.; Jouan, L.; Bilodeau, M.; Laramee, L.; Couture, F.; Sinnett, D.; Gendron, P.; Hebert, J.; et al. Cryptic recurrent ACIN1-NUTM1 fusions in non-KMT2A-rearranged infant acute lymphoblastic leukemia. Genes Chromosomes Cancer 2020, 59, 125–130. [Google Scholar] [CrossRef]
- Maffini, F.; French, C.A.; Cameron, M.J.; Stufano, V.; Barberis, M.; Pisa, E.; Manzotti, M.; Cattaneo, A.; De Fiori, E.; Viale, G. A case of NUT midline carcinoma with no HPV infection, slight EWSR1 rearrangement and strong expression of EGFR. Tumori 2013, 99, e152–e155. [Google Scholar] [CrossRef] [PubMed]
- French, C.A. Demystified molecular pathology of NUT midline carcinomas. J. Clin. Pathol. 2010, 63, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEvoy, C.R.; Holliday, H.; Thio, N.; Mitchell, C.; Choong, D.Y.; Yellapu, B.; Leong, H.S.; Xu, H.; Lade, S.; Browning, J.; et al. A MXI1-NUTM1 fusion protein with MYC-like activity suggests a novel oncogenic mechanism in a subset of NUTM1-rearranged tumors. Lab. Investig. 2021, 101, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Reynoird, N.; Schwartz, B.E.; Delvecchio, M.; Sadoul, K.; Meyers, D.; Mukherjee, C.; Caron, C.; Kimura, H.; Rousseaux, S.; Cole, P.A.; et al. Oncogenesis by sequestration of CBP/p300 in transcriptionally inactive hyperacetylated chromatin domains. EMBO J. 2010, 29, 2943–2952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Louzada, S.; An, Y.; Kim, S.Y.; Kim, S.; Youk, J.; Park, S.; Koo, S.H.; Keam, B.; Jeon, Y.K.; et al. Complex chromosomal rearrangements by single catastrophic pathogenesis in NUT midline carcinoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Stirnweiss, A.; Oommen, J.; Kotecha, R.S.; Kees, U.R.; Beesley, A.H. Molecular-genetic profiling and high-throughput in vitro drug screening in NUT midline carcinoma-an aggressive and fatal disease. Oncotarget 2017, 8, 112313–112329. [Google Scholar] [CrossRef] [Green Version]
- Tamura, R.; Nakaoka, H.; Yoshihara, K.; Mori, Y.; Yachida, N.; Nishikawa, N.; Motoyama, T.; Okuda, S.; Inoue, I.; Enomoto, T. Novel MXD4–NUTM1 fusion transcript identified in primary ovarian undifferentiated small round cell sarcoma. Genes Chromosomes Cancer 2018, 57, 557–563. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Granada, C.; Morlote, D.; Pavlidakey, P.; Rodriguez-Waitkus, P.; Ramirez, C.; Florento, E.; Swensen, J.; Gatalica, Z.; Stevens, T.M. Poroid adnexal skin tumors with YAP1 fusions exhibit similar histopathologic features: A series of six YAP1-rearranged adnexal skin tumors. J. Cutan. Pathol. 2021. [Google Scholar] [CrossRef]
- Agaimy, A.; Togel, L.; Haller, F.; Zenk, J.; Hornung, J.; Markl, B. YAP1-NUTM1 Gene Fusion in Porocarcinoma of the External Auditory Canal. Head Neck Pathol. 2020, 14, 982–990. [Google Scholar] [CrossRef]
- Stathis, A.; Bertoni, F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018, 8, 24–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.C.; Simon, B.; Rybin, V.; Grotsch, H.; Curtet, S.; Khochbin, S.; Carlomagno, T.; Muller, C.W. A bromodomain-DNA interaction facilitates acetylation-dependent bivalent nucleosome recognition by the BET protein BRDT. Nat. Commun. 2016, 7, 13855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Cao, X.J.; Kulej, K.; Liu, W.; Ma, T.; MacDonald, M.; Chiang, C.M.; Garcia, B.A.; You, J. Uncovering BRD4 hyperphosphorylation associated with cellular transformation in NUT midline carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, E5352–E5361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; You, J. Mechanistic analysis of the role of bromodomain-containing protein 4 (BRD4) in BRD4-NUT oncoprotein-induced transcriptional activation. J. Biol. Chem. 2015, 290, 2744–2758. [Google Scholar] [CrossRef] [Green Version]
- Alekseyenko, A.A.; Walsh, E.M.; Wang, X.; Grayson, A.R.; Hsi, P.T.; Kharchenko, P.V.; Kuroda, M.I.; French, C.A. The oncogenic BRD4-NUT chromatin regulator drives aberrant transcription within large topological domains. Genes Dev. 2015, 29, 1507–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Diaz, J.; Jiao, J.; Wang, R.; You, J. Perturbation of BRD4 protein function by BRD4-NUT protein abrogates cellular differentiation in NUT midline carcinoma. J. Biol. Chem. 2011, 286, 27663–27675. [Google Scholar] [CrossRef] [Green Version]
- Shiota, H.; Elya, J.E.; Alekseyenko, A.A.; Chou, P.M.; Gorman, S.A.; Barbash, O.; Becht, K.; Danga, K.; Kuroda, M.I.; Nardi, V.; et al. “Z4” Complex Member Fusions in NUT Carcinoma: Implications for a Novel Oncogenic Mechanism. Mol. Cancer Res. 2018, 16, 1826–1833. [Google Scholar] [CrossRef] [Green Version]
- Alekseyenko, A.A.; Walsh, E.M.; Zee, B.M.; Pakozdi, T.; Hsi, P.; Lemieux, M.E.; Dal Cin, P.; Ince, T.A.; Kharchenko, P.V.; Kuroda, M.I.; et al. Ectopic protein interactions within BRD4-chromatin complexes drive oncogenic megadomain formation in NUT midline carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, E4184–E4192. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Wang, J.; Rousseaux, S.; Tan, M.; Pan, L.; Peng, L.; Wang, S.; Xu, W.; Ren, J.; Liu, Y.; et al. Metabolically controlled histone H4K5 acylation/acetylation ratio drives BRD4 genomic distribution. Cell Rep. 2021, 36, 109460. [Google Scholar] [CrossRef] [PubMed]
- Sakamaki, J.I.; Wilkinson, S.; Hahn, M.; Tasdemir, N.; O’Prey, J.; Clark, W.; Hedley, A.; Nixon, C.; Long, J.S.; New, M.; et al. Bromodomain Protein BRD4 Is a Transcriptional Repressor of Autophagy and Lysosomal Function. Mol. cell 2017, 66, 517–532.e519. [Google Scholar] [CrossRef]
- Stirnweiss, A.; McCarthy, K.; Oommen, J.; Crook, M.L.; Hardy, K.; Kees, U.R.; Wilton, S.D.; Anazodo, A.; Beesley, A.H. A novel BRD4-NUT fusion in an undifferentiated sinonasal tumor highlights alternative splicing as a contributing oncogenic factor in NUT midline carcinoma. Oncogenesis 2015, 4, e174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, Y.W.; Hsieh, T.H.; Chu, P.Y.; Hsieh, S.M.; Liu, M.L.; Lee, J.C.; Liu, Y.R.; Ku, J.W.; Kao, Y.C. Primary malignant epithelioid and rhabdoid tumor of bone harboring ZNF532-NUTM1 fusion: The expanding NUT cancer family. Genes Chromosomes Cancer 2019, 58, 809–814. [Google Scholar] [CrossRef]
- Xie, X.H.; Wang, L.Q.; Qin, Y.Y.; Lin, X.Q.; Xie, Z.H.; Liu, M.; Zhang, J.X.; Ouyang, M.; Liu, J.; Gu, Y.Y.; et al. Clinical features, treatment, and survival outcome of primary pulmonary NUT midline carcinoma. Orphanet. J. Rare Dis. 2020, 15, 183. [Google Scholar] [CrossRef]
- Cheng, Z.; Luo, Y.; Zhang, Y.; Wang, Y.; Chen, Y.; Xu, Y.; Peng, H.; Zhang, G. A novel NAP1L4/NUTM1 fusion arising from translocation t(11;15)(p15;q12) in a myeloid neoplasm with eosinophilia and rearrangement of PDGFRA highlights an unusual clinical feature and therapeutic reaction. Ann. Hematol. 2020, 99, 1561–1564. [Google Scholar] [CrossRef]
- Watson, S.; Perrin, V.; Guillemot, D.; Reynaud, S.; Coindre, J.M.; Karanian, M.; Guinebretiere, J.M.; Freneaux, P.; Le Loarer, F.; Bouvet, M.; et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J. Pathol. 2018, 245, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Howard, H.C.; Mount, D.B.; Rochefort, D.; Byun, N.; Dupre, N.; Lu, J.; Fan, X.; Song, L.; Riviere, J.B.; Prevost, C.; et al. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat. Genet. 2002, 32, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Hormann, F.M.; Hoogkamer, A.Q.; Beverloo, H.B.; Boeree, A.; Dingjan, I.; Wattel, M.M.; Stam, R.W.; Escherich, G.; Pieters, R.; den Boer, M.L.; et al. NUTM1 is a recurrent fusion gene partner in B-cell precursor acute lymphoblastic leukemia associated with increased expression of genes on chromosome band 10p12.31-12.2. Haematologica 2019, 104, e455–e459. [Google Scholar] [CrossRef] [Green Version]
- De Luca, V.; Wang, H.; Squassina, A.; Wong, G.W.; Yeomans, J.; Kennedy, J.L. Linkage of M5 muscarinic and alpha7-nicotinic receptor genes on 15q13 to schizophrenia. Neuropsychobiology 2004, 50, 124–127. [Google Scholar] [CrossRef]
- Macagno, N.; Kervarrec, T.; Sohier, P.; Poirot, B.; Haffner, A.; Carlotti, A.; Balme, B.; Castillo, C.; Jullie, M.L.; Osio, A.; et al. NUT Is a Specific Immunohistochemical Marker for the Diagnosis of YAP1-NUTM1-rearranged Cutaneous Poroid Neoplasms. Am. J. Surg. Pathol. 2021, 45, 1221–1227. [Google Scholar] [CrossRef]
- Stathis, A.; Zucca, E.; Bekradda, M.; Gomez-Roca, C.; Delord, J.P.; de La Motte Rouge, T.; Uro-Coste, E.; de Braud, F.; Pelosi, G.; French, C.A. Clinical Response of Carcinomas Harboring the BRD4-NUT Oncoprotein to the Targeted Bromodomain Inhibitor OTX015/MK-8628. Cancer Discov. 2016, 6, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; Maertens, O.; Cichowski, K.; Elledge, S.J. Genetic modifiers of the BRD4-NUT dependency of NUT midline carcinoma uncovers a synergism between BETis and CDK4/6is. Genes Dev. 2018, 32, 1188–1200. [Google Scholar] [CrossRef] [Green Version]
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P.; et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol. 2016, 3, e186–e195. [Google Scholar] [CrossRef]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef]
- Zhang, X.; Zegar, T.; Lucas, A.; Morrison-Smith, C.; Knox, T.; French, C.A.; Knapp, S.; Muller, S.; Siveke, J.T. Therapeutic targeting of p300/CBP HAT domain for the treatment of NUT midline carcinoma. Oncogene 2020, 39, 4770–4779. [Google Scholar] [CrossRef]
- Morrison-Smith, C.D.; Knox, T.M.; Filic, I.; Soroko, K.M.; Eschle, B.K.; Wilkens, M.K.; Gokhale, P.C.; Giles, F.; Griffin, A.; Brown, B.; et al. Combined Targeting of the BRD4-NUT-p300 Axis in NUT Midline Carcinoma by Dual Selective Bromodomain Inhibitor, NEO2734. Mol. Cancer Ther. 2020, 19, 1406–1414. [Google Scholar] [CrossRef]
- Maur, M.; Toss, A.; Dominici, M.; Frassoldati, A.; Corradini, P.; Maiorana, A.; Fontana, A.; Conte, P. Impressive Response to Dose-Dense Chemotherapy in a Patient with NUT Midline Carcinoma. Am. J. Case. Rep. 2015, 16, 424–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Atoyan, R.; Borek, M.A.; Dellarocca, S.; Samson, M.E.; Ma, A.W.; Xu, G.X.; Patterson, T.; Tuck, D.P.; Viner, J.L.; et al. Dual HDAC and PI3K Inhibitor CUDC-907 Downregulates MYC and Suppresses Growth of MYC-dependent Cancers. Mol. Cancer Ther. 2017, 16, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteves, G.; Ferreira, J.; Afonso, R.; Martins, C.; Zagalo, C.; Felix, A. HDAC Overexpression in a NUT Midline Carcinoma of the Parotid Gland with Exceptional Survival: A Case Report. Head Neck Pathol. 2020, 14, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Orr, B.A.; Toprak, U.H.; Hovestadt, V.; Jones, D.T.W.; Capper, D.; Sill, M.; Buchhalter, I.; Northcott, P.A.; Leis, I.; et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 2016, 164, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangray, S.; Kelly, D.R.; LeGuellec, S.; Fridman, E.; Aggarwal, S.; Shago, M.; Matoso, A.; Madison, R.; Pramanik, S.; Zhong, S.; et al. Clinicopathologic Features of a Series of Primary Renal CIC-rearranged Sarcomas With Comprehensive Molecular Analysis. Am. J. Surg. Pathol. 2018, 42, 1360–1369. [Google Scholar] [CrossRef]
- Schaefer, I.M.; Dal Cin, P.; Landry, L.M.; Fletcher, C.D.; Hanna, G.J.; French, C.A. CIC-NUTM1 fusion: A case which expands the spectrum of NUT-rearranged epithelioid malignancies. Genes Chromosomes Cancer 2018, 57, 446–451. [Google Scholar] [CrossRef]
- Siegfried, A.; Masliah-Planchon, J.; Roux, F.E.; Larrieu-Ciron, D.; Pierron, G.; Nicaise, Y.; Gambart, M.; Catalaa, I.; Pericart, S.; Dubucs, C.; et al. Brain tumor with an ATXN1-NUTM1 fusion gene expands the histologic spectrum of NUTM1-rearranged neoplasia. Acta. Neuropathol. Commun. 2019, 7, 220. [Google Scholar] [CrossRef] [Green Version]
- Klossowski, S.; Miao, H.; Kempinska, K.; Wu, T.; Purohit, T.; Kim, E.; Linhares, B.M.; Chen, D.; Jih, G.; Perkey, E.; et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J. Clin. Investig. 2020, 130, 981–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Chastain, K. NUT midline carcinoma with leukemic presentation mimicking CD34-positive acute leukemia. Blood 2018, 132, 456. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Partner | Primary Tumor Site | Histology | Specimen Site |
|---|---|---|---|
| BRD4 | Skin, NOS | NUT carcinoma | Liver |
| BRD4 | Upper lobe, lung | Adenocarcinoma, metastatic, NOS | Arm, NOS |
| BRD4 | Lower lobe, lung | Squamous cell carcinoma, NOS | Mediastinum, NOS |
| NSD3 | Lung, NOS | Basaloid carcinoma | Inguinal region, NOS |
| BRD4 | Paranasal sinus | Carcinoma, NOS, midline carcinoma of children and young adults with NUT rearrangement | Eyelid |
| NSD3 | Lung, NOS | Squamous cell carcinoma, NOS | Lung, NOS |
| NSD3 | Upper lobe, lung | Squamous cell carcinoma, NOS | Pleura, NOS |
| BRD4 | Lower lobe, lung | NUT midline carcinoma | Mesentery |
| BRD4 | Upper lobe, lung | Squamous cell carcinoma, NOS | Upper lobe, lung |
| NSD3 | Lower lobe, lung | NUT midline carcinoma | Mediastinal lymph node |
| BRD4 | Nasal cavity | NUT carcinoma | Nasal cavity |
| BRD4 | Nasopharynx, NOS | Squamous cell carcinoma, NOS | Nasal cavity |
| BRD4 | Upper lobe, lung | Squamous cell carcinoma, metastatic, NOS | Mediastinal lymph node |
| BRD4 | Frontal sinus | NUT midline carcinoma | Frontal sinus |
| BRD4 | Upper lobe, lung | Carcinoma, NOS | Pleura, NOS |
| NSD3 | Lower lobe, lung | Non-small cell carcinoma | Lower lobe, lung |
| BRD4 | Upper lobe, lung | NUT midline carcinoma | Upper lobe, lung |
| NSD3 | Maxillary sinus | NUT carcinoma | Lymph node, NOS |
| NSD3 | Lung, NOS | Carcinoma, undifferentiated, NOS | Lung, NOS |
| NSD3 | Unknown primary site | NUT carcinoma | Mediastinal lymph node |
| BRD4 | Mediastinum, NOS | Neoplasm, malignant | Mediastinum, NOS |
| NSD3 | Lung, NOS | NUT midline carcinoma | Supraclavicular lymph node |
| BRD3 | Lower lobe, lung | NUT carcinoma | Lower lobe, lung |
| BRD4 | Ethmoid sinus | Malignancy, NUT carcinoma | Eye, NOS, Orbit, NOS |
| BRD4 | Upper lobe, lung | NUT carcinoma | Subclavicular lymph node |
| BRD3 | Upper lobe, lung | NUT carcinoma | Upper lobe, lung |
| NSD3 | Thyroid, NOS | Carcinoma, metastatic, NOS | Pretracheal lymph node |
| BRD4 | Upper lobe, lung | NUT midline carcinoma | Upper lobe, lung |
| BRD4 | Nasal cavity | NUT carcinoma | Skull, NOS |
| BRD4 | Upper lobe, lung | Squamous cell carcinoma, NOS | Upper lobe, lung |
| NSD3 | Lung, NOS | Squamous cell carcinoma, NOS | Main bronchus |
| NSD3 | Thyroid, NOS | Carcinoma, NOS | Thyroid gland |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, W.; Stevens, T.M.; Stafford, P.; Miettinen, M.; Gatalica, Z.; Vranic, S. NUTM1-Rearranged Neoplasms—A Heterogeneous Group of Primitive Tumors with Expanding Spectrum of Histology and Molecular Alterations—An Updated Review. Curr. Oncol. 2021, 28, 4485-4503. https://doi.org/10.3390/curroncol28060381
Luo W, Stevens TM, Stafford P, Miettinen M, Gatalica Z, Vranic S. NUTM1-Rearranged Neoplasms—A Heterogeneous Group of Primitive Tumors with Expanding Spectrum of Histology and Molecular Alterations—An Updated Review. Current Oncology. 2021; 28(6):4485-4503. https://doi.org/10.3390/curroncol28060381
Chicago/Turabian StyleLuo, Wenyi, Todd M. Stevens, Phillip Stafford, Markku Miettinen, Zoran Gatalica, and Semir Vranic. 2021. "NUTM1-Rearranged Neoplasms—A Heterogeneous Group of Primitive Tumors with Expanding Spectrum of Histology and Molecular Alterations—An Updated Review" Current Oncology 28, no. 6: 4485-4503. https://doi.org/10.3390/curroncol28060381
APA StyleLuo, W., Stevens, T. M., Stafford, P., Miettinen, M., Gatalica, Z., & Vranic, S. (2021). NUTM1-Rearranged Neoplasms—A Heterogeneous Group of Primitive Tumors with Expanding Spectrum of Histology and Molecular Alterations—An Updated Review. Current Oncology, 28(6), 4485-4503. https://doi.org/10.3390/curroncol28060381