
Integrative Analysis of Transcriptomic and Proteomic Changes Related to Cytoplasmic Male Sterility in Spring Stem Mustard (Brassica juncea var. tumida Tsen et Lee)

 ,
,  ,
,
Abstract
1. Introduction
2. Results
2.1. Phenotypic and Cytological Characterization in 09-05A/B Lines of B. juncea
2.2. Transcriptomic Analysis and Assembly
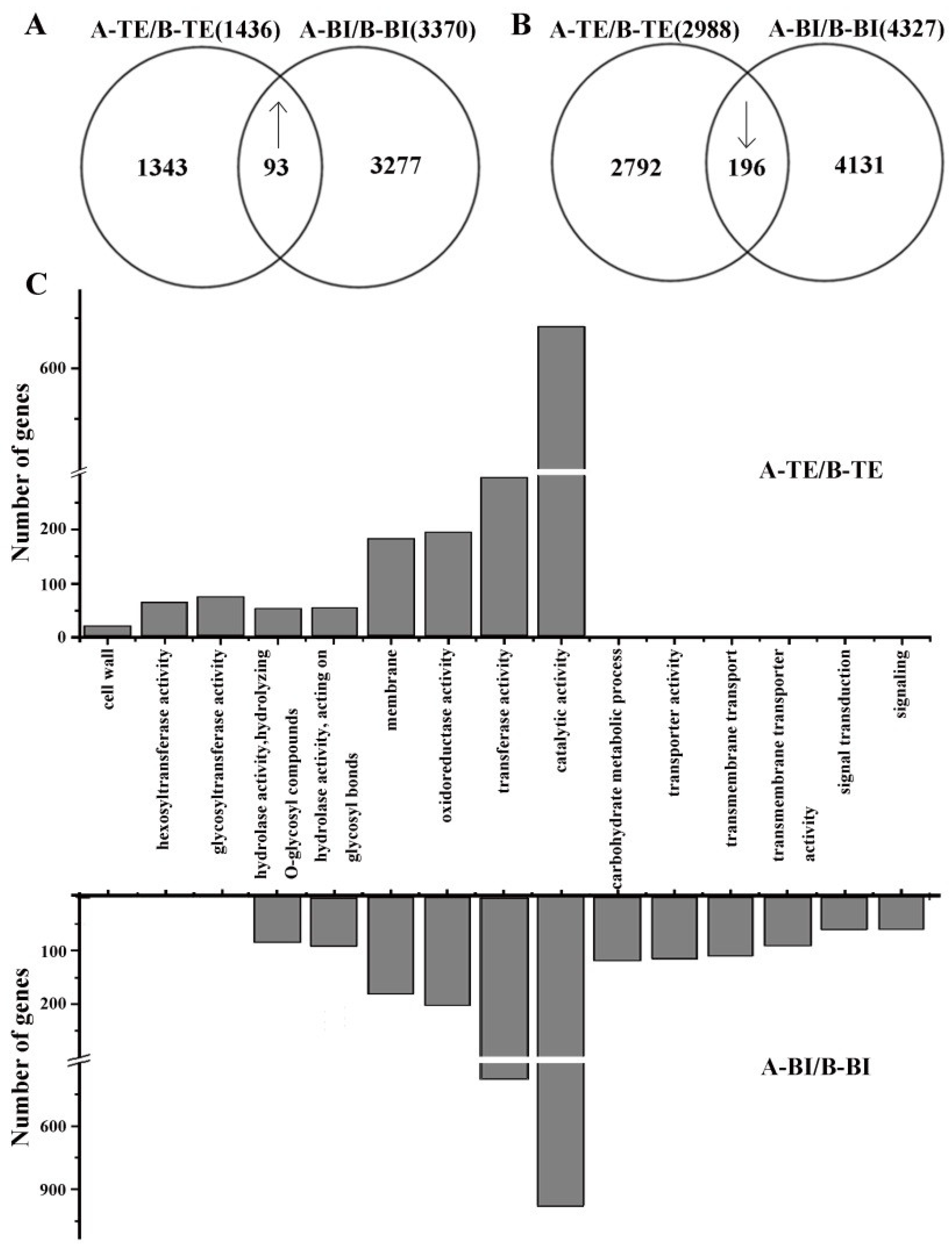
2.3. Functional Distribution of DEGs in 09-05A/B Lines of B. juncea
2.4. Functional Distribution of DAPs in 09-05A/B Lines of B. juncea
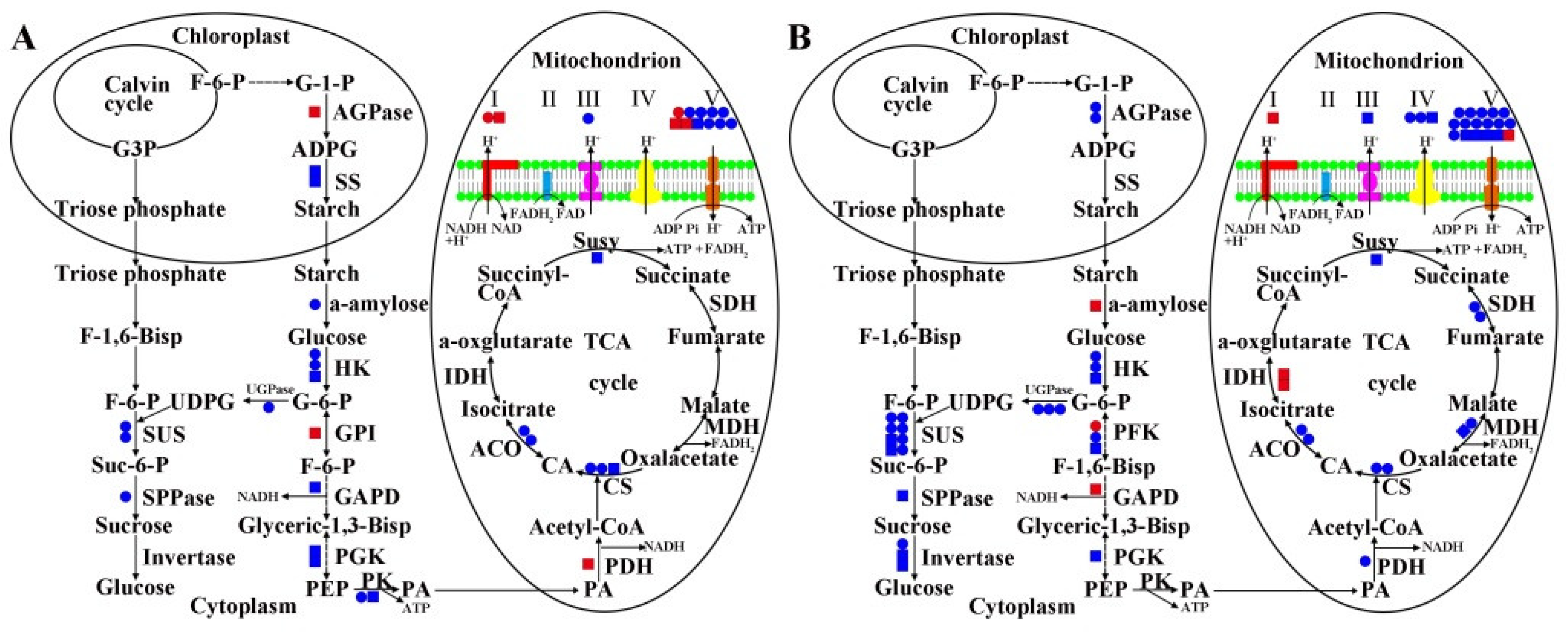
2.5. MS Related DEGs and DAPs in Carbohydrate Metabolism and Energy Metabolism of 09-05A/B Lines of B. juncea
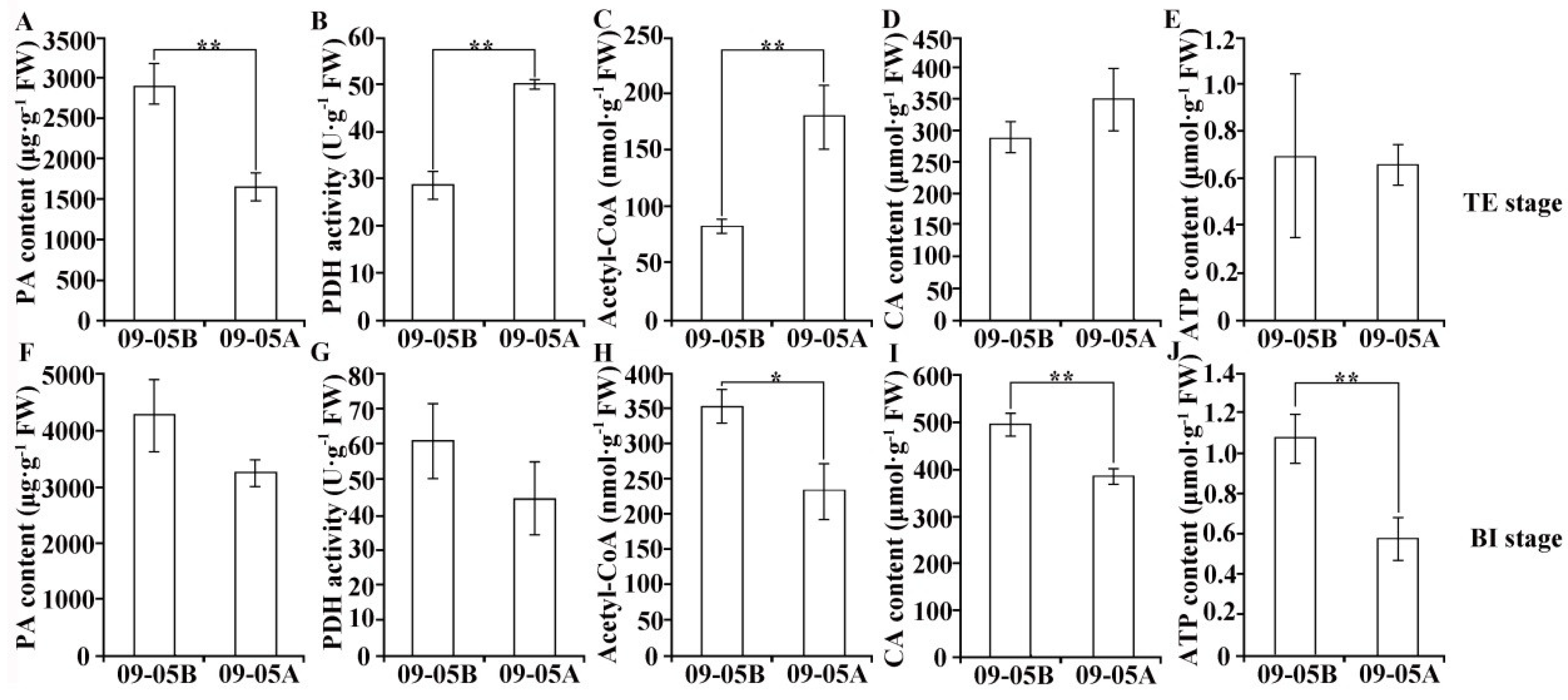
2.6. Metabolic Products and Enzyme Activity Analyses in 09-05A/B Lines of B. juncea
3. Discussion
3.1. Microspores Are Defective with Degraded Cellular Components and Altered Pollen Wall in 09-05A
3.2. Damage to Starch–Sucrose Metabolism and Glycolysis Pathway May Inhibit the Production of Respiratory Substrate in 09-05A
3.3. Damage to the TCA Cycle and Respiratory Chain May Inhibit the Production of ATP in 09-05A
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Morphological and Cytological Observation
4.3. RNA Extraction for Transcriptomic Analysis
4.4. Transcriptomic Sequencing, Data Processing, and Transcriptomic Analysis
4.5. Total Protein Extraction and Peptide Preparation
4.6. LC–MS/MS Analysis
4.7. Protein Identification from Mass Spectrometry Data
4.8. Functional Annotation
4.9. Metabolic Products and Enzyme Activity Analyses
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hu, J.; Lan, M.; Xu, X.; Yang, H.; Zhang, L.; Lv, F.; Yang, H.; Yang, D.; Li, C.; He, J. Transcriptome Profiling Reveals Molecular Changes during Flower Development between Male Sterile and Fertile Chinese Cabbage (Brassica rapa ssp. pekinensis) Lines. Life 2021, 11, 525. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, B.; Khan, A.; Zheng, J.; Dawar, F.U.; Akhtar, K.; Zhou, R. Reactive Oxygen Species Accumulation Strongly Allied with Genetic Male Sterility Convertible to Cytoplasmic Male Sterility in Kenaf. Int. J. Mol. Sci. 2021, 22, 1107. [Google Scholar] [CrossRef]
- Sang, S.; Cheng, H.; Hao, M.; Ding, B.; Hu, Q. Mitochondrial localization of orf346 causes pollen abortion in alloplasmic male sterility. Crop J. 2021, 9, 1320–1329. [Google Scholar] [CrossRef]
- Xu, F.; Yang, X.; Zhao, N.; Hu, Z.; Mackenzie, S.A.; Zhang, M.; Yang, J. Exploiting sterility and fertility variation in cytoplasmic male sterile vegetable crops. Horticul. Res. 2022, 9, uhab039. [Google Scholar] [CrossRef] [PubMed]
- Jing, B.; Heng, S.; Tong, D.; Wan, Z.; Fu, T.; Tu, J.; Ma, C.; Yi, B.; Wen, J.; Shen, J. A male sterility-associated cytotoxic protein ORF288 in Brassica juncea causes aborted pollen development. J. Exp. Bot. 2012, 63, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Yang, Z.; Yi, B.; Wen, J.; Shen, J.; Tu, J.; Ma, C.; Fu, T. Comparative transcript profiling of the fertile and sterile flower buds of pol CMS in B. napus. BMC Genom. 2014, 15, 258. [Google Scholar] [CrossRef]
- Du, K.; Liu, Q.; Wu, X.; Jiang, J.; Wu, J.; Fang, Y.; Li, A.; Wang, Y. Morphological structure and transcriptome comparison of the cytoplasmic male sterility line in Brassica napus (SaNa-1A) derived from somatic hybridization and its maintainer line saNa-1B. Front. Plant Sci. 2016, 7, 1313. [Google Scholar] [CrossRef]
- Heng, S.; Liu, S.; Xia, C.; Tang, H.; Xie, F.; Fu, T.; Wan, Z. Morphological and genetic characterization of a new cytoplasmic sterile line system (oxa CMS) in stem mustard (Brassica juncea). Theor. Appl. Genet. 2018, 131, 59–66. [Google Scholar] [CrossRef]
- Li, P.; Zhang, D.; Su, T.; Wang, W.; Yu, Y.; Zhao, X.; Li, Z.; Yu, S.; Zhang, F. Genome-wide analysis of mRNA and lncRNA expression and mitochondrial genome sequencing provide insights into the mechanisms underlying a novel cytoplasmic male sterility system, BVRC-CMS96, in Brassica rapa. Theor. Appl. Genet. 2020, 133, 2157–2170. [Google Scholar] [CrossRef]
- Chase, C. Cytoplasmic male sterility: A window to the world of plant mitochondrial-nuclear interactions. Trends Genet. 2007, 23, 81–90. [Google Scholar] [CrossRef]
- Zhao, N.; Li, Z.; Zhang, L.; Yang, X.; Mackenzie, S.; Hu, Z.; Zhang, M.; Yang, J. Muts homolog1 mediates fertility reversion from cytoplasmic male sterile Brassica juncea in response to environment. Plant Cell Environ. 2021, 44, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Yao, P.; Li, H.; Han, Y.; Xu, K.; Heng, S.; Fu, T.; Wan, Z. Genetic mapping reveals BjRf as a candidate gene controlling fertility restoration of the oxa CMS in Brassica juncea. Theor. Appl. Genet. 2021, 134, 2355–2365. [Google Scholar] [CrossRef] [PubMed]
- Heng, S.; Chen, F.; Wei, C.; Li, X.; Yi, B.; Ma, C.; Tu, J.; Shen, J.; Fu, T.; Wen, J. Cytological and iTRAQ-based quantitative proteomic analyses of hau CMS in Brassica napus L. J. Proteomics. 2019, 193, 230–238. [Google Scholar] [CrossRef]
- Du, K.; Xiao, Y.; Liu, Q.; Wu, X.; Jiang, J.; Wu, J.; Fang, Y.; Xiang, Y.; Wang, Y. Abnormal tapetum development and energy metabolism associated with sterility in SaNa-1A CMS of Brassica napus L. Plant Cell Rep. 2019, 38, 545–558. [Google Scholar] [CrossRef]
- Ji, J.; Huang, J.; Yang, L.; Fang, Z.; Zhang, Y.; Zhuang, M.; Lv, H.; Wang, Y.; Liu, Y.; Li, Z.; et al. Advances in research and application of male sterility in Brassica oleracea. Horticulturae 2020, 6, 101. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, D.; Ke, F.; Zhu, M.; Xu, J.; Zhang, M. Seedless mutant ‘Wuzi Ougan’ (Citrus suavissima Hort. ex Tanaka ‘seedless’) and the wild type were compared by iTRAQ-based quantitative proteomics and integratedly analyzed with transcriptome to improve understanding of male sterility. BMC Genet. 2018, 19, 106. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Jin, J.; Zhang, H.; Wang, Y.; Li, Q.; Zou, Y.; Huang, X.; Zhou, B.; Zhou, R.; Ding, Y. Comparative analysis of proteomics and transcriptomics during fertility transition in a two-line hybrid rice line Wuxiang S. Int. J. Mol. Sci. 2019, 20, 4542. [Google Scholar] [CrossRef]
- Dai, F.; Wang, Z.; Li, Z.; Luo, G.; Wang, Y.; Tang, C. Transcriptomic and proteomic analyses of mulberry (Morus atropurpurea) fruit response to Ciboria carunculoides. J. Proteom. 2019, 193, 142–153. [Google Scholar] [CrossRef]
- Ji, J.; Yang, L.; Fang, Z.; Zhuang, M.; Zhang, Y.; Lv, H.; Liu, Y.; Li, Z. Complementary transcriptome and proteome profiling in cabbage buds of a recessive male sterile mutant provides new insights into male reproductive development. J. Proteom. 2018, 179, 80–91. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, J.; Qu, Y.; Peng, R.; Magwanga, R.O.; Liu, F.; Huang, J. Transcriptomic and proteomic analyses of a new cytoplasmic male sterile line with a wild Gossypium bickii genetic background. BMC Genom. 2020, 21, 859. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, X.; Chen, Z.; Qin, L.; Li, B.; Ouyang, L.; Peng, X.; He, H. Quantitative Proteomics and transcriptomics reveals differences in proteins during anthers development in Oryza longistaminata. Front. Plant Sci. 2021, 12, 744792. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Sun, C.; Li, H.; Hu, S.; Lei, L.; Kang, J. Integrated analysis of transcriptome and proteome changes related to the ogura cytoplasmic male sterility in cabbage. PLoS ONE 2018, 13, e0193462. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Liu, W.; Zhang, W.; Tang, D. Integrative analysis of transcriptomic and proteomic changes related to male sterility in Tagetes erecta. Physiol. Mol. Biol. Plants 2020, 26, 2061–2074. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Yang, W.; Li, T.; Shoaib, M.; Sun, J.; Liu, D.; Li, X.; Nie, Y.; Tian, X.; Zhang, A. Combined transcriptome and proteome analysis of anthers of AL-type cytoplasmic male sterile line and its maintainer line reveals new insights into mechanism of male sterility in common wheat. Front. Genet. 2021, 12, 762332. [Google Scholar] [CrossRef]
- Ning, L.; Wang, H.; Li, D.; Lin, Z.; Li, Y.; Zhao, W.; Chao, H.; Miao, L.; Li, M. Transcriptomic and proteomic analysis of Shaan2A cytoplasmic male sterility and its maintainer line in Brassica napus. Front. Plant Sci. 2019, 10, 252. [Google Scholar] [CrossRef]
- Wu, X.; Chen, B.; Lu, G.; Wang, H.; Xu, K.; Gao, G.; Song, Y. Genetic diversity in oil and vegetable mustard (Brassica juncea) landraces revealed by SRAP markers. Genet. Resour. Crop Evol. 2009, 56, 1011–1022. [Google Scholar] [CrossRef]
- Rhythm; Sharma, P.; Sardana, V. Physiological and biochemical traits of drought tolerance in Brassica juncea (L.). Czern Coss. 2022, 146, 509–520. [Google Scholar]
- Tian, Y.; Deng, F.; Zhao, L.; Du, H.; Li, T.; Lai, D.; Zhou, T.; Qing, Z. Characterization of extractable components of fresh and fermented huarong large-leaf mustard and their inhibitory effects on human colon cancer cells. Food Biosci. 2021, 43, 101280. [Google Scholar] [CrossRef]
- Linke, B.; Börner, T. Mitochondrial effects on flower and pollen development. Mitochondrion 2005, 5, 389–402. [Google Scholar] [CrossRef]
- Hanson, M.R.; Bentolila, S. Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell. 2004, 16, S154–S169. [Google Scholar] [CrossRef]
- Wu, Z.; Cheng, J.; Qin, C.; Hu, Z.; Yin, C.; Hu, K. Differential proteomic analysis of anthers between cytoplasmic male sterile and maintainer lines in Capsicum annuum L. Int. J. Mol. Sci. 2013, 14, 22982–22996. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, Z.; Wu, S.; Wan, X. The essential roles of sugar metabolism for pollen development and male fertility in plants. Crop J. 2021, 9, 1223–1236. [Google Scholar] [CrossRef]
- Li, J.; Ding, X.; Han, S.; He, T.; Zhang, H.; Yang, L.; Yang, S.; Gai, J. Differential proteomics analysis to identify proteins and pathways associated with male sterility of soybean using iTRAQ-based strategy. J. Protein Chem. 2016, 138, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, G.; Zhang, Y.; Song, Q.; Chen, Z.; Wang, J.; Guo, J.; Niu, N.; Wang, J.; Ma, S. Comparative studies of mitochondrial proteomics reveal an intimate protein network of male sterility in wheat (Triticum aestivum L.). J. Exp. Bot. 2015, 66, 6191–6203. [Google Scholar] [CrossRef]
- Dauphinee, A.N.; Fletcher, J.I.; Denbigh, G.L.; Lacroix, C.R.; Gunawardena, A.H. Remodelling of lace plant leaves: Antioxidants and ROS are key regulators of programmed cell death. Planta 2017, 246, 133–147. [Google Scholar] [CrossRef]
- Han, Y.; Zhou, S.; Fan, J.; Zhou, L.; Shi, Q.; Zhang, Y.; Liu, X.; Chen, X.; Zhu, J.; Yang, Z. OsMS188 is a key regulator of tapetum development and sporopollenin synthesis in rice. Rice 2021, 14, 4. [Google Scholar] [CrossRef]
- Lei, T.; Zhang, L.; Feng, P.; Liu, Y.; Yin, W.; Shang, L.; He, G.; Wang, N. OsMYB103 is essential for tapetum degradation in rice. Theor. Appl. Genet. 2022, 135, 929–945. [Google Scholar] [CrossRef]
- Li, Q.; Zhao, Y.; Xie, Y. Paeonol disrupts the integrity of aspergillus flavus cell walls via releasing surface proteins, inhibiting the biosynthesis of β-1,3-Glucan and promoting the degradation of chitin, and an identification of cell surface proteins. Foods 2021, 10, 2951. [Google Scholar] [CrossRef]
- Khan, R.M.; Yu, P.; Sun, L.; Abbas, A.; Shah, L.; Xiang, X.; Wang, D.; Sohail, A.; Zhang, Y.; Liu, Q.; et al. DCET1 controls male sterility through callose regulation, exine formation, and tapetal programmed cell death in rice. Front. Genet. 2021, 12, 790789. [Google Scholar] [CrossRef]
- Che, R.; Hu, B.; Wang, W.; Xiao, Y.; Liu, D.; Yin, W.; Tong, H.; Chu, C. POLLEN STERILITY, a novel suppressor of cell division, is required for timely tapetal programmed cell death in rice. Sci. China Life Sci. 2021. [Google Scholar] [CrossRef]
- Wei, X.; Lv, Y.; Zhao, Y.; Nath, U.K.; Yuan, Y.; Wang, Z.; Yang, S.; Jia, H.; Wei, F.; Zhang, X. Comparative transcriptome analysis in Chinese cabbage (Brassica rapa ssp. pekinesis) for DEGs of Ogura-, Polima-CMS and their shared maintainer. Physiol. Mol. Biol. Plants 2020, 26, 719–731. [Google Scholar] [CrossRef]
- Mamun, E.A.; Alfred, S.; Cantrill, L.C.; Overall, R.L.; Sutton, B.G. Effects of chilling on male gametophyte development in rice. Cell Biol. Int. 2006, 30, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Eom, J.S.; Hwang, S.K.; Shin, D.; An, G.; Okita, T.W.; Jeon, J.S. Plastidic phosphoglucomutase and ADP-glucose pyrophosphorylase mutants impair starch synthesis in rice pollen grains and cause male sterility. J. Exp. Bot. 2016, 67, 5557–5569. [Google Scholar] [CrossRef]
- Suzuki, T.; Narciso, J.O.; Zeng, W.; van de Meene, A.; Yasutomi, M.; Takemura, S.; Lampugnani, E.R.; Doblin, M.S.; Bacic, A.; Ishiguro, S. Kns4/upex1: A type II arabinogalactan beta-(1, 3)-galactosyltransferase required for pollen exine development. Plant Physiol. 2017, 173, 183–205. [Google Scholar] [CrossRef]
- Reichert, A.S.; Neupert, W. Mitochondriomics or what makes us breathe. Trends Genet. 2004, 20, 555–562. [Google Scholar] [CrossRef]
- Logan, D.C. The mitochondrial compartment. J. Exp. Bot. 2006, 57, 1225–1243. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Ye, J.; Yang, X.; Li, S.; Zhang, L.; Song, X. Identification of proteins involved in carbohydrate metabolism and energy metabolism pathways and their regulation of cytoplasmic male sterility in wheat. Int. J. Mol. Sci. 2018, 19, 324. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Bao, S.; Zhou, X.; Liu, J.; Yong, Z. The key genes and pathways related to male sterility of eggplant revealed by comparative transcriptome analysis. BMC Plant Biol. 2018, 18, 209. [Google Scholar]
- Hu, L.; Liang, W.; Yin, C.; Cui, X.; Zong, J.; Wang, X.; Hu, J.; Zhang, D. Rice MADS3 regulates ROS homeostasis during late anther development. Plant Cell. 2011, 23, 515–533. [Google Scholar] [CrossRef]
- Duroc, Y.; Hiard, S.; Vrielynck, N.; Ragu, S.; Budar, F. The Ogura sterility-inducing protein forms a large complex without interfering with the oxidative phosphorylation components in rapeseed mitochondria. Plant Mol. Biol. 2009, 70, 123–137. [Google Scholar] [CrossRef]
- Wen, L.; Liu, G.; Zhang, Z.; Tao, J.; Wan, C.; Zhu, Y. Preliminary proteomics analysis of the total proteins of HL Type cytoplasmic male sterility rice anther. Yi Chuan 2006, 28, 311–316. [Google Scholar] [PubMed]
- Min, L.; Zhu, L.; Tu, L.; Deng, F.; Yuan, D.; Zhang, X. Cotton ghcki disrupts normal male reproduction by delaying tapetum programmed cell death via inactivating starch synthase. Plant J. 2013, 75, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhao, X.; Shao, Z.; Wei, Z.; Wang, Y.; Zhu, L.; Zhao, J.; Sun, M.; He, R.; He, G. Rice UDP-glucose pyrophosphorylase1 is essential for pollen callose deposition and its cosuppression results in a new type of thermosensitive genic male sterility. Plant Cell. 2007, 19, 847–861. [Google Scholar] [CrossRef]
- Woo, M.-O.; Ham, T.-H.; Ji, H.-S.; Choi, M.-S.; Jiang, W.; Chu, S.-H.; Piao, R.; Chin, J.-H.; Kim, J.-A.; Park, B.S.; et al. Inactivation of the UGPase1 gene causes genic male sterility and endosperm chalkiness in rice (Oryza sativa L.). Plant J. 2008, 54, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Ba, Q.; Zhang, L.; Li, G.; Zhang, G.; Liu, H.; Fu, Z. Altered sucrose metabolism and gene regulation in wheat (Triticum aestivum) k-type cytoplasmic male sterility. Crop Pasture Sci. 2019, 70, 204–210. [Google Scholar] [CrossRef]
- Giegé, J.L.P.; Heazlewood, U.; Roessner-Tunali, A.H.; Millar, A.R.; Fernie, A.R.; Leaver, C.J.; Sweetlove, L.J. Enzymes of glycolysis are functionally associated with the mitochondrion in Arabidopsis cells. Plant Cell. 2003, 15, 2140–2151. [Google Scholar] [CrossRef]
- Lee, S.K.; Kim, H.; Cho, J.I.; Nguyen, C.D.; Moon, S.; Park, J.E.; Park, H.R.; Huh, J.H.; Jung, K.H.; Guiderdoni, E.; et al. Deficiency of rice hexokinase HXK5 impairs synthesis and utilization of starch in pollen grains and causes male sterility. J. Exp. Bot. 2020, 71, 116–125. [Google Scholar] [CrossRef]
- Israelsen, W.J.; Vander Heiden, M.G. Pyruvate kinase: Function, regulation and role in cancer. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2015; Volume 43, pp. 43–51. [Google Scholar]
- Pei, X.; Jing, Z.; Tang, Z.; Zhu, Y. Comparative transcriptome analysis provides insight into differentially expressed genes related to cytoplasmic male sterility in broccoli (Brassica oleracea var. italica). Sci. Hortic. 2017, 217, 234–242. [Google Scholar] [CrossRef]
- Sun, Q.P.; Hu, C.F.; Hu, J.; Li, S.Q.; Zhu, Y.G. Quantitative proteomic analysis of CMS-related changes in Honglian CMS rice anther. J. Protein Chem. 2009, 28, 341–348. [Google Scholar] [CrossRef]
- Yui, R.; Iketani, S.; Mikami, T.; Kubo, T. Antisense inhibition of mitochondrial pyruvate dehydrogenase E1alpha subunit in anther tapetum causes male sterility. Plant J. 2010, 34, 57–66. [Google Scholar] [CrossRef]
- Ruprich-Robert, G.; Zickler, D.; Berteaux-Lecellier, V.; Vélot, C.; Picard, M. Lack of mitochondrial citrate synthase discloses a new meiotic checkpoint in a strict aerobe. EMBO J. 2002, 21, 6440–6451. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fang, X.; Fu, H.F.; Gong, Z.H.; Chai, W.G. Involvement of a universal amino acid synthesis impediment in cytoplasmic male sterility in pepper. Sci. Rep. 2016, 6, 23357. [Google Scholar] [CrossRef] [PubMed]
- Leon, G.; Holuigue, L.; Jordana, X. Mitochondrial complex II is essential for gametophyte development in Arabidopsis. Plant Physiol. 2007, 143, 1534–1546. [Google Scholar] [CrossRef]
- Petrussa, E.; Bertolini, A.; Casolo, V.; KrajnÁková, J.; Macrì, F.; Vianello, A. Mitochondrial bioenergetics linked to the manifestation of programmed cell death during somatic embryogenesis of Abies alba. Planta 2009, 231, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.H.; Liu, W.; Zhang, G.S.; Ye, J.X. Mitochondrial proteomic analysis of cytoplasmic male sterility line and its maintainer in wheat (Triticum aestivum L.). Agric. Sci. China 2010, 9, 771–782. [Google Scholar] [CrossRef]
- Li, J.J.; Pandeya, D.; Jo, Y.D.; Liu, W.Y.; Kang, B.C. Reduced activity of ATP synthase in mitochondria causes cytoplasmic male sterility in chili pepper. Planta 2013, 237, 1097–1109. [Google Scholar] [CrossRef]
- Liu, Y.; Qu, J.; Zhang, L.; Xu, X.; Wei, G.; Zhao, Z.; Ren, M.; Cao, M. Identification and characterization of the TCA cycle genes in maize. BMC Plant Biol. 2019, 19, 592. [Google Scholar] [CrossRef]
- Ducos, E.; Touzet, P.; Boutry, M. The male sterile G cytoplasm of wild beet displays modified mitochondrial respiratory complexes. Plant J. 2001, 26, 171–180. [Google Scholar] [CrossRef]
- Teixeira, R.T.; Knorpp, C.; Glimelius, K. Modified sucrose, starch and ATP levels in two alloplasmic male-sterile lines of B. napus. J. Exp. Bot. 2005, 56, 1245–1253. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TE Stage | Gene ID | KO ID | KO Description | Annotation | Regulation |
|---|---|---|---|---|---|
| DEG | BjuA029507 | K01176 | alpha-amylase | - | DOWN |
| DEG | BjuB037515 | K00695 | sucrose synthase | AT5G49190 | DOWN |
| DEG | BjuB030962 | K00695 | sucrose synthase | - | DOWN |
| DEG | BjuO008945 | K00695 | sucrose synthase | - | DOWN |
| DEG | BjuA046136 | K00963 | UTP--glucose-1-phosphate uridylyltransferase | - | DOWN |
| DEG | BjuB005071 | K07024 | sucrose-6-phosphatase | -- | DOWN |
| DEG | BjuB047043 | K00844 | hexokinase | AT1G50460 | DOWN |
| DEG | BjuA021412 | K00844 | hexokinase | - | DOWN |
| DEG | novel.400 | K00850 | 6-phosphofructokinase 1 | AT4G26270 | DOWN |
| DEG | BjuB040267 | K00873 | pyruvate kinase | AT3G49160 | DOWN |
| DAP | BjuA041438 | K00975 | Glucose-1-phosphate adenylyltransferase large subunit 1 | AT5G19220 | UP |
| DAP | BjuA023577 | K00703 | Starch synthase, chloroplastic/amyloplastic | AT5G24300 | DOWN |
| DAP | BjuA037309 | K00844 | Phosphotransferase | AT1G47840 | DOWN |
| DAP | BjuB028137 | K01810 | Glucose-6-phosphate isomerase | AT5G42740 | UP |
| DAP | BjuA006306 | K00134 | Glyceraldehyde-3-phosphate dehydrogenase | AT1G13440 | DOWN |
| DAP | BjuA032999 | K00927 | Phosphoglycerate kinase | AT1G79550 | DOWN |
| DAP | BjuB022100 | K00927 | Phosphoglycerate kinase | AT1G79550 | DOWN |
| DAP | BjuA006685 | K00873 | Pyruvate kinase | AT5G63680 | DOWN |
| BI Stage | Gene ID | KO ID | KO Description | Annotation | Regulation |
| DEG | BjuB038490 | K00975 | glucose-1-phosphate adenylyltransferase | AT4G39210 | DOWN |
| DEG | BjuB030220 | K00695 | sucrose synthase | AT1G73370 | DOWN |
| DEG | BjuO006586 | K00695 | sucrose synthase | AT4G02280 | DOWN |
| DEG | BjuB015313 | K00695 | sucrose synthase | AT5G20830 | DOWN |
| DEG | BjuB037515 | K00695 | sucrose synthase | AT5G49190 | DOWN |
| DEG | BjuB030962 | K00695 | sucrose synthase | - | DOWN |
| DEG | BjuO008945 | K00695 | sucrose synthase | - | DOWN |
| DEG | BjuA041856 | K00963 | UTP--glucose-1-phosphate uridylyltransferase | AT5G17310 | DOWN |
| DEG | BjuA046136 | K00963 | UTP--glucose-1-phosphate uridylyltransferase | - | DOWN |
| DEG | BjuO002531 | K01193 | beta-fructofuranosidase | AT2G36190 | DOWN |
| DEG | BjuB047043 | K00844 | hexokinase | AT1G50460 | DOWN |
| DEG | BjuA021412 | K00844 | hexokinase | - | DOWN |
| DEG | novel.6397 | K00850 | 6-phosphofructokinase 1 | AT5G56630 | UP |
| DEG | BjuB016068 | K00850 | 6-phosphofructokinase 1 | - | DOWN |
| DAP | BjuB038082 | K00703 | Starch synthase, chloroplastic/amyloplastic | AT5G24300 | DOWN |
| DAP | BjuB030962 | K00695 | Sucrose synthase | AT1G73370 | DOWN |
| DAP | BjuO008945 | K00695 | Sucrose synthase | AT5G20830 | DOWN |
| DAP | BjuO007590 | K07024 | SPP1 | AT1G51420 | DOWN |
| DAP | BjuO002531 | K01193 | CwINV4 | AT2G36190 | DOWN |
| DAP | BjuB026743 | K01193 | Beta-fructofuranosidase | AT2G36190 | DOWN |
| DAP | BjuB042433 | K00134 | Gp_dh_N domain-containing protein | AT3G04120 | UP |
| DAP | BjuB022100 | K00927 | Phosphoglycerate kinase | AT1G79550 | DOWN |
| DAP | BjuB048068 | K19893 | X8 domain-containing protein | AT5G58090 | UP |
| TE Stage | Gene ID | KO ID | KO Description | Annotation | Regulation |
|---|---|---|---|---|---|
| DEG | BjuA041635 | K03940 | NADH dehydrogenase (ubiquinone) | AT5G11770 | UP |
| DEG | BjuA017841 | K01647 | citrate synthase | - | DOWN |
| DEG | novel.10587 | K01647 | citrate synthase | AT2G42790 | DOWN |
| DEG | BjuA012893 | K01681 | aconitate hydratase | - | DOWN |
| DEG | BjuB043684 | K01681 | aconitate hydratase | AT4G26970 | DOWN |
| DEG | BjuA015174 | K00417 | ubiquinol-cytochrome c reductase subunit 7 | AT5G25450 | DOWN |
| DEG | novel.1027 | K01535 | H+—transporting ATPase | - | DOWN |
| DEG | BjuA047355 | K02133 | F-type H+—transporting ATPase subunit beta | - | DOWN |
| DEG | BjuB036256 | K02133 | F-type H+—transporting ATPase subunit beta | AT5G08690 | DOWN |
| DEG | BjuA033276 | K02150 | V-type H+—transporting ATPase subunit E | - | DOWN |
| DEG | BjuB029476 | K02150 | V-type H+—transporting ATPase subunit E | AT3G08560 | DOWN |
| DEG | BjuO006984 | K02154 | V-type H+—transporting ATPase subunit a | - | DOWN |
| DEG | novel.8077 | K02154 | V-type H+—transporting ATPase subunit a | - | DOWN |
| DAP | BjuB029356 | K00627 | Acetyltransferase component of pyruvate | AT1G54220 | UP |
| DAP | BjuB040953 | K01899 | dehydrogenase complex | AT5G08300 | DOWN |
| DAP | BjuO005963 | K01214 | Succinate—CoA ligase | AT2G39930 | DOWN |
| DAP | BjuA013768 | K02267 | ISA1 | AT5G57815 | UP |
| DAP | BjuB028190 | K03953 | Cytochrome c oxidase subunit | AT2G20360 | UP |
| DAP | BjuB004697 | K02154 | V-type proton ATPase subunit a | AT4G39080 | DOWN |
| DAP | BjuA015211 | K02138 | ATP synthase subunit d, mitochondrial | AT3G52300 | UP |
| DAP | BjuO008600 | K02154 | V-type proton ATPase subunit a | AT4G39080 | UP |
| BI Stage | Gene ID | KO ID | KO Description | Annotation | Regulation |
| DEG | BjuA038905 | K00627 | pyruvate dehydrogenase E2 component | - | DOWN |
| DEG | BjuA017841 | K01647 | citrate synthase | - | DOWN |
| DEG | novel.10587 | K01647 | citrate synthase | AT2G42790 | DOWN |
| DEG | BjuA015968 | K00026 | malate dehydrogenase | AT2G22780 | DOWN |
| DEG | BjuA023314 | K00234 | succinate dehydrogenase | AT2G18450 | DOWN |
| DEG | BjuA026416 | K00235 | succinate dehydrogenase | AT5G40650 | DOWN |
| DEG | BjuB043684 | K01681 | aconitate hydratase | AT4G26970 | DOWN |
| DEG | BjuA012893 | K01681 | aconitate hydratase | - | DOWN |
| DEG | novel.1027 | K01535 | H+—transporting ATPase | - | DOWN |
| DEG | BjuA047355 | K02133 | F-type H+—transporting ATPase subunit beta | - | DOWN |
| DEG | BjuB036256 | K02133 | F-type H+—transporting ATPase subunit beta | AT5G08690 | DOWN |
| DEG | BjuA033276 | K02150 | V-type H+—transporting ATPase subunit E | - | DOWN |
| DEG | BjuB029476 | K02150 | V-type H+—transporting ATPase subunit E | AT3G08560 | DOWN |
| DEG | BjuO006984 | K02154 | V-type H+—transporting ATPase subunit a | - | DOWN |
| DEG | novel.8077 | K02154 | V-type H+—transporting ATPase subunit a | - | DOWN |
| DEG | novel.10278 | K02146 | V-type H+—transporting ATPase subunit d | - | DOWN |
| DEG | novel.4259 | K02147 | V-type H+—transporting ATPase subunit B | - | DOWN |
| DEG | BjuA013499 | K02152 | V-type H+—transporting ATPase subunit G | - | DOWN |
| DEG | BjuB014179 | K02152 | V-type H+—transporting ATPase subunit G | - | DOWN |
| DEG | BjuB048951 | K02152 | V-type H+—transporting ATPase subunit G | - | DOWN |
| DEG | novel.397 | K02152 | V-type H+—transporting ATPase subunit G | - | DOWN |
| DAP | BjuA004114 | K02152 | V-type H+—transporting ATPase subunit G | - | DOWN |
| DAP | BjuA029659 | K00030 | isocitrate dehydrogenase (NAD+) | AT4G35650 | DOWN |
| DAP | BjuB024174 | K00026 | Malate dehydrogenase | AT2G22780 | DOWN |
| DAP | BjuA003018 | K00030 | IDH1 | AT4G35260 | UP |
| DAP | BjuA003018 | K00030 | IDH1 | AT4G35260 | UP |
| DAP | BjuB019661 | K00026 | Malate dehydrogenase, chloroplastic | AT3G47520 | UP |
| DAP | BjuB040953 | K01899 | Succinate—CoA ligase | AT5G08300 | DOWN |
| DAP | BjuB027452 | K03963 | Uncharacterized protein | AT2G02050 | UP |
| DAP | BjuA012602 | K03966 | Uncharacterized protein | AT3G18410 | UP |
| DAP | BjuA004528 | K03966 | Uncharacterized protein | AT3G18410 | UP |
| DAP | BjuB003649 | K02266 | Uncharacterized protein | AT4G37830 | DOWN |
| DAP | BjuA001747 | K02152 | V-type proton ATPase subunit G | AT4G23710 | UP |
| DAP | BjuB004697 | K02154 | V-type proton ATPase subunit a | AT4G39080 | DOWN |
| DAP | BjuO008600 | K02144 | V-type proton ATPase subunit a | AT4G39080 | DOWN |
| DAP | BjuA034730 | K02150 | VHA-E2 | AT3G08560 | DOWN |
| DAP | BjuB029476 | K02150 | VHA-E2 | AT3G08560 | DOWN |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Y.; Wang, J.; Xia, R.; Tong, M.; Huang, Y.; Xu, L.; Zhu, Z.; Meng, Q.; Yu, Y. Integrative Analysis of Transcriptomic and Proteomic Changes Related to Cytoplasmic Male Sterility in Spring Stem Mustard (Brassica juncea var. tumida Tsen et Lee). Int. J. Mol. Sci. 2022, 23, 6248. https://doi.org/10.3390/ijms23116248
Shen Y, Wang J, Xia R, Tong M, Huang Y, Xu L, Zhu Z, Meng Q, Yu Y. Integrative Analysis of Transcriptomic and Proteomic Changes Related to Cytoplasmic Male Sterility in Spring Stem Mustard (Brassica juncea var. tumida Tsen et Lee). International Journal of Molecular Sciences. 2022; 23(11):6248. https://doi.org/10.3390/ijms23116248
Chicago/Turabian StyleShen, Ying, Jie Wang, Rui Xia, Minyang Tong, Yunping Huang, Liai Xu, Zhujun Zhu, Qiufeng Meng, and Youjian Yu. 2022. "Integrative Analysis of Transcriptomic and Proteomic Changes Related to Cytoplasmic Male Sterility in Spring Stem Mustard (Brassica juncea var. tumida Tsen et Lee)" International Journal of Molecular Sciences 23, no. 11: 6248. https://doi.org/10.3390/ijms23116248
APA StyleShen, Y., Wang, J., Xia, R., Tong, M., Huang, Y., Xu, L., Zhu, Z., Meng, Q., & Yu, Y. (2022). Integrative Analysis of Transcriptomic and Proteomic Changes Related to Cytoplasmic Male Sterility in Spring Stem Mustard (Brassica juncea var. tumida Tsen et Lee). International Journal of Molecular Sciences, 23(11), 6248. https://doi.org/10.3390/ijms23116248