Possible Therapeutic Potential of Disulfiram for Multiple Myeloma


Abstract
1. Characteristic of Multiple Myeloma
1.1. Development of B-Lymphocytes and Plasma Cells Is Crucial for the Pathogenesis
1.2. Role of Plasma Cells in Disease Progression
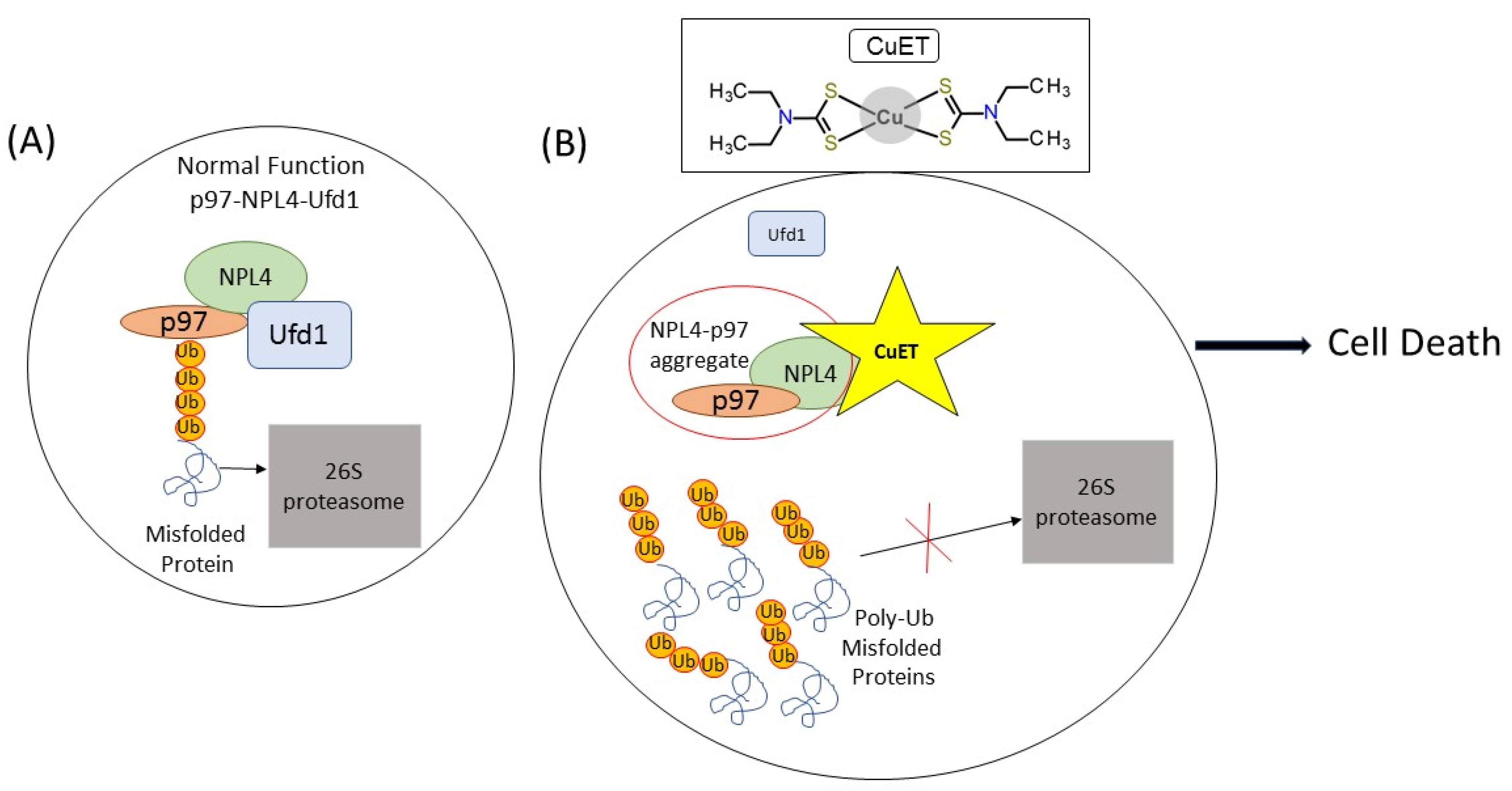
2. Disulfiram
Disulfiram Treatment Potential
3. Conclusions
Funding
Conflicts of Interest
References
- Alexander, D.D.; Mink, P.J.; Adami, H.-O.; Cole, P.; Mandel, J.S.; Oken, M.M.; Trichopoulos, D. Multiple myeloma: A review of the epidemiologic literature. Int. J. Cancer 2007, 120 (Suppl. 12), 40–61. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple myeloma: 2018 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2018, 93, 1091–1110. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Kuehl, W.M. Chromosome translocations in multiple myeloma. Oncogene 2001, 20, 5611–5622. [Google Scholar] [CrossRef]
- Novosadová, M. Léčba mnohočetného myelomu včera, dnes a zítra—repetitorium pro lékárníky. Prakt. Lékarenství 2016, 12, e25–e37. Available online: https://www.praktickelekarenstvi.cz/pdfs/lek/2016/92/05.pdf (accessed on 30 March 2021). [CrossRef]
- International Myeloma Foundation, © 1990–2020. What Are MGUS, Smoldering Myeloma, and MM? Available online: https://www.myeloma.org/what-are-mgus-smm-mm (accessed on 30 March 2021).
- Rajkumar, S.V. Multiple myeloma: 2016 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2016, 91, 719–734. [Google Scholar] [CrossRef]
- Ho, M.; Patel, A.; Hanley, C.; Murphy, A.; McSweeney, T.; Zhang, L.; McCann, A.; O’Gorman, P.; Bianchi, G. Exploiting autophagy in multiple myeloma. J. Cancer Metastasis Treat. 2019, 2019. [Google Scholar] [CrossRef]
- International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: A report of the International Myeloma Working Group. Br. J. Haematol. 2003, 121, 749–757. [Google Scholar] [CrossRef]
- Wu, H.; Huang, T.; Ye, Z.; Fu, X.; Hu, K.; Yang, X. Correlation of MicroRNA 17-92 Cluster Host Gene (MIR17HG) Polymorphisms with Susceptibility and Prognosis for Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, e359–e366. [Google Scholar] [CrossRef] [PubMed]
- Gerecke, C.; Fuhrmann, S.; Strifler, S.; Schmidt-Hieber, M.; Einsele, H.; Knop, S. The Diagnosis and Treatment of Multiple Myeloma. Dtsch. Aerzteblatt Online 2016, 113, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Naymagon, L.; Abdul-Hay, M. Novel agents in the treatment of multiple myeloma: A review about the future. J. Hematol. Oncol. 2016, 9, 1–20. [Google Scholar] [CrossRef]
- Shelef, M.; Calame, K. Regulation of plasma-cell development. Nat. Rev. Immunol. 2005, 5, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Busslinger, M. Transcriptional Control of Early B Cell Development. Annu. Rev. Immunol. 2004, 22, 55–79. [Google Scholar] [CrossRef] [PubMed]
- Pieper, K.; Grimbacher, B.; Eibel, H. B-cell biology and development. J. Allergy Clin. Immunol. 2013, 131, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Mackay, I.R.; Rose, N.R. The Autoimmune Diseases, 5th ed.; Elsevier Inc: Amsterdam, The Netherlands, 2013. [Google Scholar] [CrossRef]
- Pilzecker, B.; Jacobs, H. Mutating for Good: DNA Damage Responses during Somatic Hypermutation. Front. Immunol. 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- LeBien, T.W. B Cell Development. Fetal Neonatal Physiol. 2017, 1202–1207. [Google Scholar] [CrossRef]
- Bird, S.A.; Boyd, K. Multiple myeloma: An overview of management. Palliat. Care Soc. Pract. 2019, 13, 13. [Google Scholar] [CrossRef]
- Hideshima, T.; Bergsagel, P.L.; Kuehl, W.M.; Anderson, K.C. Advances in biology of multiple myeloma: Clinical applications. Blood 2004, 104, 607–618. [Google Scholar] [CrossRef]
- Roulland, S.; Suarez, F.; Hermine, O.; Nadel, B. Pathophysiological aspects of memory B-cell development. Trends Immunol. 2008, 29, 25–33. [Google Scholar] [CrossRef]
- Rose, N.R.; Mackay, I.R. (Eds.) The Autoimmune Diseases; Elsevier: Amsterdam, The Netherlands, 2006; ISBN 9780125959612. [Google Scholar] [CrossRef]
- Calame, K.L.; Lin, K.-I.; Tunyaplin, C. Regulatory mechanisms that determine the development and function of plasma cells. Annu. Rev. Immunol. 2003, 21, 205–230. [Google Scholar] [CrossRef]
- Shelef, M.; Lin, K.-I.; Savitsky, D.; Liao, J.; Calame, K. Blimp-1 is required for maintenance of long-lived plasma cells in the bone marrow. J. Exp. Med. 2005, 202, 1471–1476. [Google Scholar] [CrossRef]
- Klein, B.; Tarte, K.; Jourdan, M.; Mathouk, K.; Moreaux, J.; Jourdan, E.; Legouffe, E.; De Vos, J.; Rossic, J.F. Survival and Proliferation Factors of Normal and Malignant Plasma Cells. Int. J. Hematol. 2003, 78, 106–113. [Google Scholar] [CrossRef]
- Oracki, S.A.; Walker, J.A.; Hibbs, M.L.; Corcoran, L.M.; Tarlinton, D.M. Plasma cell development and survival. Immunol. Rev. 2010, 237, 140–159. [Google Scholar] [CrossRef]
- Lightman, S.M.; Utley, A.; Lee, K.P. Survival of Long-Lived Plasma Cells (LLPC): Piecing Together the Puzzle. Front. Immunol. 2019, 10, 965. [Google Scholar] [CrossRef] [PubMed]
- Brynjolfsson, S.F.; Berg, L.P.; Ekerhult, T.O.; Rimkute, I.; Wick, M.-J.; Mårtensson, I.-L.; Grimsholm, O. Long-Lived Plasma Cells in Mice and Men. Front. Immunol. 2018, 9, 2673. [Google Scholar] [CrossRef] [PubMed]
- Davenport, E.L.; Moore, H.E.; Dunlop, A.S.; Sharp, S.Y.; Workman, P.; Morgan, G.J.; Davies, F.E. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood 2007, 110, 2641–2649. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef]
- Manier, S.; Huynh, D.; Shen, Y.J.; Zhou, J.; Yusufzai, T.; Salem, K.Z.; Ebright, R.Y.; Shi, J.; Park, J.; Glavey, S.V.; et al. Inhibiting the oncogenic translation program is an effective therapeutic strategy in multiple myeloma. Sci. Transl. Med. 2017, 9, eaal2668. [Google Scholar] [CrossRef]
- Pawlyn, C.; Morgan, G. Evolutionary biology of high-risk multiple myeloma. Nat. Rev. Cancer 2017, 17, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Touzeau, C.; Maciag, P.; Amiot, M.; Moreau, P. Targeting Bcl-2 for the treatment of multiple myeloma. Leukemia 2018, 32, 1899–1907. [Google Scholar] [CrossRef]
- Nikesitch, N.; Lee, J.M.; Ling, S.; Roberts, T.L. Endoplasmic reticulum stress in the development of multiple myeloma and drug resistance. Clin. Transl. Immunol. 2018, 7, e1007. [Google Scholar] [CrossRef]
- Gabrea, A.; Bergsagel, P.; Chesi, M.; Shou, Y.; Kuehl, W. Insertion of Excised IgH Switch Sequences Causes Overexpression of Cyclin D1 in a Myeloma Tumor Cell. Mol. Cell 1999, 3, 119–123. [Google Scholar] [CrossRef]
- Burger, R.; Günther, A.; Klausz, K.; Staudinger, M.; Peipp, M.; Penas, E.M.M.; Rose-John, S.; Wijdenes, J.; Gramatzki, M. Due to interleukin-6 type cytokine redundancy only glycoprotein 130 receptor blockade efficiently inhibits myeloma growth. Haematol. 2016, 102, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Harmer, D.; Falank, C.; Reagan, M.R. Interleukin-6 Interweaves the Bone Marrow Microenvironment, Bone Loss, and Multiple Myeloma. Front. Endocrinol. 2019, 9, 788. [Google Scholar] [CrossRef] [PubMed]
- Vrábel, D.; Pour, L.; Ševčíková, S. The impact of NF-κB signaling on pathogenesis and current treatment strategies in multiple myeloma. Blood Rev. 2019, 34, 56–66. [Google Scholar] [CrossRef]
- Wong, A.H.-H.; Shin, E.M.; Tergaonkar, V.; Chng, W.-J. Targeting NF-kB Signaling for Multiple Myeloma. Cancers 2020, 12, 2203. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.C. Progress and Paradigms in Multiple Myeloma. Clin. Cancer Res. 2016, 22, 5419–5427. [Google Scholar] [CrossRef]
- Borjan, B.; Kern, J.; Steiner, N.; Gunsilius, E.; Wolf, D.; Untergasser, G. Spliced XBP1 Levels Determine Sensitivity of Multiple Myeloma Cells to Proteasome Inhibitor Bortezomib Independent of the Unfolded Protein Response Mediator GRP78. Front. Oncol. 2020, 9, 1530. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef]
- Michallet, A.-S.; Mondiere, P.; Taillardet, M.; Leverrier, Y.; Genestier, L.; Defrance, T. Compromising the Unfolded Protein Response Induces Autophagy-Mediated Cell Death in Multiple Myeloma Cells. PLoS ONE 2011, 6, e25820. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Shen, J.; Yan, H.; Gao, X.; Dong, T.; Wang, P.; Zhou, J. The Evolving Role of Disulfiram in Radiobiology and the Treatment of Breast Cancer. OncoTargets Ther. 2020, 13, 10441–10446. [Google Scholar] [CrossRef]
- Meraz-Torres, F.; Plöger, S.; Garbe, C.; Niessner, H.; Sinnberg, T. Disulfiram as a Therapeutic Agent for Metastatic Malignant Melanoma—Old Myth or New Logos? Cancers 2020, 12, 3538. [Google Scholar] [CrossRef]
- Skrott, Z.; Mistrik, M.; Andersen, K.K.; Friis, S.; Majera, D.; Gursky, J.; Oždian, T.; Bartkova, J.; Turi, Z.; Moudry, P.; et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nat. Cell Biol. 2017, 552, 194–199. [Google Scholar] [CrossRef]
- Center for Substance Abuse Treatment. Incorporating Alcohol Pharmacotherapies Into Medical Practice; Treatment Improvement Protocol (TIP) Series, No. 49; Chapter 3—Disulfiram; Substance Abuse and Mental Health Services Administration (US): Rockville, MD, USA, 2009. Available online: https://www.ncbi.nlm.nih.gov/books/NBK64036/ (accessed on 21 March 2021).
- PubChem [Internet]. PubChem Compound Summary for CID 3117, Disulfiram; National Library of Medicine (US), National Center for Biotechnology Information: Bethesda, MD, USA, 2004. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Disulfiram (accessed on 21 May 2021).
- Barth, K.S.; Malcolm, R.J. Disulfiram: An Old Therapeutic with New Applications. CNS Neurol. Disord. Drug Targets 2010, 9, 5–12. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 3117, Disulfiram. 2021. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Disulfiram (accessed on 31 March 2021).
- Skrott, Z.; Majera, D.; Gursky, J.; Buchtova, T.; Hajduch, M.; Mistrik, M.; Bartek, J. Disulfiram’s anti-cancer activity reflects targeting NPL4, not inhibition of aldehyde dehydrogenase. Oncogene 2019, 38, 6711–6722. [Google Scholar] [CrossRef]
- Kranzler, H.R. (Ed.) The Pharmacology of Alcohol Abuse. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 1995; ISBN 978-3-642-78437-8. [Google Scholar] [CrossRef]
- Pye, V.E.; Beuron, F.; Keetch, C.A.; McKeown, C.; Robinson, C.V.; Meyer, H.H.; Zhang, X.; Freemont, P.S. Structural insights into the p97-Ufd1-Npl4 complex. Proc. Natl. Acad. Sci. USA 2007, 104, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Yamada, Y.; Enokida, H.; Osako, Y.; Tsuruda, M.; Kuroshima, K.; Sakaguchi, T.; Sugita, S.; Tatarano, S.; Nakagawa, M. Targeting NPL4 via drug repositioning using disulfiram for the treatment of clear cell renal cell carcinoma. PLoS ONE 2020, 15, e0236119. [Google Scholar] [CrossRef] [PubMed]
- Masaki, R. Mechanism of action of bortezomib in multiple myeloma therapy. Int. J. Myeloma 2016, 6, 1–6. Available online: http://www.jsm.gr.jp/files/journalpdf/2016_6_1_ri-final.pdf (accessed on 30 March 2021).
- Hideshima, T.; Richardson, P.G.; Anderson, K.C. Mechanism of Action of Proteasome Inhibitors and Deacetylase Inhibitors and the Biological Basis of Synergy in Multiple Myeloma. Mol. Cancer Ther. 2011, 10, 2034–2042. [Google Scholar] [CrossRef]
- Conticello, C.; Martinetti, D.; Adamo, L.; Buccheri, S.; Giuffrida, R.; Parrinello, N.L.; Lombardo, L.; Anastasi, G.; Amato, G.; Cavalli, M.; et al. Disulfiram, an old drug with new potential therapeutic uses for human hematological malignancies. Int. J. Cancer 2012, 131, 2197–2203. [Google Scholar] [CrossRef]
- Hassani, S.; Ghaffari, P.; Chahardouli, B.; Alimoghaddam, K.; Ghavamzadeh, A.; Alizadeh, S.; Ghaffari, S.H. Disulfiram/copper causes ROS levels alteration, cell cycle inhibition, and apoptosis in acute myeloid leukaemia cell lines with modulation in the expression of related genes. Biomed. Pharmacother. 2018, 99, 561–569. [Google Scholar] [CrossRef]
- Liu, P.; Brown, S.; Goktug, T.; Channathodiyil, P.; Kannappan, V.; Hugnot, J.-P.; Guichet, P.-O.; Bian, X.; Armesilla, A.L.; Darling, J.L.; et al. Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br. J. Cancer 2012, 107, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhou, Q.; Feng, X.; Dai, Y.; Jiang, Y.; Jiang, W.; Liu, X.; Xing, X.; Wang, Y.; Ni, Y.; et al. Disulfiram/copper markedly induced myeloma cell apoptosis through activation of JNK and intrinsic and extrinsic apoptosis pathways. Biomed. Pharmacother. 2020, 126, 110048. [Google Scholar] [CrossRef] [PubMed]
- Jin, N.; Zhu, X.; Cheng, F.; Zhang, L. Disulfiram/copper targets stem cell-like ALDH + population of multiple myeloma by inhibition of ALDH1A1 and Hedgehog pathway. J. Cell. Biochem. 2018, 119, 6882–6893. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| FEATURE | DIAGNOSTIC CRITERIA |
|---|---|
| C—Calcium | serum calcium >0.25 mmol/L (>1 mg/dL) higher than the upper limit of normal or >2.75 mmol/L (>11 mg/dL) |
| R—Renal Insufficienci | creatinine clearance <40 mL per minute or serum creatinine >177 mol/L (>2 mg/dL) |
| A—Anemia | hemoglobin valure of >20 g/L below the lowest limit of normal, or a hemoglobin value <100 g/L |
| B—Bone Disease | one or more osteolytic lesion on skeletal radiography, CT, or PET/CT |
| Source: International Myeloma Working Group (IMWG) Br. J. Haematol., 2003, 121:749–757 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiser Drozdkova, D.; Smesny Trtkova, K. Possible Therapeutic Potential of Disulfiram for Multiple Myeloma. Curr. Oncol. 2021, 28, 2087-2096. https://doi.org/10.3390/curroncol28030193
Weiser Drozdkova D, Smesny Trtkova K. Possible Therapeutic Potential of Disulfiram for Multiple Myeloma. Current Oncology. 2021; 28(3):2087-2096. https://doi.org/10.3390/curroncol28030193
Chicago/Turabian StyleWeiser Drozdkova, Denisa, and Katerina Smesny Trtkova. 2021. "Possible Therapeutic Potential of Disulfiram for Multiple Myeloma" Current Oncology 28, no. 3: 2087-2096. https://doi.org/10.3390/curroncol28030193
APA StyleWeiser Drozdkova, D., & Smesny Trtkova, K. (2021). Possible Therapeutic Potential of Disulfiram for Multiple Myeloma. Current Oncology, 28(3), 2087-2096. https://doi.org/10.3390/curroncol28030193