
Celecoxib Microparticles for Inhalation in COVID-19-Related Acute Respiratory Distress Syndrome

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Development of CXB-Loaded PLGA Microparticles
2.3. Characterization of Microparticles
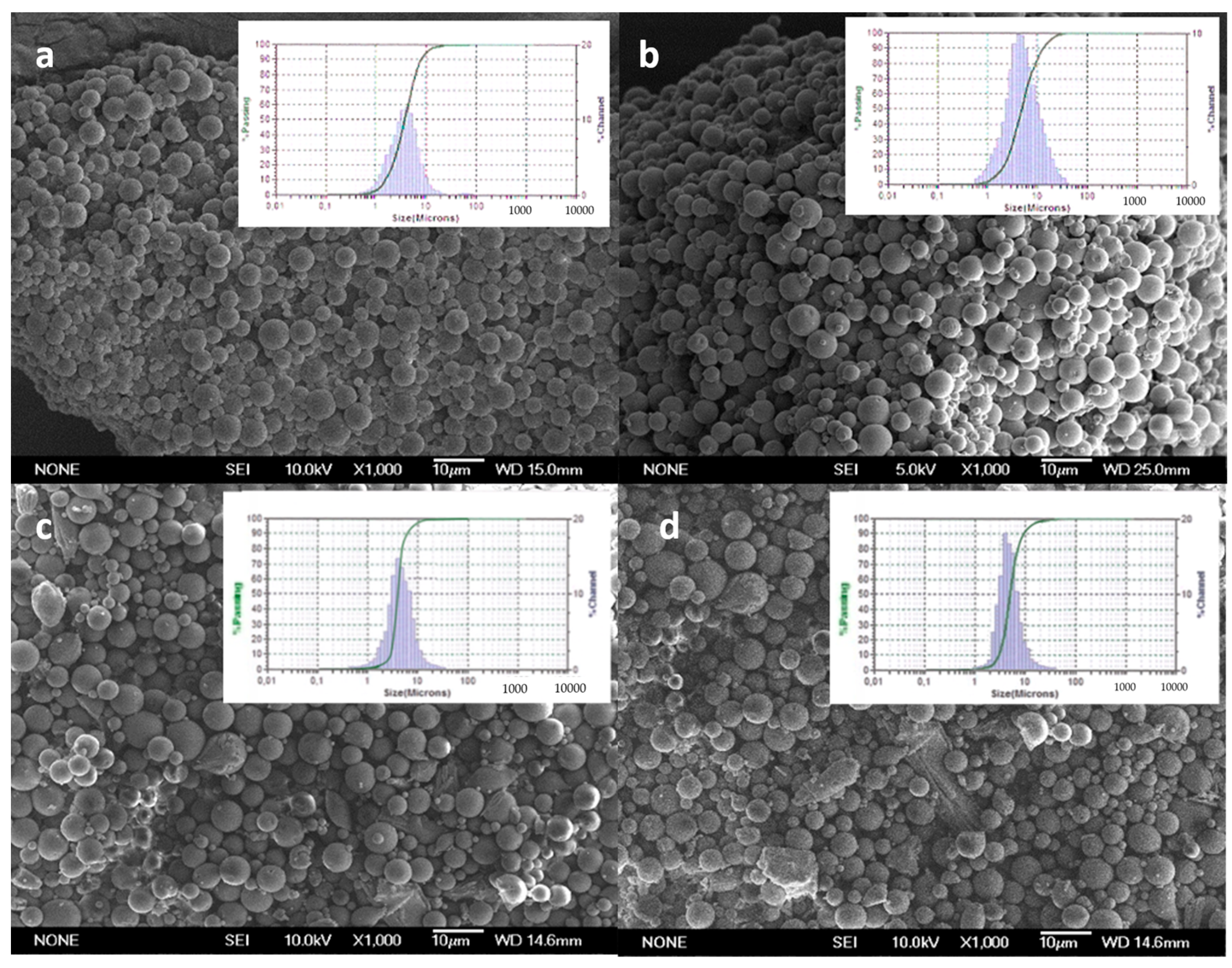
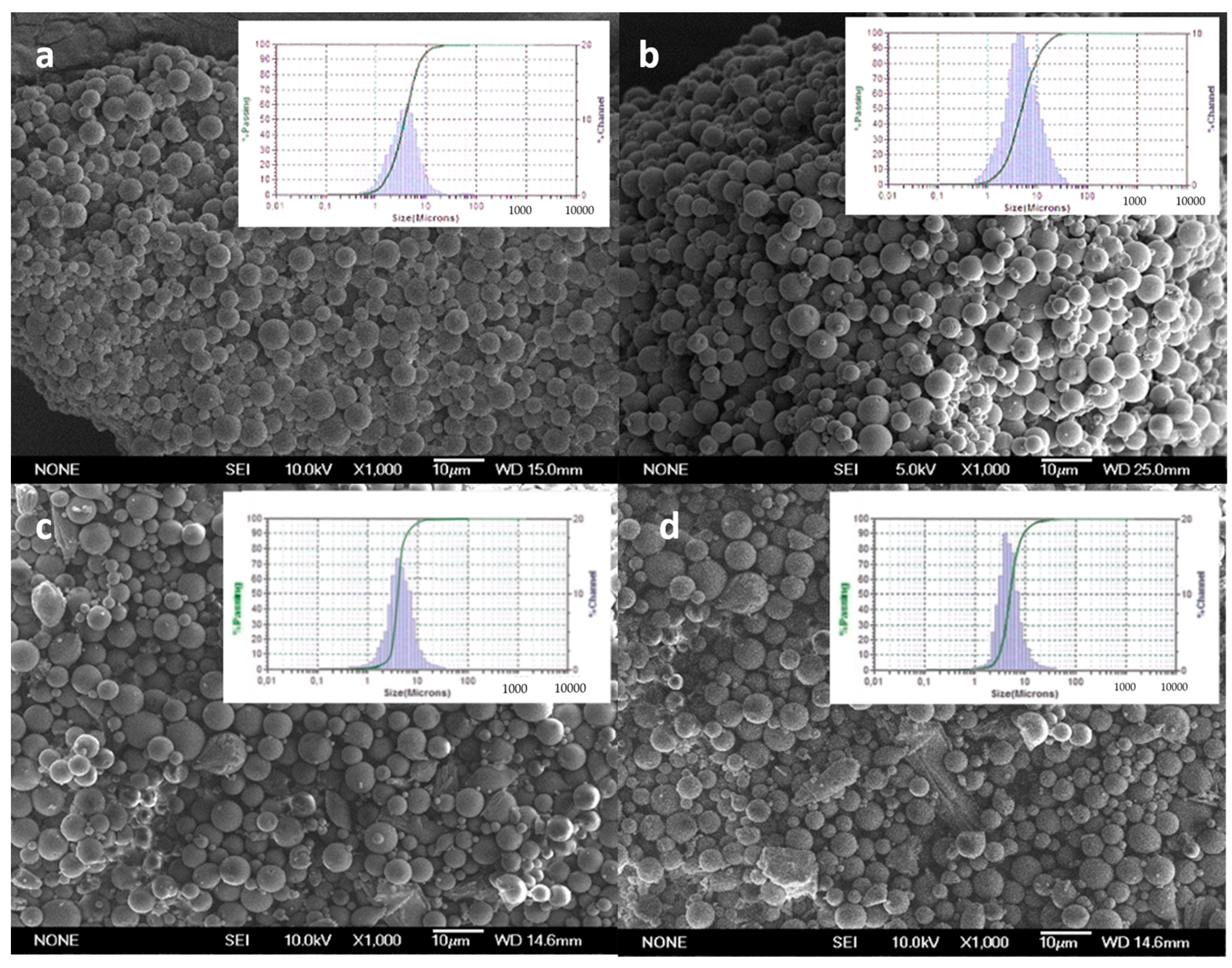
2.3.1. Morphology and Size Distribution
2.3.2. Encapsulation Efficiency and Drug Loading
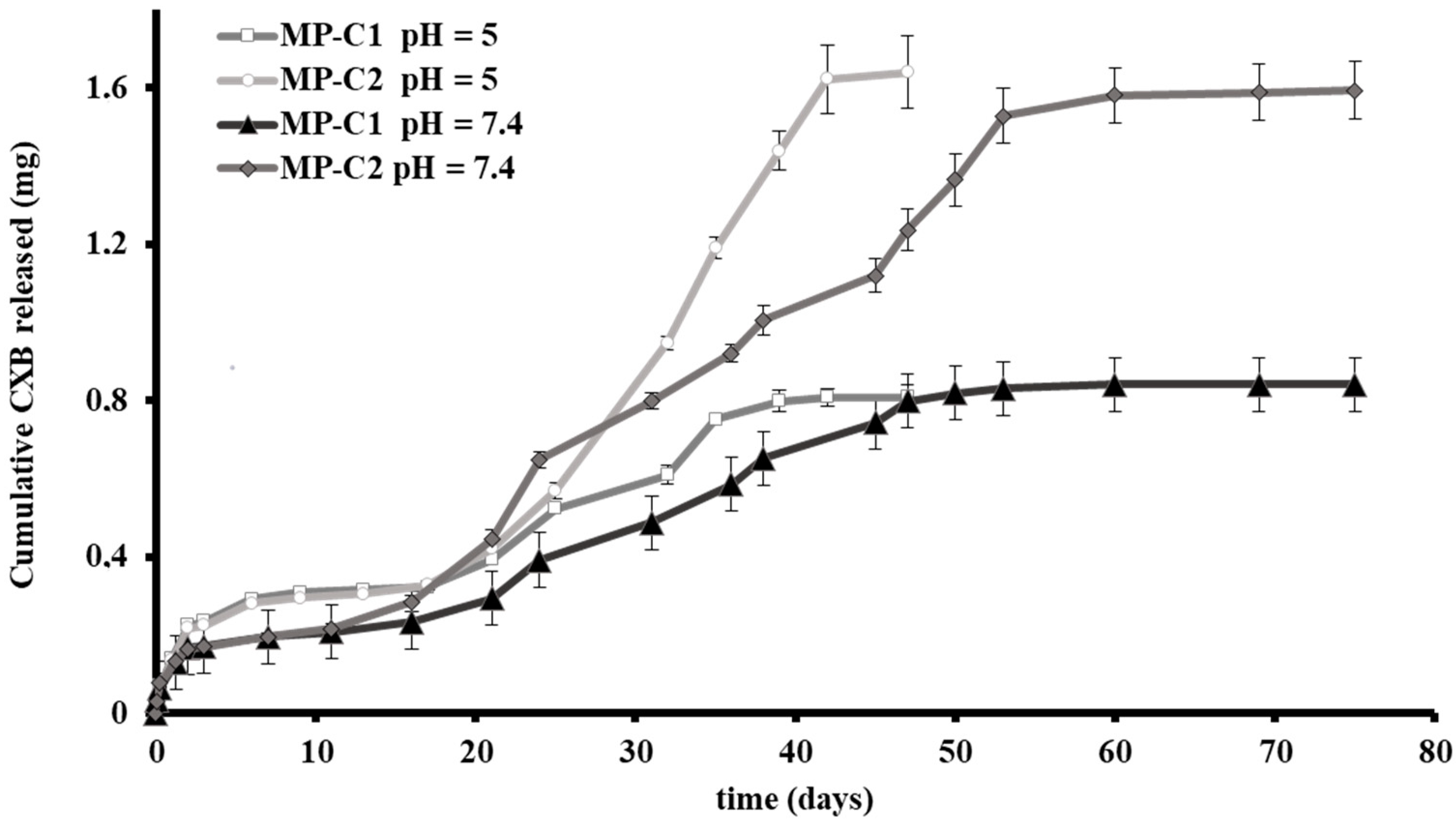
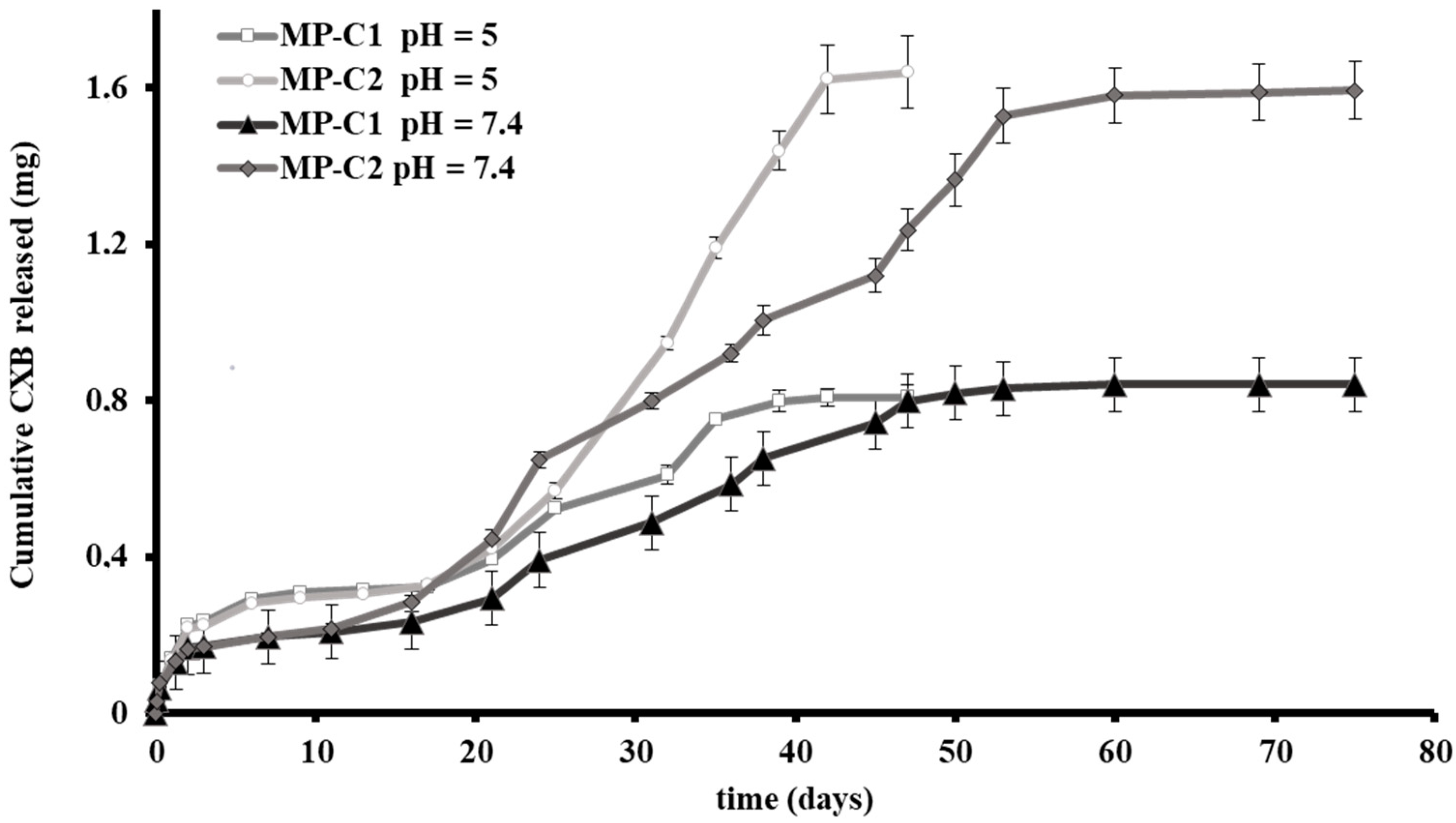
2.3.3. In Vitro Release Studies
2.3.4. Dry Powder Studies for Inhalation
2.4. Studies in Cell Macrophages
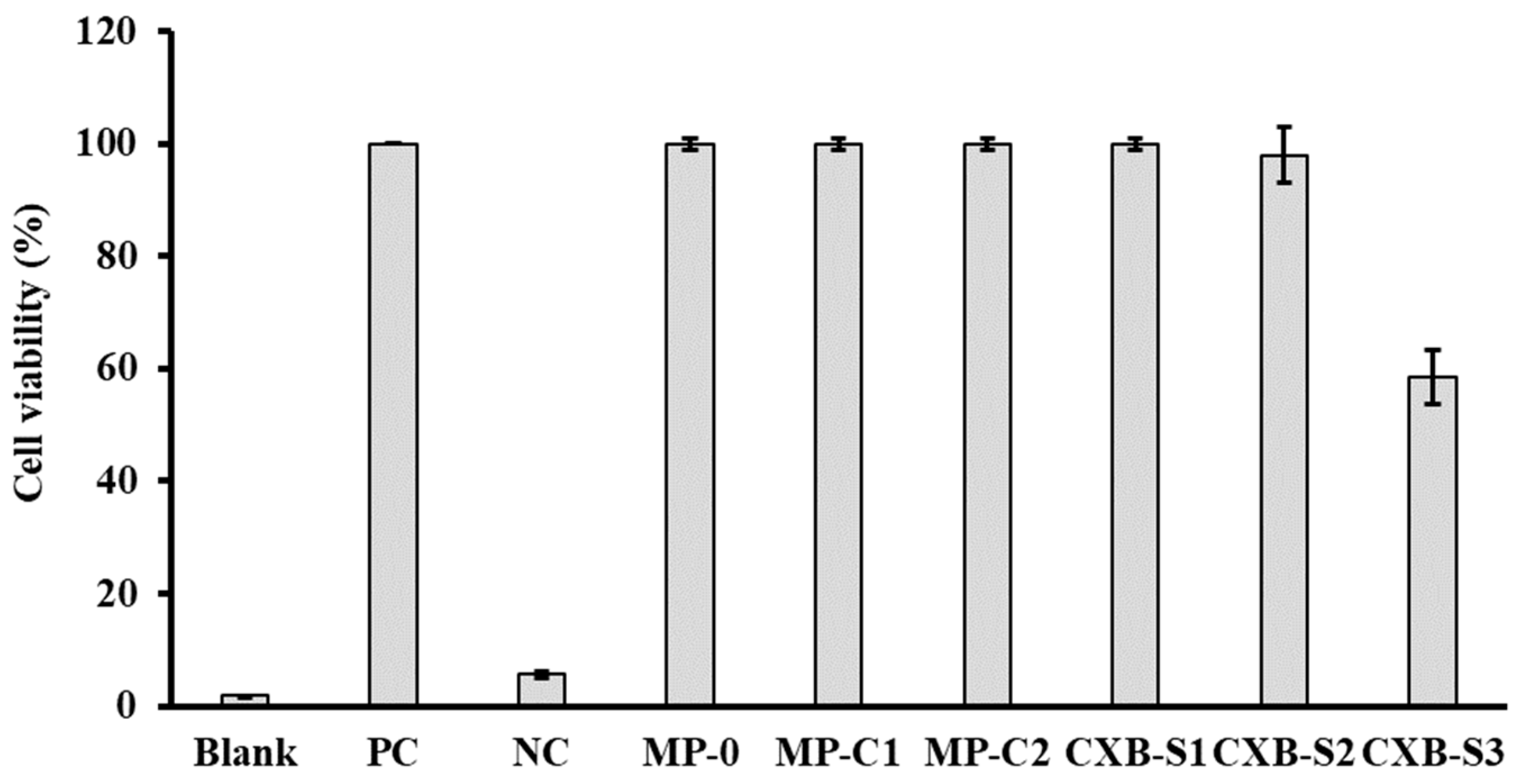
2.4.1. Cell Viability
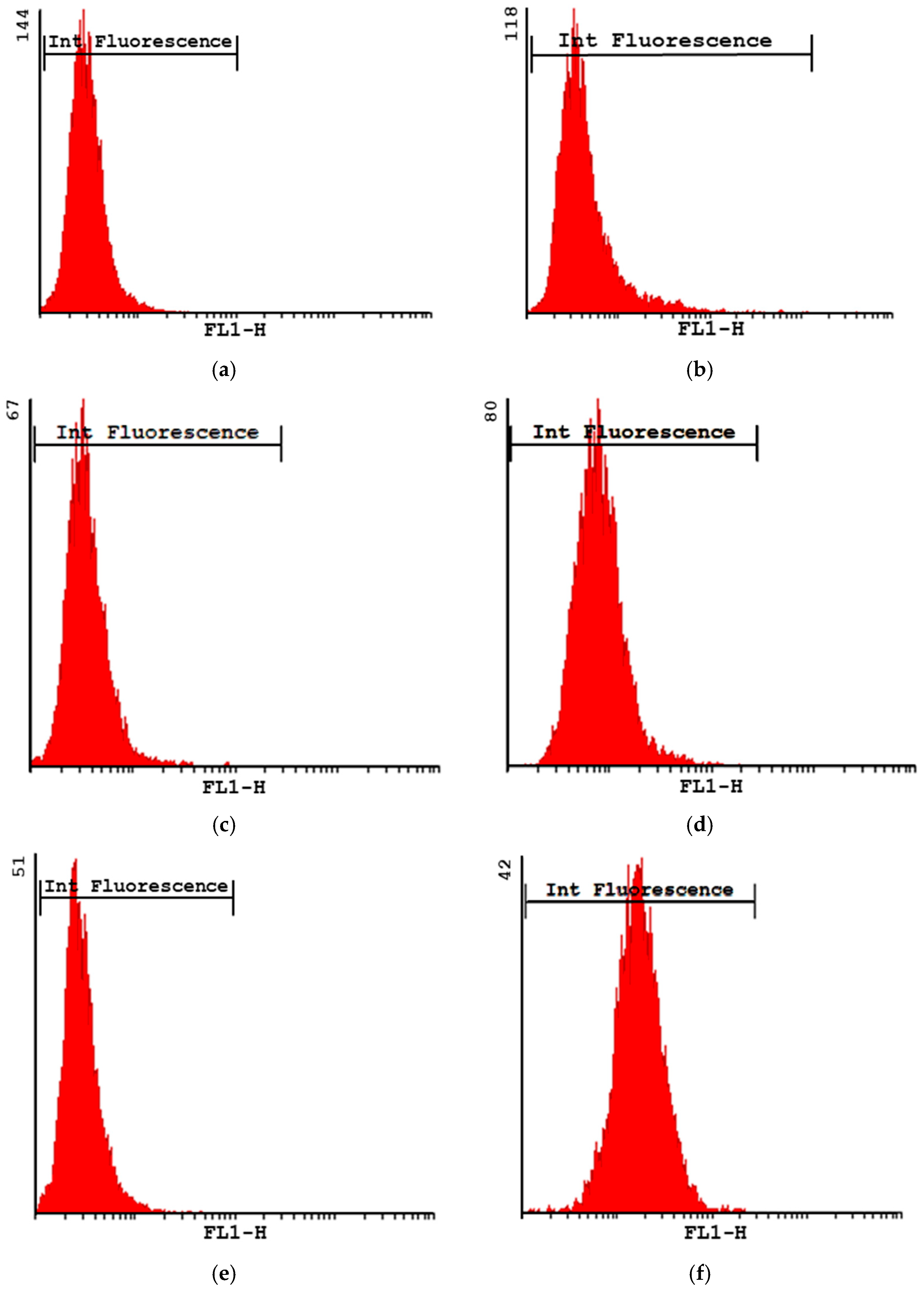
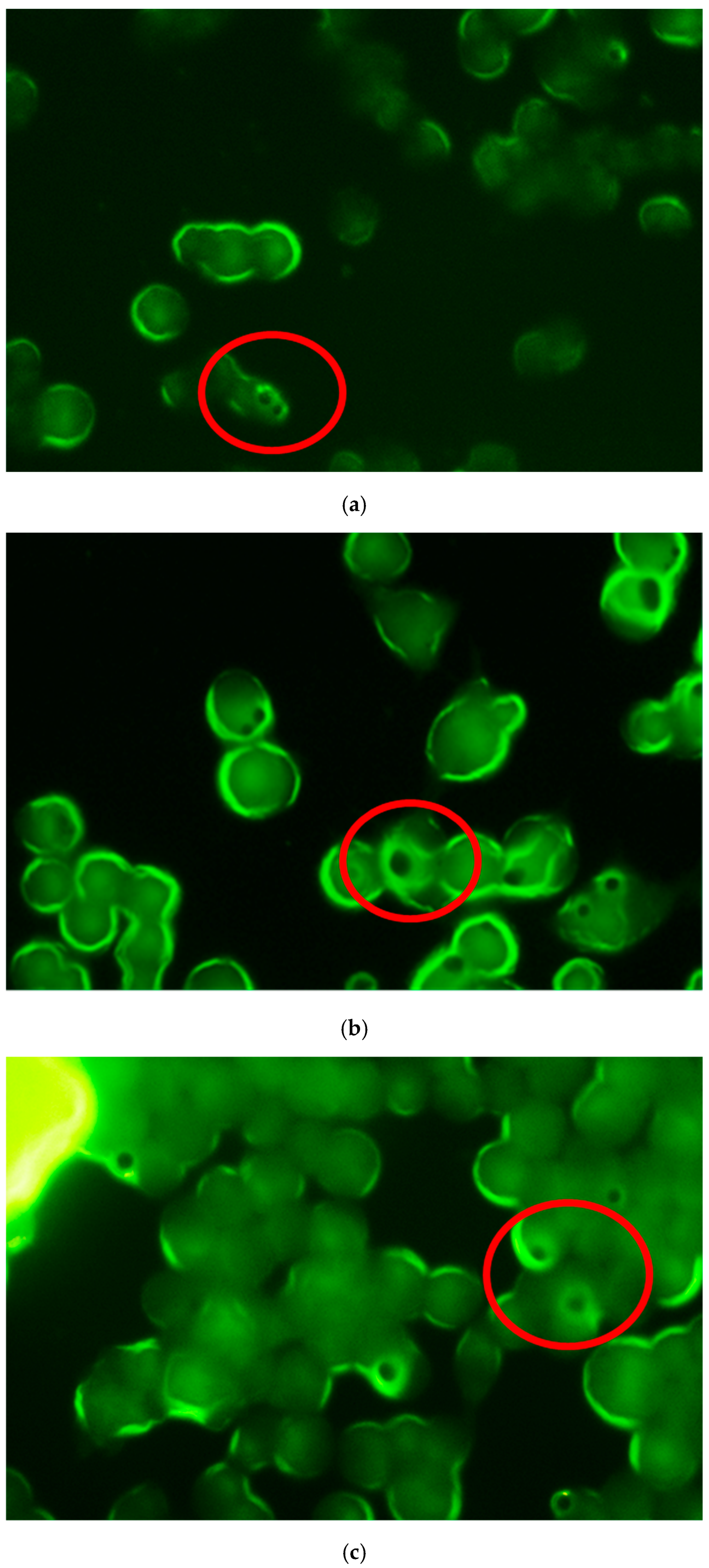
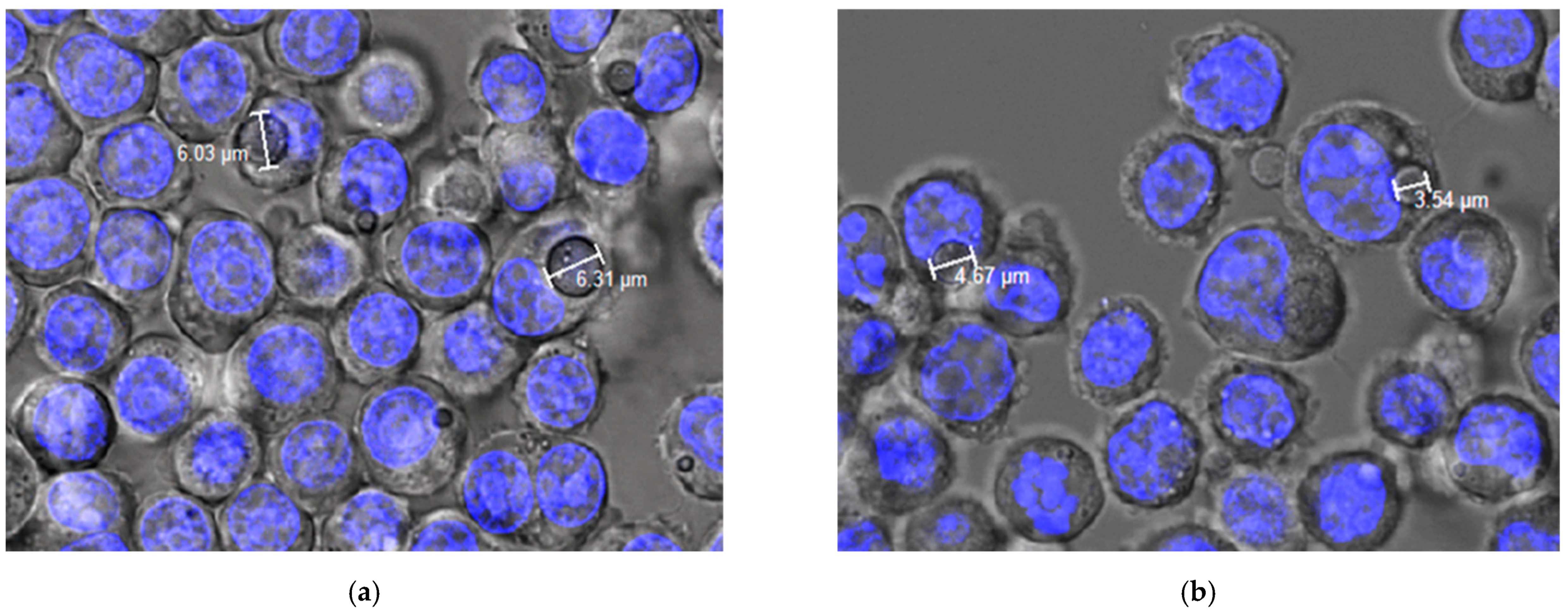
2.4.2. Phagocytosis Studies of Microparticles
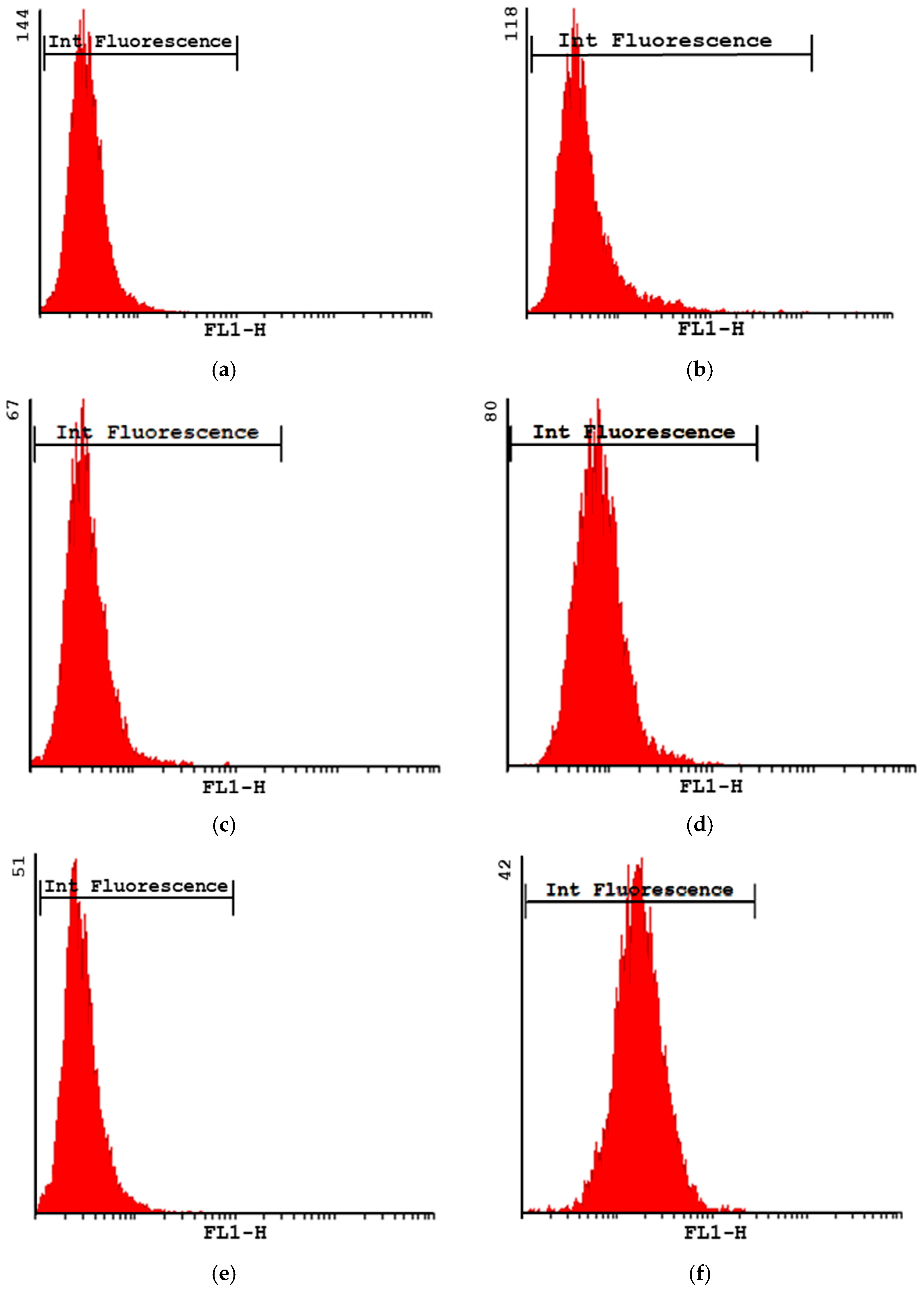
Phagocytosis via Flow Cytometry
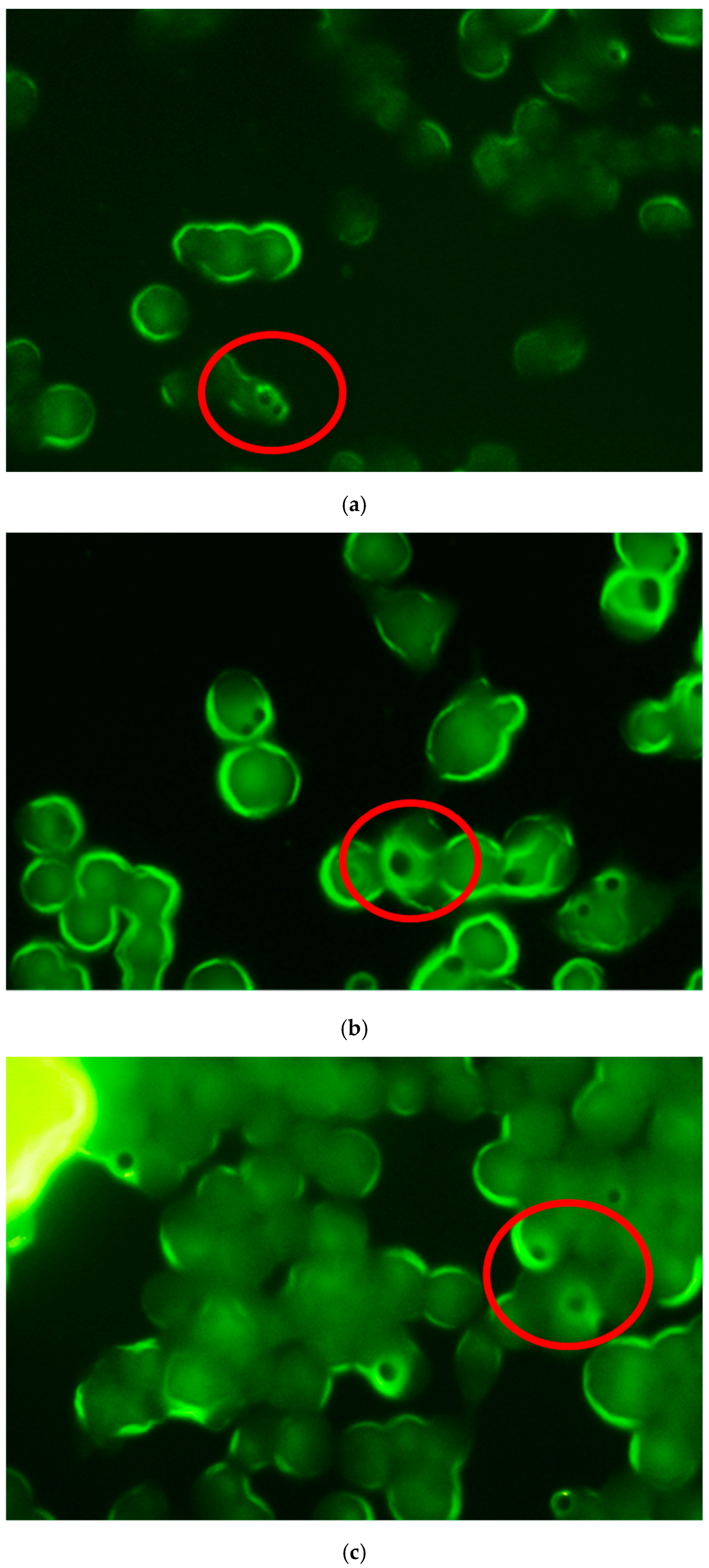
Phagocytosis by Fluorescence Microscopy
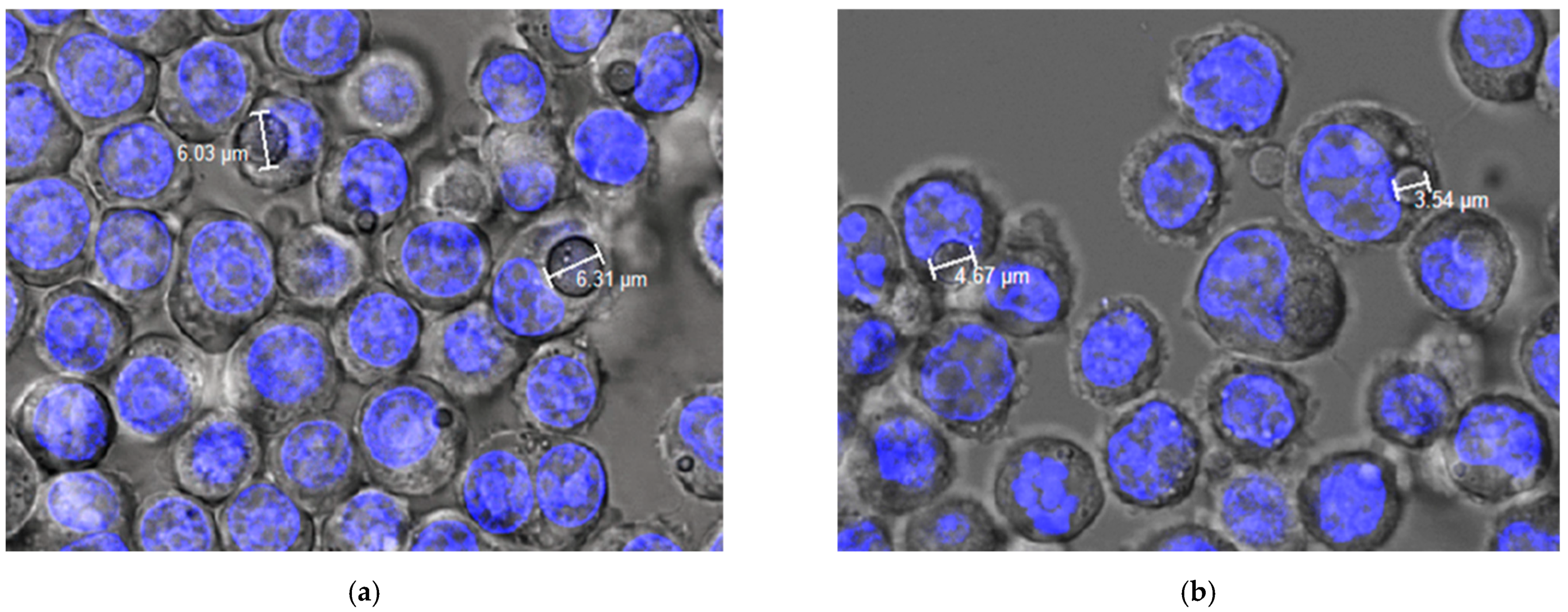
Phagocytosis via Confocal Microscopy
2.5. Anti-Inflammatory Activity of CXB-Loaded PLG MPs
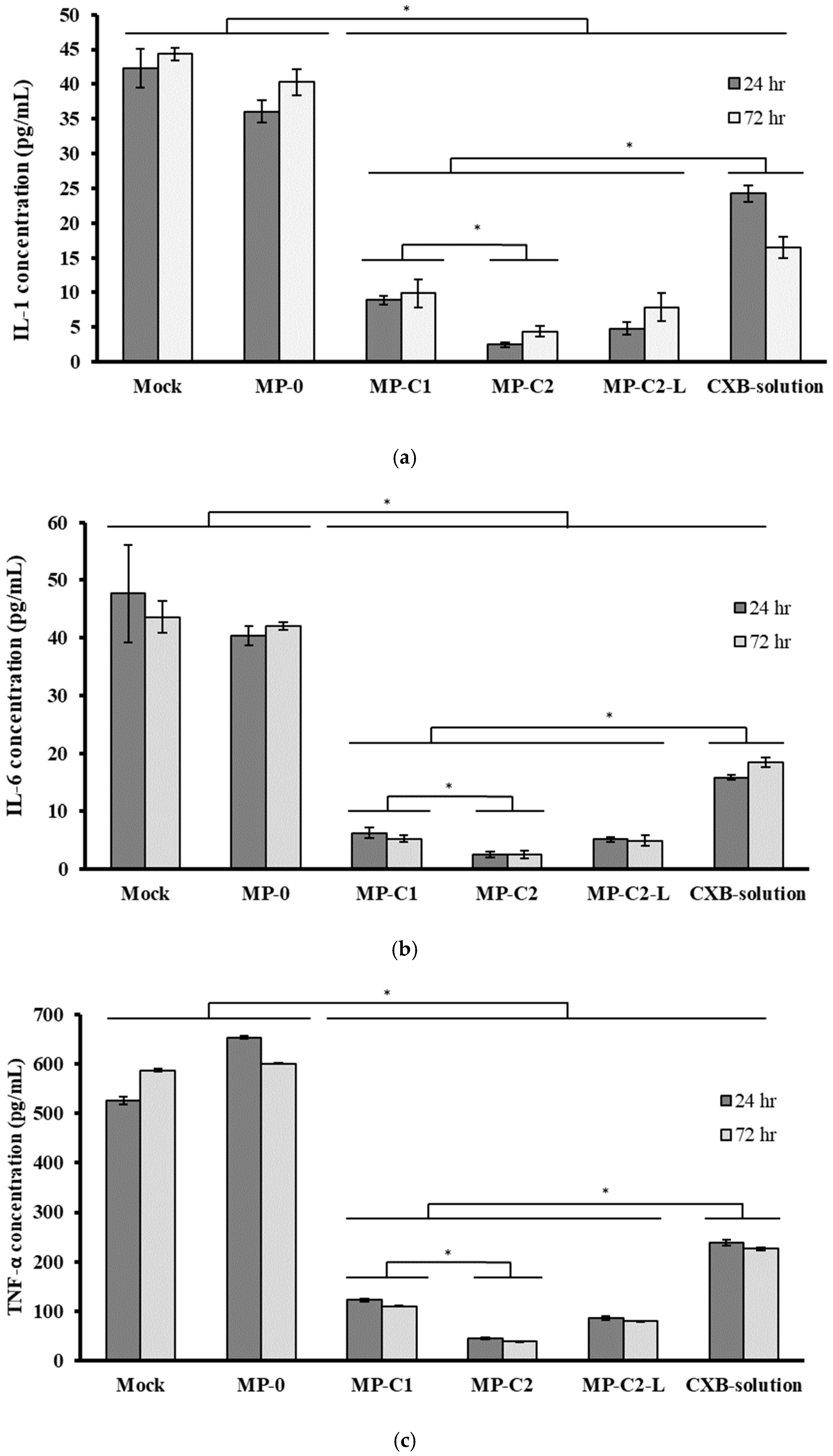
2.5.1. Determination of Inflammatory Cytokines
2.5.2. Determination of Gene Expression
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.6. Statistical Analysis
3. Results and Discussion
3.1. Characterization of Microparticles
3.2. Studies in Cell Macrophages
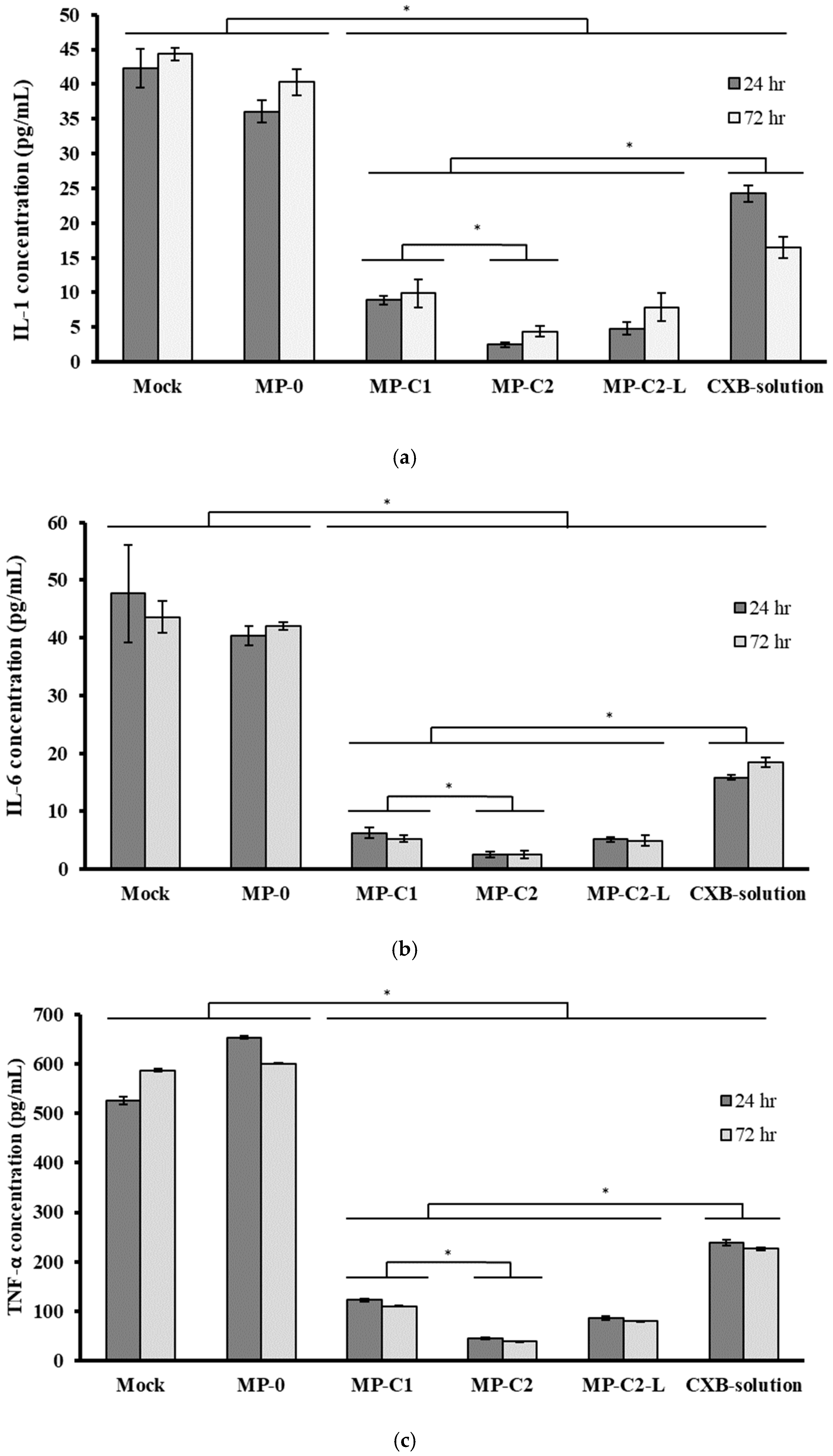
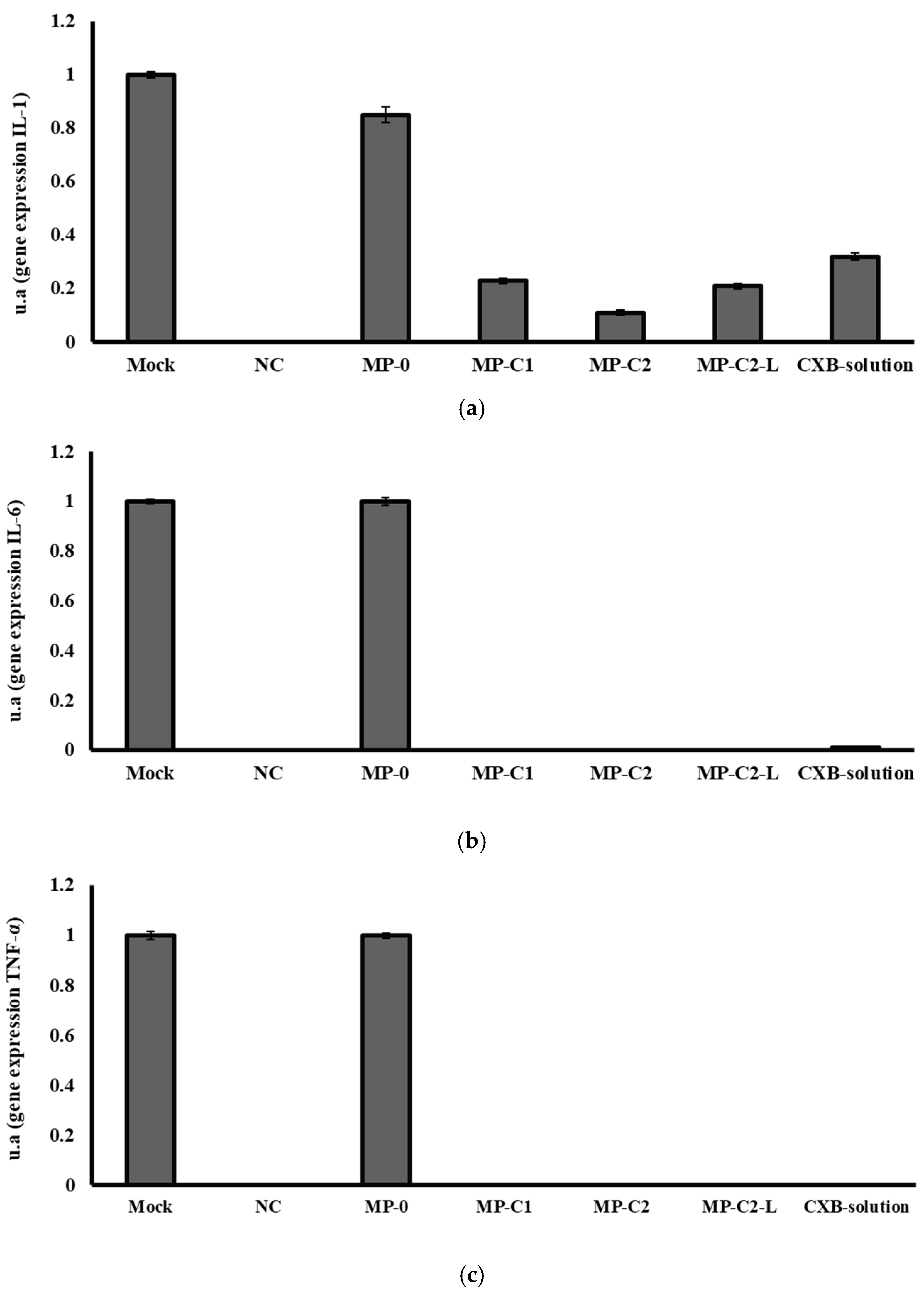
3.3. Anti-Inflammatory Activity of CXB-Loaded PLGA MPs
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hongliu, C.; Yu, C.; Zuobing, C.; Qiang, F.; Weili, H.; Shaohua, H.; Jianping, L.; Tong, L.; Xiaoyang, L.; Tingting, Q.; et al. Handbook of COVID-19 Prevention and Treatment. Compiled According to Clinical Experience; The First Afliated Hospital, Zhejiang University School of Medicine: Hangzhou, China, 2020; pp. 1–65. Available online: https://www.alibabacloud.com/Handbook_of_COVID_19_Prevention_en_Mobile.pdf (accessed on 15 February 2022).
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; McGoogan, J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72,314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Frisoni, P.; Neri, M.; D’Errico, S.; Alfieri, L.; Bonuccelli, D.; Cingolani, M.; Di Paolo, M.; Gaudio, R.M.; Lestani, M.; Marti, M.; et al. Cytokine storm and histopathological findings in 60 cases of COVID-19-related death: From viral load research to immunohistochemical quantification of major players IL-1β, IL-6, IL-15 and TNF-α. Forensic. Sci. Med. Pathol. 2021, 1, 4–19. [Google Scholar] [CrossRef]
- Manik, M.; Singh, R.K. Role of toll-like receptors in modulation of cytokine storm signaling in SARS-CoV-2-induced COVID-19. J. Med. Virol. 2021, 94, 869–877. [Google Scholar] [CrossRef]
- Morgulchik, N.; Athanasopoulou, F.; Chu, E.; Lam, Y.; Kamaly, N. Potential therapeutic approaches for targeted inhibition of inflammatory cytokines following COVID-19 infection-induced cytokine storm. Interface Focus 2021, 12, 20210006. [Google Scholar] [CrossRef]
- Lai, C.C.; Ko, W.C.; Lee, P.I.; Jean, S.S.; Hsueh, P.R. Extra-respiratory manifestations of COVID-19. Int. J. Antimicrob. Agents 2020, 56, 106024. [Google Scholar] [CrossRef]
- Li, H.; Zhou, Y.; Zhang, M.; Wang, H.; Zhao, Q.; Liu, J. Updated approaches against SARS-CoV-2. Antimicrob. Agents Chemother. 2020, 64, e00483-20. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liao, X.; Qian, S.; Yuan, J.; Wang, F.; Liu, Y.; Zhang, Z. Community transmission of severe acute respiratory syndrome coronavirus 2, Shenzhen, China. Emerg. Infect. Dis. 2020, 26, 1320–1323. [Google Scholar] [CrossRef]
- Ahn, D.G.; Shin, H.J.; Kim, M.H.; Lee, S.; Kim, H.S.; Myoung, J.; Kim, B.T.; Kim, S.J. Current status of epidemiology, diagnosis, therapeutics, and vaccines for novel coronavirus disease 2019 (COVID-19). J. Microbiol. Biotechnol. 2020, 30, 313–324. [Google Scholar] [CrossRef]
- Ari, A. Practical strategies for a safe and effective delivery of aerosolized medications to patients with COVID-19. Respir. Med. 2020, 167, 105987. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; Mestres-Truyol, J.; Ojeda-Montes, M.J.; Macip, G.; Saldivar-Espinoza, B.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallvé, S. Prediction of novel inhibitors of the main protease (M-pro) of SARS-CoV-2 through consensus docking and drug reposition. Int. J. Mol. Sci. 2020, 21, 3793. [Google Scholar] [CrossRef] [PubMed]
- Khupse, R.; Dixit, P. Potential antiviral mechanism of hydroxychloroquine in COVID-19 and further extrapolation to celecoxib (celebrex) for future clinical trials. SSRN Electron. J. 2020, 1–8. [Google Scholar] [CrossRef]
- Kumar, G.V.; Jeyanthi, V.; Ramakrishnan, S. A short review on antibody therapy for COVID-19. New Microbes New Infect. 2020, 35, 100682. [Google Scholar] [CrossRef] [PubMed]
- Halpin, D.; Singh, D.; Hadfield, R.M. Inhaled corticosteroids and COVID-19: A systematic review and clinical perspective. Eur. Respir. J. 2020, 55, 2001009. [Google Scholar] [CrossRef] [PubMed]
- Han, F.Y.; Thurecht, K.J.; Whittaker, A.K.; Smith, M.T. Bioerodable PLGA-Based Microparticles for Producing Sustained-Release Drug Formulations and Strategies for Improving Drug Loading. Front. Pharmacol. 2016, 7, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshavarz, A.; Kadry, H.; Alobaida, A.; Ahsan, F. Newer approaches and novel drugs for inhalational therapy for pulmonary arterial hypertension. Expert Opin. Drug Deliv. 2020, 17, 439–461. [Google Scholar] [CrossRef]
- Fidahic, M.; Jelicic Kadic, A.; Radic, M.; Puljak, L. Celecoxib for rheumatoid arthritis. Cochrane Database Syst. Rev. 2017, 6, CD012095. [Google Scholar] [CrossRef]
- Puljak, L.; Marin, A.; Vrdoljak, D.; Markotic, F.; Utrobicic, A.; Tugwell, P. Celecoxib for osteoarthritis. Cochrane Database Syst. Rev. 2017, 5, CD009865. [Google Scholar] [CrossRef]
- Hong, W.; Chen, Y.; You, K.; Tan, S.; Wu, F.; Tao, J.; Xiaoyang, L.; Tingting, Q.; Yihong, S.; Jifang, S.; et al. Celebrex adjuvant therapy on COVID-19: An experimental study. Front. Pharmacol. 2020, 11, 561674. [Google Scholar] [CrossRef]
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012.
- Masclee, G.M.C.; Straatman, H.; Arfè, A.; Castellsague, J.; Garbe, E.; Herings, R.; Kollhorst, B.; Lucchi, S.; Perez-Gutthann, S.; Romio, S.; et al. Risk of acute myocardial infarction during use of individual NSAIDs: A nested case-control study from the SOS project. PLoS ONE 2018, 13, e0204746. [Google Scholar] [CrossRef] [Green Version]
- Bao, L.; Deng, W.; Huang, B.; Gao, H.; Liu, J.; Ren, L.; Wei, Q.; Yu, P.; Xu, Y.; Qi, F.; et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 2020, 583, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.; Cervantes, M.; Pham, L.; Rothchild, A. Alveolar macrophages: Novel therapeutic targets for respiratory diseases. Expert Rev. Mol. Diagn. 2021, 23, E18. [Google Scholar] [CrossRef]
- Shi, S.; Hickey, A.J. PLGA microparticles in respirable sizes enhance an in vitro T cell response to recombinant Mycobacterium tuberculosis antigen TB10.4-Ag85B. Pharm. Res. 2010, 27, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Gupta, N.; Ahsan, F. Particle engineering to enhance or lessen particle uptake by alveolar macrophages and to influence the therapeutic outcome. Eur. J. Pharm. Biopharm. 2015, 89, 163–174. [Google Scholar] [CrossRef]
- Marcianes, P.; Negro, S.; Barcia, E.; Montejo, C.; Fernandez-Carballido, A. Potential active targeting of gatifloxacin to macrophages by means of surface-modified PLGA microparticles destined to treat tuberculosis. AAPS PharmSciTech 2020, 21, 15. [Google Scholar] [CrossRef]
- Mathaes, R.; Winter, G.; Besheer, A.; Engert, J. Influence of particle geometry and PEGylation on phagocytosis of particulate carriers. Int. J. Pharm. 2014, 465, 1–6. [Google Scholar] [CrossRef]
- Garapaty, A.; Champion, J.A. Tunable particles alter macrophage uptake based on combinatorial effects of physical properties. Bioeng. Transl. Med. 2017, 2, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Makino, K.; Yamamoto, N.; Higuchi, K.; Harada, N.; Ohshima, H.; Terada, H. Phagocytic uptake of polystyrene microspheres by alveolar macrophages: Effects of the size and surface properties of the microspheres. Colloid. Surf. B 2003, 27, 33–39. [Google Scholar] [CrossRef]
- Hirota, K.; Hasegawa, T.; Hinata, H.; Ito, F.; Inagawa, H.; Kochi, C.; Soma, G.; Makino, K.; Terada, H. Optimum conditions for efficient phagocytosis of rifampicin loaded PLGA microspheres by alveolar macrophages. J. Control. Release 2007, 119, 69–76. [Google Scholar] [CrossRef]
- Hirota, K.; Terada, H. Endocytosis of particles formulations by macrophages and its application to clinical treatment. In Molecular Regulation of Endocytosis; Ceresa, B., Ed.; IntechOpen: London, UK, 2012; Chapter 16; pp. 413–428. [Google Scholar] [CrossRef] [Green Version]
- Nyberg, K.; Johansson, U.; Johansson, A.; Camner, P. Phagolysosomal pH in alveolar macrophages. Environ. Health Perspect. 1992, 97, 149–152. [Google Scholar] [CrossRef] [PubMed]
- European Directorate for Quality in Medicines (EDQM). Preparations for Inhalations: Aerodynamic Assessment of Fine Particles. In European Pharmacopoeia, 10th ed.; Monograph 2.9.18; European Directorate for Quality in Medicines (EDQM): Strasburg, France, 2021. [Google Scholar]
- Wilfinger, W.W.; Mackey, K.; Chomczynski, P. Effect of pH and ionic strength on the spectrophotometric assessment of nucleic acid purity. Biotechniques 1997, 22, 478–481. [Google Scholar] [CrossRef]
- Green, M.R.; Sambrook, J. Agarose gel electrophoresis. Cold Spring Harb. Protoc. 2019, 1. [Google Scholar] [CrossRef] [PubMed]
- Rasband, W.S. Image J. U.S. National Institutes of Health, Bethesda, MD, USA. 1997–2018. Available online: https://imagej.nih.gov/ij/ (accessed on 20 May 2021).
- Hirota, K.; Hasegawa, T.; Nakajima, T.; Inagawa, H.; Kohchi, C.; Soma, G.; Makino, K.; Terada, H. Delivery of rifampicin-PLGA microspheres into alveolar macrophages is promising for treatment of tuberculosis. J. Control. Release 2010, 142, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Deniz, A.; Sade, A.; Severcan, F.; Keskin, D.; Tezcaner, A.; Banerjee, S. Celecoxib-loaded liposomes: Effect of cholesterol on encapsulation and in vitro release characteristics. Biosci. Rep. 2010, 30, 365–373. [Google Scholar] [CrossRef] [Green Version]
- Kozlu, S.; Sahin, A.; Ultav, G.; Yerlikaya, F.; Calis, S.; Capan, Y. Development and in vitro evaluation of doxorubicin and celecoxib co-loaded bone targeted nanoparticles. J. Drug Deliv. Sci. Technol. 2018, 45, 213–219. [Google Scholar] [CrossRef]
- Chandel, A.; Goyal, A.K.; Ghosh, G.; Rath, G. Recent advances in aerosolised drug delivery. Biomed. Pharm. 2019, 112, 108601. [Google Scholar] [CrossRef]
- Emami, J.; Hamishehkar, H.; Najafabadi, A.R.; Gilani, K.; Minaiyan, M.; Mahdavi, H.; Mirzadeh, H.; Fakhari, A.; Nokhodchi, A. Particle size design of PLGA microspheres for potential pulmonary drug delivery using response surface methodology. J. Microencapsul. 2009, 26, 1–8. [Google Scholar] [CrossRef]
- Baranov, M.V.; Kumar, M.; Sacanna, S.; Thutupalli, S.; van den Bogaart, G. Modulation of immune responses by particle size and shape. Front. Immunol. 2021, 11, 607945. [Google Scholar] [CrossRef]
- Hines, D.J.; Kaplan, D.L. Poly(lactic-co-glycolic) acid-controlled-release systems: Experimental and modeling insights. Crit. Rev. Ther. Drug Carrier. Syst. 2013, 30, 257–276. [Google Scholar] [CrossRef]
- Fernández, A.; Casan, P. Deposition of inhaled particles in the lungs. Arch. Bronconeumol. 2012, 48, 240–246. [Google Scholar] [CrossRef]
- Quinlan, T.R.; BeruBe, K.A.; Hacker, M.P.; Taatjes, D.J.; Timblin, C.R.; Goldberg, J.; Kimberley, P.; O’Shaughnessy, P.; Hemenway, D.; Torino, J.; et al. Mechanisms of asbestos-induced nitric oxide production by rat alveolar macrophages in inhalation and in vitro models. Free Radic. Biol. Med. 1998, 24, 778–788. [Google Scholar] [CrossRef]
- Yu, W.; Liu, C.; Liu, Y.; Xu, W. Mannan-modified solid lipid nanoparticles for targeted gene delivery to alveolar macrophages. Pharm. Res. 2010, 27, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Shenoy, S.; Gautam, A.; Munjal, S.; Niu, J.; Gopalakrishnan, M.; Gobburru, J. Pharmacokinetics of DFN-15, a novel oral solution of celecoxib, versus celecoxib 400-mg capsules: A randomized crossover study in fasting healthy volunteers. Clin. Drug Investig. 2017, 937–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomashefski, J.; Farver, C. Anatomy and histology of the lung. In Dail and Hammar’s Pulmonary Pathology: Volume I: Nonneoplastic Lung Disease; Tomashefski, J., Cagle, P., Farver, C., Fraire, A., Eds.; Springer Science Business: New York, NY, USA, 2008; pp. 19–48. [Google Scholar]
- Martins, S.; Costa-Lima, S.; Carneiro, T.; Cordeiro-da-Silva, A.; Souto, E.B.; Ferreira, D.C. Solid lipid nanoparticles as intracellular drug transporters: An investigation of the uptake mechanism and pathway. Int. J. Pharm. 2012, 430, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.J.; Pradel, L.C.; Chasson, L.; Van Rooijen, N.; Grau, G.E.; Hunt, N.H.; Chimini, G. Technical Advance: Autofluorescence as a tool for myeloid cell analysis. J. Leukoc. Biol. 2010, 88, 597–603. [Google Scholar] [CrossRef]
- Sekiryu, T.; Oguchi, Y.; Arai, S.; Wada, I.; Iida, T. Autofluorescence of the cells in human subretinal fluid. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8534–8541. [Google Scholar] [CrossRef]
- Misharin, A.; Morales-Nebreda, L.; Mutlu, G.M.; Budinger, G.R.; Perlman, H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 2013, 49, 503–510. [Google Scholar] [CrossRef] [Green Version]
- Villa-Hermosilla, M.C.; Fernández-Carballido, A.; Hurtado, C.; Barcia, E.; Montejo, C.; Alonso, M.; Negro, S. Sulfasalazine microparticles targeting macrophages for the treatment of inflammatory diseases affecting the synovial cavity. Pharmaceutics 2021, 13, 951. [Google Scholar] [CrossRef]
- Mantovani, G.; Macciò, A.; Madeddu, C.; Serpe, R.; Antoni, G.; Massa, E.; Dessì, M.; Panzone, F. Phase II nonrandomized study of the efficacy and safety of COX-2 inhibitor celecoxib on patients with cancer cachexia. J. Mol. Med. 2010, 88, 85–92. [Google Scholar] [CrossRef]
- Mills, C.D. Anatomy of a discovery: M1 and M2 macrophages. Front. Immunol. 2015, 6, 212. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.H.; Choi, J.H.; Byun, M.S.; Jue, D.M. Chloroquine inhibits production of TNF-alpha, IL-1beta and IL-6 from lipopolysaccharide-stimulated human monocytes/macrophages by different modes. Rheumatology 2006, 45, 703–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.B.; Zhang, X.L.; Lv, M.Y.; Hu, X.F.; Li, Y. Detection of IL-1β IL-6 and TNF-α in Sprague-Dawely rats’ atrophic thymus induced by lipopolysaccharide. Pol. J. Vet. Sci. 2018, 21, 589–597. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef]
- Nicolete, R.; dos Santos, D.F.; Faccioli, L.H. The uptake of PLGA micro or nanoparticles by macrophages provokes distinct in vitro inflammatory response. Int. Immunopharmacol. 2011, 11, 1557–1563. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, C.; O’Connor, G.; O’Leary, S.; Gallagher, P.J.; Cryan, S.A.; Keane, J.; O’Sullivan, M.P. Treatment of Mycobacterium tuberculosis-infected macrophages with poly(lactic-co-glycolic acid) microparticles drives NFκB and autophagy dependent bacillary killing. PLoS ONE 2016, 11, e0149167. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | CXB (%) | FITC (%) | Ratio Drug:Polymer | EE ± DS (%) | DL ± DS (mg of CXB in 100 mg of MPs) | Particle Size ± DS (μm) |
|---|---|---|---|---|---|---|
| MP-C1 | 5 | - | 1:20 | 89.35 ± 4.94 | 4.28 ± 0.23 | 4.73 ± 0.46 |
| MP-C2 | 10 | - | 1:10 | 95.12 ± 3.98 | 8.65 ± 0.62 | 4.40 ± 0.57 |
| MP-0 | - | - | - | - | - | 4.17 ± 0.45 |
| MP-F | - | 10 | 1:10 | 1.29 ± 0.21 | 0.12 ± 0.02 | 4.01 ± 0.89 |
| 5′-Forward-3′ | 5′-Reverse-3′ | |
|---|---|---|
| IL-1 | AGTTGACGGACCCCAAAAGAT | GTTGATGTGCTGCTGCGAGA |
| IL-6 | CTTCCATCCAGTTGCCTTCTTG | AATTAAGCCTCCGACTTGTGAAG |
| TNF-α | GATCTCAAAGACAACCAACATGTG | CTCCAGCTGGAAGACTCCTCCCAG |
| GADPH | TGAGGCCGGTGCTGAGTATGTCG | CCACAGTCTTCTGGGTGGCAGTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villa-Hermosilla, M.-C.; Negro, S.; Barcia, E.; Hurtado, C.; Montejo, C.; Alonso, M.; Fernandez-Carballido, A. Celecoxib Microparticles for Inhalation in COVID-19-Related Acute Respiratory Distress Syndrome. Pharmaceutics 2022, 14, 1392. https://doi.org/10.3390/pharmaceutics14071392
Villa-Hermosilla M-C, Negro S, Barcia E, Hurtado C, Montejo C, Alonso M, Fernandez-Carballido A. Celecoxib Microparticles for Inhalation in COVID-19-Related Acute Respiratory Distress Syndrome. Pharmaceutics. 2022; 14(7):1392. https://doi.org/10.3390/pharmaceutics14071392
Chicago/Turabian StyleVilla-Hermosilla, Monica-Carolina, Sofia Negro, Emilia Barcia, Carolina Hurtado, Consuelo Montejo, Mario Alonso, and Ana Fernandez-Carballido. 2022. "Celecoxib Microparticles for Inhalation in COVID-19-Related Acute Respiratory Distress Syndrome" Pharmaceutics 14, no. 7: 1392. https://doi.org/10.3390/pharmaceutics14071392
APA StyleVilla-Hermosilla, M.-C., Negro, S., Barcia, E., Hurtado, C., Montejo, C., Alonso, M., & Fernandez-Carballido, A. (2022). Celecoxib Microparticles for Inhalation in COVID-19-Related Acute Respiratory Distress Syndrome. Pharmaceutics, 14(7), 1392. https://doi.org/10.3390/pharmaceutics14071392