Abstract
Cardiovascular disease and in particular coronary heart disease (CHD) are major causes of mortality and low density lipoprotein-cholesterol is causally related to CHD. Risk prevention requires a global approach for patient management, including lifestyle and dietary changes for all. Pharmaceutical lipid-lowering therapy using statins and non-statin combinations significantly improves cardiovascular outcomes. Severe hypercholesterolaemia and familial forms require aggressive lipid-lowering therapy. New approaches with anti-PCSK9 antibodies enable more therapeutic goals to be achieved. Residual inflammatory assessment may help guide risk evaluation and become a therapeutic target.
Introduction
Despite a decreasing incidence rate of acute myocardial infarction (MI) per person-years of follow-up, cardiovascular disease remains a major health and economic burden to society, accounting for the greatest proportion of deaths in both men and women above the age of 65 years [1,2]. Over recent decades, research into the causes and mechanisms driving the development of cardiovascular disease has rapidly evolved from focusing purely on blood pressure or lipid management, to treating the inflammatory processes closely associated with it. In everyday real life, this translates into ever more targeted treatment strategies that can be adapted to each individual’s cardiovascular history and risk.
Why envisage intensive LDL-cholesterol lowering and inhibition of inflammation?
Some of the first epidemiological studies associating coronary heart disease (CHD) with baseline cardiovascular risk factor variables date back to the 1960s, prior to therapeutic and Mendelian studies [3], and point at both systolic blood pressure and serum cholesterol as risk factors for the development of CHD. The role of lifestyle and modifiable physical characteristics, initially investigated by Ancel Keys, was then further explored in the Framingham Heart Study, funded by the US Public Health Service [3].
The first interventional studies aiming at establishing a causal relationship between CHD and cardiovascular risk began to emerge in the 1970s. One example is the co-operative trial in the primary prevention of ischaemic heart disease using clofibrate published in 1978, which included 15 745 males aged 30 to 59 years and with high cholesterol levels [4]. Treatment with clofibrate (ethyl-chlorophenoxyisobutyrate) resulted in an overall decrease in cholesterol levels by 9% versus placebo. Furthermore, the percentage of males free from all major ischaemic heart disease or nonfatal MI was higher in the clofibrate treatment arm, albeit associated with an excess mortality rate. Although this excess death rate was later attributed to issues not related to the drug, at the time it did lead to widespread scepticism regarding the benefit of cholesterol reduction in the prevention of CHD. Controversy over the benefit of lowering cholesterol continued until the end of the 1980s as further studies on clofibrate and other lipid lowering drugs were published, and culminated with the results of a trial on the effect of partial ileal bypass surgery on CHD mortality and morbidity in patients with hypercholesterolaemia, which concluded that, in patients who had had an MI, this procedure resulted in sustained improvements in blood lipid levels while at the same time reducing subsequent morbidity due to CHD [5].
In parallel to these interventional studies, basic science research on the pathophysiology of atherosclerotic plaque formation was able to demonstrate that when the process of low-density lipoprotein-cholesterol (LDL-C) transport becomes unregulated, complex inflammatory processes cause oxidised LDL uptake by macrophages in the arterial walls. As the macrophages become engorged they form foam cells, which contribute to the process of atherosclerosis through the concomitant activation of inflammatory cascades [6]. This process ultimately leads to cardiovascular events such as thrombosis, MI and stroke [6]. Sufficient data and genetic Mendelian population studies have now been generated to confirm the causal association of LDL-C and atherosclerosis, and are laid out in a consensus statement by the European Society of Cardiology (ESC) [7]. In summary, LDL-C is not only causally related to the risk of cardiovascular disease (CVD), but also displays a cumulative effect proportional to the duration of exposure (Figure 1).
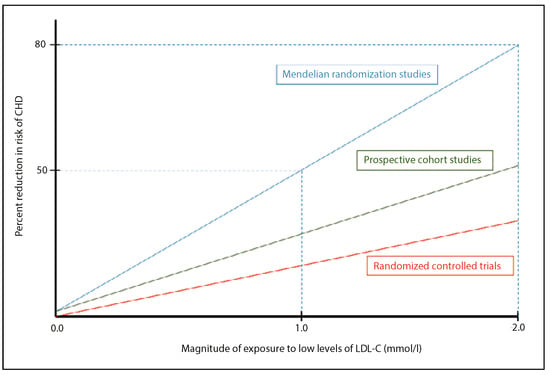
Figure 1.
Causal and cumulative effects of exposure to LDL-cholesterol (LDL-C) on the risk of coronary heart disease (CHD). (Adapted from: Eur Heart J. 2017;38:2459–72 [7]).
Intensive LDL-cholesterol lowering
In daily practice, therefore, the important task remains to identify those individuals who may benefit from intensive LDL-C lowering therapy. Among these are individuals who are at high risk of developing coronary artery disease (CAD) or of presenting recurrent cardiovascular events. The ESC and the Swiss Atherosclerosis Association (AGLA-GSLA) have defined very high risk individuals as those with CVD documented by means of invasive or noninvasive testing (such as coronary angiography, nuclear imaging, stress echocardiography, carotid plaque on ultrasound), previous MI, acute coronary syndrome (ACS), coronary revascularisation and other arterial revascularisation procedures, ischaemic stroke, peripheral arterial disease, patients with type 2 diabetes, patients with type 1 diabetes with target organ damage (such as microalbuminuria), patients with moderate to severe chronic kidney disease or a calculated 10-year risk SCORE >10% [8]. These individuals all have theoretical indications for intensive secondary cardiovascular prevention which, as a function of total CVD risk and LDL-C levels, most often requires therapeutic intervention strategies combining lifestyle advice and concomitant drug therapy.
Individuals with the severe hypercholesterolaemia phenotype (SHP) can also benefit from intensive lipid lowering strategies. SHP refers to all subjects with LDL-C levels above 5 mmol/l (190 mg/dl), regardless of the cause as the clinical consequences of extreme hypercholesterolaemia are the same [9]. The main cause remains primary dyslipidaemias, namely familial hypercholesterolaemia (FH), although secondary causes can also lead to high LDL-C levels. The major molecular causes of FH are mutations in the gene coding for the LDL receptor (LDLR) that binds to an isoform (APOB-100) of apolipoprotein B on the LDL particle [9]. The low-density lipoprotein receptor adaptor protein 1 (LDLRAP1) gene is then responsible for transferring LDLR-mediated LDL to the coated pits and to proprotein convertase subtilisin/kexin type 9 (PCSK9), which causes LDLR degradation [9]. Depending on their site of action, down- or up-regulating mutations in these genes can lead to increased levels of circulating LDL-C (Table 1).

Table 1.
Major molecular causes of familial hypercholesterolaemia.
Based on current knowledge on the effect of prolonged exposure to high levels of circulating LDL-C, it appears increasingly important to identify early individuals with heterozygous FH. Indeed, the risk of premature CHD is estimated to be 20 times higher in untreated FH patients. In an analysis of 4534 patients following an ACS, Nanchen et al. showed that patients with FH and ACS have a greater than two-fold adjusted risk of coronary event recurrence within the first year after discharge as compared with patients without FH, despite widespread use of statins [10]. The Dutch Lipid Clinic Network criteria [11,12] enable the diagnosis of FH on the basis of family and clinical history, as well as on physical examination, LDL-C levels and molecular genetic testing through DNA analysis (Table 2).
Table 2.
Dutch Lipid Clinic Network criteria for diagnosing familial hypercholesterolaemia in adults.
Drug options for intensive LDL-cholesterol reduction and achieving therapeutic goals
How then to achieve intensive LDL-C lowering? Bile acid resins or sequestrants will not be further covered in this review, nor will fibrates or nicotinic acid. In 1971, the search for microbial metabolites that would inhibit 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase began, and in 1990 the first statin was discovered [13]. This discovery, coupled with that of the LDLR pathway for which Joseph Goldsmith and Michael Brown were awarded the Nobel Prize for Medicine in 1985, provided a genetic cause for MI in FH [14]. In 1994, the first randomised trial of cholesterol lowering with simvastatin versus placebo was published. In this trial, which included 4444 patients with CHD who were followed up for a median of 5.4 years, simvastatin led to a 31% relative risk reduction of death; furthermore, the proportion of patients free of major CHD events or without any CHD event, who were free of atherosclerosis-related events or did not require revascularisation also increased [15]. This landmark trial paved the road to further statin trials, notably with pravastatin, in which a reduction of major cardiovascular event rates over 5 years was systematically associated with statin therapy [16]. These promising results rapidly led to the question as to how low LDL-C goals should be set, and whether was there a linear relationship between further lowering of LDL-C and clinical benefits, both in primary and secondary prevention [17].
In the PROVE-IT trial, intensive versus moderate lipid lowering with statins was investigated in the immediate setting after an ACS [18]. In this study, high-dose (80 mg atorvastatin) as compared with moderate (40 mg pravastatin) statin therapy led to a decrease in LDL-C levels by 51% versus 21%, respectively, and was associated with a 16% relative risk (RR) reduction for death or major cardiovascular events. In a later study, LaRosa et al. were able to demonstrate similar findings in the context of stable CAD, where high- (80 mg) vs low-dose (10 mg) atorvastatin was associated with a 2.2% absolute risk reduction and a 22% RR reduction in major cardiovascular events, at the expense, however, of increased alanine transaminase (ALT) levels [19]. In these studies, LDL-C levels were reduced to approximately 1.8 mmol/l (70 mg/dl), and the linear association between event rates and LDL-C levels was maintained.
It took almost ten more years before a major trial was able to show that the addition of a substance that reduces the absorption of cholesterol from the gastrointestinal tract, namely ezetimibe, to background statin therapy could lower LDL-C by a further 24%; these results were observed in a post-ACS patient population, whose baseline LDL-C levels already complied to guideline recommendations [20]. A practical approach to achieving LDL-C targets was summarised in the 2016 ESC/EAS guidelines for the management of dyslipidaemias [21]. In brief, baseline LDL-C levels >3.9 mmol/l (150 mg/dl) will most likely require combination therapy including statins and ezetimibe in order to reach LDL-C goals <1.8 mmol/l (~70 mg/dl); this observation is further reflected in the 2016 ESC guidelines on CVD prevention [22], which state that if the target LDL-C level is not reached with statins alone, combination with either a cholesterol absorption inhibitor, a bile acid sequestrant or nicotinic acid may be considered (class IIb, level of evidence C). But this approach has proved to be insufficient in many clinical situations for a multitude of reasons. For one, adherence to therapy has shown to be suboptimal at best, with frequent treatment discontinuations linked to a lack of motivation, side effects or overt statin intolerance. Alternative strategies, such as doubling statin doses, were only able to achieve a 6% additional LDL-C reduction, with high-dose statin at best leading to a maximum LDL-C reduction of approximately 50% compared with baseline values [23,24]. Further considerations that need to be taken into account are the counter-regulation of cholesterol absorption and synthesis by homeostatic mechanisms that can intervene, as well as patients’ genetic variability that can lead to wide differences in responses to cholesterol-lowering therapy [25,26].
In 2006, new data demonstrated that PCSK9 gene sequence variations causing moderate lifelong reductions in LDL-C plasma levels were associated with a substantial reduction in the incidence of coronary events. This observation could, furthermore, be extended to populations with a high prevalence of non-lipid-related cardiovascular risk factors [27]. The importance of these findings led to intense research into the mechanisms surrounding PCSK9 inhibition, which revealed that PCSK9 inhibition prevents LDL-receptor degradation, thereby increasing LDL-C removal from the blood stream. Humanised and fully human antibodies directed against PCSK9 have since been developed and been investigated in phase I and II trials. Currently, only the fully human molecules have been selected for further investigation, as the chimeric part of the humanised antibodies has been revealed to be linked to antidrug antibody formation, causing large variations in lipid reduction [28]. The efficacy and safety of two fully human PCSK9 inhibitors, alirocumab [29] and evolocumab [30], was recently demonstrated, with a decrease in LDL-C levels of nearly 60% beyond that accomplished with statins alone or in combination with ezetimibe. These results heralded a new era in the management of hyperlipidaemia, with the prospect of achieving extreme levels of reduction in LDL-C in patients intolerant to statins, and also in high-risk patients such as those with FH. Evolocumab was, furthermore, able to demonstrate a decrease atheroma volume, as assessed with intravascular ultrasound, with possibly even plaque regression [31].
Based on the encouraging phase I and II results achieved with PCSK9 inhibitors, large outcome trials were designed. The first evolocumab outcome trial included 27 564 patients and showed a sustained LDL-C reduction of 59% over a median follow-up of 26 months, with an attained median LDL-C level of 0.78 mmol/l (30 mg/dl), associated with a decrease in hazard ratio (HR) of 15% for the cumulative incidence of the primary study endpoint, a composite of cardiovascular death, MI, stroke, hospitalisation for unstable angina or coronary revascularisation, and a 20% HR reduction for the key secondary efficacy endpoint, a composite of cardiovascular death, MI or stroke. No benefit could be shown for mortality alone, possibly owing to the relatively short duration of follow-up for an event-driven trial. Data presented from an outcome trial with alirocumab in the setting of ACS as opposed to stable CAD showed similar LDL-C reductions and a 15% HR reduction in major adverse cardiovascular events (MACE). No safety signal emerged in this study, including for cognitive function [32,33]. Furthermore, the clinical efficacy and safety of evolocumab was demonstrated even when very low LDL-C concentrations were achieved [34]. This is important as there has been concern regarding the safety of achieving very low levels of LDL-C, in particular with regard to short- and long-term neurocognitive function, haemorrhagic stroke, incidence of cancer and incidence of glucose intolerance or new onset diabetes. Analysis of the IMPROVE-IT cohort had previously addressed this concern and showed that the safety profile of patients achieving an LDL-C level below 0.78 mmol/l (30 mg/dl) was similar to those achieving higher concentrations [35]. Additionally, the cumulative incidence of new-onset diabetes over a 3-year follow-up period in the FOURRIER trial was not statistically significantly increased in the evolocumab group as compared with the placebo group [36]. When interpreting these results, potential adverse effects of very low LDL-C must be distinguished from those attributable to specific therapeutic agents. Anti-PCSK9 antibodies have now been included in ESC/EAS guidelines as a treatment option for very high-risk patients with persistent high LDL-C levels despite maximally tolerated statin doses used in combination with ezetimibe, or in patients intolerant to statins [21,37,38].
The value of decreasing LDL-C levels through PCSK9 inhibition has prompted interest in other approaches, one of which is the use of oligonucleotide therapies targeting hepatic production of PCSK9. This method involves use of small interfering RNAs (siRNAs) which selectively and catalytically silence the translation of their complementary target messenger RNA [39]. Craig Mello and Andrew Fire were awarded a Nobel prize for discovering this method in 2006. Single subcutaneous injections have since been shown to significantly lower LDL-C for up to 6 months, reducing levels of circulating PCSK9 by up to 84% and LDL-C levels by approximately 60% [40,41].
The role of inflammation in atherosclerosis
The concomitant activation of inflammatory cascades resulting from the infiltration of oxidised LDL into endothelial cells is paramount in the development of atherosclerotic plaques. In the process of atherosclerosis, leucocyte adhesion molecules are secreted, leading to monocyte adhesion, migration and differentiation into macrophages which secrete inflammatory cytokines (interferon-γ, interleukin-1, tumour necrosis factor [TNF]), chemokines, proteases and radicals. T-cell activation and activated immune cells in plaques lead to the production of interleukin-6 (IL-6), which in turn stimulates the production of acute-phase reactants, including C-reactive protein (CRP) and fibrinogen (Figure 2) [42,43].
In a retrospective analysis of 543 men who had suffered MI or stroke, baseline plasma CRP concentrations were shown to be predictive of the risk of these events, whereas the use of aspirin was associated with a reduction in the relative risk of MI [2]. This was the first study to demonstrate a clinical benefit from anti-inflammatory agents in preventing cardiovascular disease. In a further retrospective study, CRP and LDL-C baseline measurements were made in 27 939 apparently healthy women who were then followed up for the occurrence of MI, ischaemic stroke, coronary revascularisation or death from cardiovascular disease; over a mean follow-up period of 8 years both CRP and LDL-C quintiles were proven to be associated with the probability of event-free survival, with a synergistic association between the two [44]. More recently, the IMPROVE-IT trial showed that patients on statin therapy who had achieved low levels of high-sensitive CRP (hsCRP) had better clinical outcomes than those with higher hsCRP levels, independently of the level of LDL-C reached [45]. In the Jupiter study, 17 802 apparently healthy men and women with normal LDL-C values of <3.4 mmol/l but CRP values elevated to >2.0 mg/l at baseline were randomly assigned to rosuvastatin 20 mg/d or placebo [46]. After a mean follow-up period of 1.9 years, both LDL-C and CRP levels were significantly reduced in the rosuvastatin arm (by 50% and 37%, respectively); furthermore, rosuvastatin was associated with a significant reduction in the incidence of cardiovascular events compared with placebo, thereby demonstrating the positive effect of LDL-C-lowering on inflammation [46]. The question then arose as to whether reducing inflammation alone could reduce cardiovascular events.


Figure 2.
Inflammatory cytokine cascade and novel targets for atheroprotection. (Adapted from N Engl J Med. 2005;352:1685–95 [43] and Circulation Res. 2016;118:145–56 [47]).
Figure 2.
Inflammatory cytokine cascade and novel targets for atheroprotection. (Adapted from N Engl J Med. 2005;352:1685–95 [43] and Circulation Res. 2016;118:145–56 [47]).
In inflammatory diseases, the inflammasome plays a key role in translating organic insult into an inflammatory response. Canakinumab is a high-affinity human monoclonal anti-human antibody designed to bind and functionally neutralise the bioactivity of the pro-inflammatory cytokine interleukin-1B (IL-1B), thereby reducing inflammation [47]. In two IL-1B-driven inflammatory diseases, namely cryopyrin-associated periodic syndrome and systemic juvenile idiopathic arthritis, canakinumab has already been shown to lead to rapid remission of symptoms [48,49]. In the field of cardiovascular disease, the effect of reducing inflammation without affecting lipid levels was tested in the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS), which investigated the effect of three dosages (50 mg, 150 mg and 300 mg every 3 months) of canakinumab administered subcutaneously compared with placebo in 10 061 stable CAD patients with persistently elevated hsCRP levels (>2 mg/l) over a period of 48 months. As expected, canakinumab did not lead to reductions in LDL-C levels; on the other hand, hsCRP levels were reduced by between 26% (50 mg) and 41% (300 mg) compared with baseline values [50]. At a median follow-up of 3.7 years, the primary endpoint of nonfatal MI, nonfatal stroke or cardiovascular death had been achieved for the 150 mg dose, with a risk reduction of 15% compared with placebo, an effect probably dependent on the level of hsCRP reduction. This study was able to demonstrate that anti-inflammatory therapy alone, in this case targeting the IL-1B innate immunity pathway, can positively impact on the rate of recurrence of cardiovascular events, independent of any lipid-lowering effect. In the case of canakinumab, these results were achieved at the expense of mild neutropenia, an increased risk of infection and osteoarthritis, whereas cancer mortality was significantly lower [46].
Perspectives
Current evidence favours applying risk stratification based on LDL-C levels to high-risk patients. Risk stratification might eventually be further improved by a better understanding of the role played by other serum lipids, such as lipoprotein(a) (Lp(a)). For example, if the latter proves capable of indicating residual cholesterol risk, it could become an interesting therapeutic target [51]. Plasma levels of inflammatory biomarkers do, nevertheless, also appear to be strongly correlated with recurrent cardiovascular events, independent of lipid levels [52]. Further studies are, however, needed to understand how to integrate this residual inflammatory risk, as assessed by hsCRP measurement, into patient management. In primary prevention, individuals with elevated baseline hsCRP and non-elevated LDL-C treated with statins who achieve both low CRP and low LDL-C levels seem to benefit most from statin therapy [46,47,53]. In secondary prevention, it may eventually be beneficial to distinguish patients with a residual cholesterol risk from those with a residual inflammatory risk, as the latter may also become a separate therapeutic target. Statins, as opposed to PCSK9 inhibitors, have both lipid lowering and anti-inflammatory properties and will therefore probably remain an integral part of therapy in secondary prevention, at least in the short to medium term. A more comprehensive approach to risk assessment will, moreover, be necessary in order to achieve a more personalised approach to patient management, integrating parameters such as genetic profiling, a complete lipid screening panel including Lp(a) and perhaps multiple hsCRP measurements to clearly evaluate the potential inflammatory risk.
Disclosure Statement
No financial support and no other potential conflict of interest relevant to this article was reported.
References
- Yeh, R.W.; Sidney, S.; Chandra, M.; Sorel, M.; Selby, J.V.; Go, A.S. Population trends in the incidence and outcomes of acute myocardial infarction. N Engl J Med 2010, 362, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 1997, 336, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Keys, A.; Taylor, H.L.; Blackburn, H.; Brozek, J.; Anderson, J.T.; Simonson, E. Coronary Heart Disease among Minnesota Business and Professional Men Followed Fifteen Years. Circulation 1963, 28, 381–395. [Google Scholar] [CrossRef] [PubMed]
- A co-operative trial in the primary prevention of ischaemic heart disease using clofibrate. Report from the Committee of Principal Investigators. Br Heart J 1978, 40, 1069–1118. [CrossRef]
- Buchwald, H.; Varco, R.L.; Matts, J.P.; Long, J.M.; Fitch, L.L.; Campbell, G.S.; et al. Effect of partial ileal bypass surgery on mortality and morbidity from coronary heart disease in patients with hypercholesterolemia. Report of the Program on the Surgical Control of the Hyperlipidemias (POSCH). N Engl J Med 1990, 323, 946–955. [Google Scholar] [CrossRef]
- Quehenberger, O.; Dennis, E.A. The human plasma lipidome. N Engl J Med 2011, 365, 1812–1823. [Google Scholar] [CrossRef]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2017, 38, 2459–2472. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; et al.; ESC Scientific Document Group. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts) Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur Heart J 2016, 37, 2315–2381. [Google Scholar] [CrossRef]
- Sniderman, A.D.; Tsimikas, S.; Fazio, S. The severe hypercholesterolemia phenotype: Clinical diagnosis, management, and emerging therapies. J Am Coll Cardiol 2014, 63, 1935–1947. [Google Scholar] [CrossRef]
- Nanchen, D.; Gencer, B.; Muller, O.; Auer, R.; Aghlmandi, S.; Heg, D.; et al. Prognosis of Patients With Familial Hypercholesterolemia After Acute Coronary Syndromes. Circulation 2016, 134, 698–709. [Google Scholar] [CrossRef]
- Hovingh, G.K.; Davidson, M.H.; Kastelein, J.J.; O’Connor, A.M. Diagnosis and treatment of familial hypercholesterolaemia. Eur Heart J 2013, 34, 962–971. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; et al.; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the European Atherosclerosis Society. Eur Heart J 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [PubMed]
- Endo, A. The discovery and development of HMG-CoA reductase inhibitors. J Lipid Res 1992, 33, 1569–1582. [Google Scholar] [CrossRef]
- Nabel, E.G.; Braunwald, E. A tale of coronary artery disease and myocardial infarction. N Engl J Med 2012, 366, 54–63. [Google Scholar] [CrossRef]
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 344, 1383–1389. [CrossRef]
- Genest, J.; Pedersen, T.R. Prevention of cardiovascular ischemic events: High-risk and secondary prevention. Circulation 2003, 107, 2059–2065. [Google Scholar] [CrossRef]
- Cannon, C.P. The next step in cardiovascular protection. Atheroscler Suppl 2003, 4, 3–9. [Google Scholar] [CrossRef]
- Cannon, C.P.; Braunwald, E.; McCabe, C.H.; Rader, D.J.; Rouleau, J.L.; Belder, R.; et al.; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004, 350, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- LaRosa, J.C.; Grundy, S.M.; Waters, D.D.; Shear, C.; Barter, P.; Fruchart, J.C.; et al.; Treating to New Targets (TNT) Investigators. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005, 352, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Theroux, P.; et al.; IMPROVE-IT Investigators. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med 2015, 372, 2387–2397. [Google Scholar] [CrossRef]
- Catapano, A.L.; Graham, I.; De Backer, G.; Wiklund, O.; Chapman, M.J.; Drexel, H.; et al.; ESC Scientific Document Group. 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias. Eur Heart J 2016, 37, 2999–3058. [Google Scholar] [CrossRef] [PubMed]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; et al.; ESC Scientific Document Group. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts)Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur Heart J 2016, 37, 2315–2381. [Google Scholar] [CrossRef]
- Reiner, Z.; Catapano, A.L.; De Backer, G.; Graham, I.; Taskinen, M.R.; Wiklund, O.; et al.; European Association for Cardiovascular Prevention & Rehabilitation; ESC Committee for Practice Guidelines (CPG) 2008-2010 and 2010-2012 Committees. ESC/EAS Guidelines for the management of dyslipidaemias: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J 2011, 32, 1769–1818. [Google Scholar] [CrossRef]
- Weng, T.C.; Yang, Y.H.; Lin, S.J.; Tai, S.H. A systematic review and meta-analysis on the therapeutic equivalence of statins. J Clin Pharm Ther 2010, 35, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Bosner, M.S.; Lange, L.G.; Stenson, W.F.; Ostlund, R.E., Jr. Percent cholesterol absorption in normal women and men quantified with dual stable isotopic tracers and negative ion mass spectrometry. J Lipid Res 1999, 40, 302–308. [Google Scholar] [CrossRef]
- Pedro-Botet, J.; Schaefer, E.J.; Bakker-Arkema, R.G.; Black, D.M.; Stein, E.M.; Corella, D.; et al. Apolipoprotein E genotype affects plasma lipid response to atorvastatin in a gender specific manner. Atherosclerosis 2001, 158, 183–193. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H., Jr.; Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Ridker, P.M.; Tardif, J.C.; Amarenco, P.; Duggan, W.; Glynn, R.J.; Jukema, J.W.; et al.; SPIRE Investigators. Lipid-Reduction Variability and Antidrug-Antibody Formation with Bococizumab. N Engl J Med 2017, 376, 1517–1526. [Google Scholar] [CrossRef]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; et al.; ODYSSEY LONG TERM Investigators. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med 2015, 372, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Wiviott, S.D.; Raal, F.J.; Blom, D.J.; Robinson, J.; et al.; Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 2015, 372, 1500–1509. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Puri, R.; Anderson, T.; Ballantyne, C.M.; Cho, L.; Kastelein, J.J.; et al. Effect of Evolocumab on Progression of Coronary Disease in Statin-Treated Patients: The GLAGOV Randomized Clinical Trial. JAMA 2016, 316, 2373–2384. [Google Scholar] [CrossRef]
- Giugliano, R.P.; Mach, F.; Zavitz, K.; Kurtz, C.; Schneider, J.; Wang, H.; et al.; EBBINGHAUS Investigators. Design and rationale of the EBBINGHAUS trial: A phase 3, double-blind, placebo-controlled, multicenter study to assess the effect of evolocumab on cognitive function in patients with clinically evident cardiovascular disease and receiving statin background lipid-lowering therapy-A cognitive study of patients enrolled in the FOURIER trial. Clin Cardiol 2017, 40, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, R.P.; Sabatine, M.S.; Ott, B.R. Cognitive Function in a Randomized Trial of Evolocumab. N Engl J Med 2017, 377, 1997. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, R.P.; Pedersen, T.R.; Park, J.G.; De Ferrari, G.M.; Gaciong, Z.A.; Ceska, R.; et al.; FOURIER Investigators. Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: A prespecified secondary analysis of the FOURIER trial. Lancet 2017, 390, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, R.P.; Wiviott, S.D.; Blazing, M.A.; De Ferrari, G.M.; Park, J.G.; Murphy, S.A.; et al. Long-term Safety and Efficacy of Achieving Very Low Levels of Low-Density Lipoprotein Cholesterol : A Prespecified Analysis of the IMPROVE-IT Trial. JAMA Cardiol 2017, 2, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Leiter, L.A.; Wiviott, S.D.; Giugliano, R.P.; Deedwania, P.; De Ferrari, G.M.; et al. Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new-onset diabetes: A prespecified analysis of the FOURIER randomised controlled trial. Lancet Diabetes Endocrinol 2017, 5, 941–950. [Google Scholar] [CrossRef]
- Landmesser, U.; Chapman, M.J.; Farnier, M.; Gencer, B.; Gielen, S.; Hovingh, G.K.; et al.; European Society of Cardiology (ESC); European Atherosclerosis Society (EAS). European Society of Cardiology/European Atherosclerosis Society Task Force consensus statement on proprotein convertase subtilisin/kexin type 9 inhibitors: Practical guidance for use in patients at very high cardiovascular risk. Eur Heart J 2017, 38, 2245–2255. [Google Scholar] [CrossRef]
- Landmesser, U.; Chapman, M.J.; Stock, J.K.; Amarenco, P.; Belch, J.J.F.; Borén, J.; et al. 2017 Update of ESC/EAS Task Force on practical clinical guidance for proprotein convertase subtilisin/kexin type 9 inhibition in patients with atherosclerotic cardiovascular disease or in familial hypercholesterolaemia. Eur Heart J 2018, 39, 1131–1143. [Google Scholar] [CrossRef]
- Khvorova, A. Oligonucleotide Therapeutics—A New Class of Cholesterol-Lowering Drugs. N Engl J Med 2017, 376, 4–7. [Google Scholar] [CrossRef]
- Fitzgerald, K.; White, S.; Borodovsky, A.; Bettencourt, B.R.; Strahs, A.; Clausen, V.; et al. A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N Engl J Med 2017, 376, 41–51. [Google Scholar] [CrossRef]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; et al. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N Engl J Med 2017, 376, 1430–1440. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis--an inflammatory disease. N Engl J Med 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 2005, 352, 1685–1695. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Rose, L.; Buring, J.E.; Cook, N.R. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med 2002, 347, 1557–1565. [Google Scholar] [CrossRef]
- Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; et al.; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 (PROVE IT-TIMI 22) Investigators. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005, 352, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M.; Jr Kastelein, J.J.; et al.; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream To Identify Novel Targets for Atheroprotection. Circ Res 2016, 118, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, H.J.; Kone-Paut, I.; Kuemmerle-Deschner, J.B.; Leslie, K.S.; Hachulla, E.; Quartier, P.; et al.; Canakinumab in CAPS Study Group. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med 2009, 360, 2416–2425. [Google Scholar] [CrossRef]
- Ruperto, N.; Brunner, H.I.; Quartier, P.; Constantin, T.; Wulffraat, N.; Horneff, G.; et al.; PRINTO; PRCSG. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012, 367, 2396–2406. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; et al.; CANTOS Trial Group. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Gencer, B.; Kronenberg, F.; Stroes, E.S.; Mach, F. Lipoprotein(a): The revenant. Eur Heart J 2017, 38, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Lüscher, T.F. Anti-inflammatory therapies for cardiovascular disease. Eur Heart J 2014, 35, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. Residual inflammatory risk: Addressing the obverse side of the atherosclerosis prevention coin. Eur Heart J 2016, 37, 1720–1722. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the author. Attribution - Non-Commercial - NoDerivatives 4.0.