Analysis of Endocrine Disrupting Pesticides by Capillary GC with Mass Spectrometric Detection

Abstract
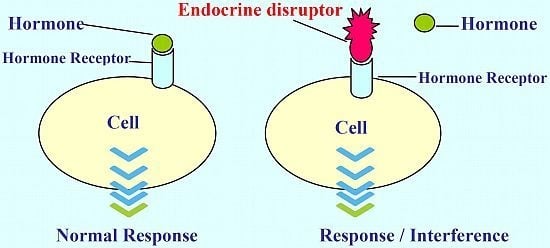
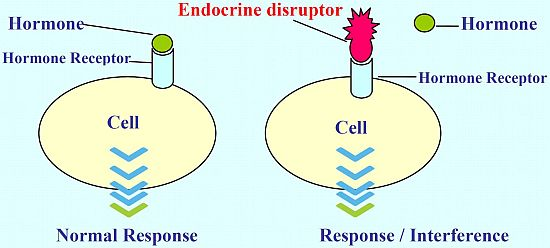
:
1. Introduction
2. Definition and Characteristics of EDPs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical group | Pesticide | CAS | Year | Exposure |
|---|---|---|---|---|
| Benzoic acid derivatives | Methyl p-Hydroxybenzoate | 99-76-3 | DHI 2006 | Medium |
| Ethyl 4-hydroxybenzoate | 120-47-8 | DHI 2006 | Medium | |
| n-Propyl p-hydroxybenzoate | 94-13-3 | DHI 2006 | Medium | |
| Carbamates | Carbaryl | 63-25-2 | BKH 2002 | High |
| DDT derivatives and metabolites | DDT (technical), (clofenotane) | 50-29-3 | EM 1999 | High |
| o,p’-DDT | 789-02-6 | BKH 2002 | High | |
| 3-OH- o,p’-DDT | 43216-70-2 | BKH 2002 | High | |
| 4-MeO- o,p’-DDT | 65148-72-3 | BKH 2002 | High | |
| 5-OH- o,p’-DDT | 65148-73-4 | BKH 2002 | High | |
| 5-MeO- o,p’-DDT | 65148-74-5 | BKH 2002 | High | |
| p,p’-DDT (clofenotane) | 50-29-3 | EM 1999 | High | |
| o,p’-DDD | 53-19-0 | BKH 2002 | High | |
| 5-MeO- o,p’-DDD | 65148-75-6 | BKH 2002 | High | |
| p,p’-DDD | 72-54-8 | BKH 2002 | High | |
| m,p’-DDD | 4329-12-8 | BKH 2002 | High | |
| o,p’-DDE | 3424-82-6 | BKH 2002 | High | |
| 3-MeO- o,p’-DDE | 65148-80-3 | BKH 2002 | High | |
| 4-MeO- o,p’-DDE | 65148-81-4 | BKH 2002 | High | |
| 5-MeO- o,p’-DDE | 65148-82-5 | BKH 2002 | High | |
| p,p’-DDE | 72-55-9 | BKH 2002 | High | |
| o,p’-DDA-glycinate | 65148-83-6 | BKH 2002 | High | |
| o,p’-DDMU | 14835-94-0 | BKH 2002 | High | |
| 1,1,1,2-Tetrachloro-2,2-bis(4-chlorophenyl) ethane (tetrachloro DDT) | 3563-45-9 | EM 1999 | High | |
| 1,1,1-Trichloro-2,2-bis(4-chlorophenyl) ethane | 2971-22-4 | BKH 2002 | High | |
| Dicarboximides | Procymidon | 32809-16-8 | BKH 2002 | High |
| Vinclozolin | 50471-44-8 | EM 1999 | High | |
| Dinitroanilines | Trifluralin | 1582-09-8 | DHI 2006 | High |
| Diphenyl ether | Nitrofen | 1836-75-5 | EM 1999 | Medium |
| Dithiocarbamates | Mancozeb | 8018-01-7 | BKH 2002 | High |
| Maneb | 12427-38-2 | EM 1999 | High | |
| Metam Natrium | 137-42-8 | EM 1999 | High | |
| Dithiocarbamates | Metiram (Metiram-complex) | 9006-42-2 | BKH 2002 | High |
| Thiram | 137-26-8 | EM 1999 | High | |
| Zineb | 12122-67-7 | EM 1999 | High | |
| Formamidine | Chlordimeform | 6164-98-3 | DHI 2006 | Low |
| Chlorinated Phenol | Pentachlorophenol (PCP) | 87-86-5 | EM 2002 | High |
| Chloroacetanilide | Acetochlor | 34256-82-1 | EM 1999 | High |
| Alachlor | 15972-60-8 | EM 1999 | High | |
| Chlorophenoxy acid | 2,4-dichlorophenoxybutyric acid (2,4-DB) | 94-82-6 | BKH 2002 | - |
| Halogenated organic | Dibromoethane (EDB) | 106-93-4 | BKH 2002 | Medium |
| Dibromochloropropane (DBCP) | 96-12-8 | DHI 2006 | - | |
| HCH and isomers | Hexachlorocyclohexane | 608-73-1 | BKH 2002 | - |
| Beta-HCH | 319-85-7 | BKH 2002 | High | |
| Gamma-HCH (Lindane) | 58-89-9 | EM 1999 | High | |
| Hydroxybenzonitrile | Ioxynil | 1689-83-4 | BKH 2002 | Medium |
| Methoxychlor and derivatives | Methoxychlor | 72-43-5 | BKH 2002 | High |
| p,p’-Methoxychlor | 72-43-5 | BKH 2002 | - | |
| Bis-OH-Methoxychlor | 2971-36-0 | BKH 2002 | High | |
| 1,3-Dichloro-2,2-bis(4-methoxy-3-methylphenyl)propane | 30668-06-5 | BKH 2002 | - | |
| Organochlorine | Chlordane | 12789-03-6 | EM 1999 | High |
| Chlordane ( cis- and trans-) | 57-74-9 | EM 1999 | High | |
| Cis-Nonachlor | 5103-73-1 | BKH 2002 | - | |
| Hexachlorobenzene (HCB) | 118-74-1 | EM 1999 | High | |
| Kepone (Chlordecone) | 143-50-0 | EM 1999 | High | |
| Mirex | 2385-85-5 | EM 1999 | High | |
| Toxaphene (Camphechlor) | 8001-35-2 | EM 1999 | High | |
| Trans-Nonachlor | 39765-80-5 | BKH 2002 | - | |
| Organotin | 2-propenoic acid, 2-methyl-, methyl ester (Stannane, tributylmeacrylate) | 26354-18-7 | EM 1999 | High |
| Fentin acetate (triphenyltin acetate) | 900-95-8 | EM 1999 | High | |
| Stannane, tributyl[(1-oxo-9,12-octadeca dienyl)oxy]-, (Z,Z)- | 24124-25-2 | EM 1999 | High | |
| Stannane, tributyl[[[1,2,3,4,4a,4b,5,6,10,10a-decahydro-1,4a-dimethyl-7-(1-methylethyl)-1-phenanthrenyl]carbonyl]oxy]-,[1R-(1a,4ab,4ba,10aa)]- | 26239-64-5 | EM 1999 | High | |
| Stannane, (benzoyloxy)tributyl- | 4342-36-3 | EM 1999 | High | |
| Stannane, tributylfluoro- | 1983-10-4 | EM 1999 | High | |
| Phenol, 2-[(tributylstannyl)oxy]carbony | 4342-30-7 | EM 1999 | High | |
| Tributyl[(2-methyl-1-oxo-2-propenyl)oxy] stannane | 2155-70-6 | EM 1999 | High | |
| Organophosphorous | Fenitrothion | 122-14-5 | EM 1999 | High |
| Omethoate | 1113-02-6 | DHI 2006 | Low | |
| Organothiophosphor | Quinalphos (Chinalphos) | 13593-03-8 | DHI 2006 | Medium |
| Pyrethroids | Bifenthrin | 82657-04-3 | BKH 2002 | High |
| Cyhalothrin | 91465-08-6 | BKH 2002 | High | |
| Deltamethrin | 52918-63-5 | BKH 2002 | High | |
| Resmethrin | 10453-86-8 | BKH 2002 | High | |
| Pyrimidines and Pyridines | Fenarimol | 60168-88-9 | BKH 2002 | High |
| Pyridinecarboxylic acid | Picloram | 1918-02-1 | BKH 2002 | Medium |
| Triazines and Triazoles | Amitrol (Aminotriazole) | 61-82-5 | EM 1999 | Medium |
| Atrazine | 1912-24-9 | EM 1999 | - | |
| Metribuzin | 21087-64-9 | BKH 2002 | High | |
| Ketoconazol | 65277-42-1 | BKH 2002 | High | |
| Terbutryn | 886-50-0 | BKH 2002 | Medium | |
| Urea | Linuron (Lorox) | 330-55-2 | EM 1999 | High |
| Other pesticides | Ethylene Thiourea (ETU) | 96-45-7 | DHI 2006 | Low |
| Chemical group | Pesticide | CAS | Year |
|---|---|---|---|
| Alkyl Phthalate | Diisobutylphthalate | 84-69-5 | DHI 2006 |
| Anilide | Propanil | 709-98-8 | EM 1999 |
| Azole | Etridiazole | 2593-15-9 | BKH 2002 |
| Prochloraz | 67747-09-5 | EM 1999 | |
| Triadimefon | 43121-43-3 | EM 1999 | |
| Triadimenol | 123-88-6 | BKH 2002 | |
| Benzimidazole | Carbendazim | 10605-21-7 | EM 1999 |
| Carbamates | Aldicarb | 116-06-3 | BKH 2002 |
| Carbofuran | 1563-66-2 | BKH 2002 | |
| Fenoxycarb | 72490-01-8 | BKH 2002 | |
| Methomyl | 16752-77-5 | BKH 2002 | |
| DDT derivatives and metabolites | p,p’-DDA | 83-05-6 | DHI 2006 |
| Dicarboximide | Iprodione | 36734-19-7 | EM 1999 |
| Dithiocarbamate | Ziram | 137-30-4 | EM 1999 |
| Halogenated organic | Methyl bromide (bromomethane) | 74-83-9 | EM 1999 |
| HCH isomers | Delta-HCH | 319-86-8 | BKH 2002 |
| Hydroxybenzonitrils | Bromoxynil | 1689-84-5 | BKH 2002 |
| Chlorinated Phenol | 4-Chloro-3-methylphenol | 59-50-7 | EM 1999 |
| 4-Chloro-2-methylphenol | 1570-64-5 | EM 1999 | |
| Chlorophenoxy acid | 2,4,5-Trichlorophenoxyacetic acid (2,4,5-T) | 93-76-5 | BKH 2002 |
| 2,4-Dichlorophenoxyacetic acid (2,4-D) | 94-75-7 | EM 1999 | |
| Organochlorine | Aldrin | 309-00-2 | EM 1999 |
| Dicofol (Kelthane) | 115-32-2 | EM 1999 | |
| Dieldrin | 60-57-1 | EM 1999 | |
| Endrin | 72-20-8 | EM 1999 | |
| Endosulfan | 115-29-7 | EM 1999 | |
| Endosulfan-alpha | 959-98-8 | EM 1999 | |
| Endosulfan-beta | 33213-65-9 | EM 1999 | |
| Heptachlor | 76-44-8 | EM 1999 | |
| Oxychlordane | 27304-13-8 | EM 1999 | |
| Organophosphorous | Acephate | 30560-19-1 | BKH 2002 |
| Elsan (Dimephenthoate) | 2597-03-7 | DHI 2006 | |
| Diazinon | 333-41-5 | EM 1999 | |
| Dimethoate | 60-51-5 | EM 1999 | |
| Chlorfenvinphos | 470-90-6 | BKH 2002 | |
| Malathion | 121-75-5 | EM 1999 | |
| Methylparathion | 298-00-0 | EM 1999 | |
| Mevinphos (Phosdrin) | 7786-34-7 | BKH 2002 | |
| Parathion (Parathion-ethyl) | 56-38-2 | EM 1999 | |
| Phosophamidon | 13171-21-6 | BKH 2002 | |
| Trichlorfon (Dipterex) | 52-68-6 | BKH 2002 | |
| Phenol | p-Cresol | 106-44-5 | DHI 2006 |
| 4-Nitrophenol | 100-02-7 | DHI 2006 | |
| o-Phenylphenol | 90-43-7 | EM 1999 | |
| Pyrethrins | Pyrethrin | 121-29-9 | DHI 2006 |
| Pyrethroids | Allethrin (d-trans-allethrin) | 584-79-2 | BKH 2002 |
| Cypermethrin | 52315-07-8 | BKH 2002 | |
| Fenothrin (sumithrin) | 26002-80-2 | BKH 2002 | |
| Fenvalerate | 51630-58-1 | BKH 2002 | |
| Fluvalinate | 69409-94-5 | BKH 2002 | |
| Permethrin | 52645-53-1 | BKH 2002 | |
| Triazines | Cyanazine | 21725-46-2 | BKH 2002 |
| Prometryn | 7287-19-6 | BKH 2002 | |
| Simazine | 122-34-9 | EM 1999 | |
| Urea | Diuron | 330-54-1 | EM 1999 |
| Other pesticides | Piperonyl butoxide | 51-03-6 | BKH 2002 |
| Photomirex | 39801-14-4 | EM 1999 |
| Chemical group | Category | Pesticide | CAS No. | Year |
|---|---|---|---|---|
| Azoles | 3b | Fenbuconazole | 114369-43-6 | DHI 2006 |
| 3a | Imazalil | 3554-44-0 | BKH 2002 | |
| 3a | Benomyl | 17804-35-2 | BKH 2002 | |
| 3b | Bitertanol | 55179-31-2 | BKH 2002 | |
| 3b | Cyproconazole | 94361-07-6 | BKH 2002 | |
| Azoles | 3b | Difenoconazole | 119446-68-3 | BKH 2002 |
| 3b | Epiconazole | 121 | BKH 2002 | |
| 3b | Epoxiconazole | 135319-73-2 | BKH 2002 | |
| 3b | Flutriafol | 76674-21-0 | BKH 2002 | |
| 3b | Myclobutanil | 88671-89-0 | BKH 2002 | |
| 3b | Penconazole | 66246-88-6 | BKH 2002 | |
| 3b | Propiconazole | 60207-90-1 | BKH 2002 | |
| 3b | Fipronil | 120068-37-3 | BKH 2002 | |
| 3b | Tebuconazole | 107534-96-3 | BKH 2002 | |
| 2,6-Dinitroanilines | 3a | Oryzalin | 19044-88-3 | BKH 2002 |
| 3a | Pendimethalin | 40487-42-1 | BKH 2002 | |
| 3b | Prodiamine | 29091-21-2 | BKH 2002 | |
| Formamidine | 3a | Amitraz | 33089-61-1 | BKH 2002 |
| Organochlorine | 3a | Heptachlor-epoxide | 1024-57-3 | BKH 2002 |
| Organophosphorus Phosphonoglycine Uracil | 3a | Demeton-s-methyl | 919-86-8 | BKH 2002 |
| 3a | Dichlorvos | 62-73-7 | BKH 2002 | |
| 3a | Chlorpyrifos | 2921-88-2 | BKH 2002 | |
| 3a | Oxydemeton-methyl | 301-12-2 | BKH 2002 | |
| 3a | Ronnel (fenchlorfos) | 299-84-3 | BKH 2002 | |
| 3a | Tetrachlorvinphos (Gardona) | 22248-79-9 | BKH 2002 | |
| 3b | Demefion | 682-80-4 | DHI 2006 | |
| 3b | Formothion | 2540-82-1 | DHI 2006 | |
| Amide | 3a | Glyphosate | 1071-83-6 | BKH 2002 |
| Aryloxyphenoxy propionic acid | 3a | Bromacil | 314-40-9 | DHI 2006 |
| Bipyridylium | 3b | Pronamide | 23950-58-5 | BKH 2002 |
| Dinitrophenol and derivatives | 3b | Fluazifop-butyl | 69806-50-4 | BKH 2002 |
| 3b | Paraquat | 4685-14-7 | BKH 2002 | |
| Dithiocarbamate Organochlorine | 3b | Dinitrophenol | 25550-58-7 | DHI 2006 |
| 3b | Dinoseb | 88-85-7 | BKH 2002 | |
| Organophosphorus | 3b | Nabam | 142-59-6 | BKH 2002 |
| 3b | Octachlorostyrene | 29082-74-4 | BKH 2002 | |
| Phosphonoglycine | 3b | Glufosinate-ammonium | 70393-85-0 | DHI 2006 |
| Pyridinecarboxylic acid | 3b | Thiazopyr | 117718-60-2 | BKH 2002 |
| Pyrethroid | 3b | Esfenvalerate | 66230-04-4 | BKH 2002 |
| Substituted Benzene | 3b | Pentachloronitrobenzene (Quintozene) | 82-68-8 | BKH 2002 |
| Tetrazine | 3b | Clofentezine (chlorfentezine) | 74115-24-5 | BKH 2002 |
| Thiocarbamate | 3b | Molinate | 2212-67-1 | BKH 2002 |
| Other pesticides | 3a | Azadirachtin | 11141-17-6 | BKH 2002 |
| 3a | Abamectin | 71751-41-2 | BKH 2002 | |
| 3a | Diphenyl | 92-52-4 | BKH 2002 | |
| 3a | Glufosinate | 51276-47-2 | BKH 2002 | |
| 3a | Chlordene | 3734-48-3 | BKH 2002 | |
| 3b | Dimethylformamide (DMFA) | 68-12-2 | BKH 2002 | |
| 3b | Ethofenprox | 80844-07-1 | BKH 2002 |
3. Analysis of EDPs
3.1. Conventional Capillary GC with MS Detection
3.1.1. Sample Matrix and Sample Preparation
| Analytes | Matrix | Sample preparation | Injection technique | LOD | Separation & detection | Ref. | |
|---|---|---|---|---|---|---|---|
| 23 pesticides | apples | QuEChERS | PTV, SVV | EI: 0.09–3.12 µg/kg | GC-QMS (SIM) | [45] | |
| NCI: 1.9–935 ng/kg | NCI, EI | ||||||
| 25 pesticides | apples | QuEChERS | PTV, SVV | EI: 0.02–6.32 µg/kg | fast GC-QMS (SIM) | [46] | |
| NCI: 0.15–619.3 ng/kg | NCI, EI | ||||||
| 20 OCPs | 9 vegetable matrices | SBSE (PDMS 47 µL) | LVI-PTV, SVV | <10 µg/kg | GC-QMS (SIM) | [47] | |
| EI | |||||||
| 29 pesticides | fruit and vegetables | QuEChERS | PTV, SVV | ≤5 µg/kg | fast GC-QMS (SIM) | [48][49] | |
| EI | |||||||
| 35 pesticides | fruit and vegetables | QuEChERS | PTV, SVV | EI: ≤5 µg/kg, | fast GC-QMS (SIM) | [50] | |
| NCI: ≤1 µg/kg | EI, NCI | ||||||
| 9 pesticides, phtalates, 1 PAH | water | on-line SPE | on-column, retaining precolumn, SVV | 0.1–20 ng/L | GC-QMS (FS) | [51] | |
| EI | |||||||
| 11 pesticides, phthalates | water | on-line SPE | LVI-PTV, SVV | 1–36 ng/L | GC-QMS (FS) | [52] | |
| EI | |||||||
| HCB, atrazine, lindane, vinclozolin, malathion, aldrin, α-endosulfan, 4,4´-DDE, dieldrin, endrin, 4,4´-DDT | river water | SBSE (PDMS 63 µL) | split/splitless, LVI-PTV, SVV | 0.01–0.24 µg/L | GC-QMS (FS), EI | [53] | |
| 15 herbicides, 7 OPPs, 17 OCPs | water | SBSE (PDMS 47 µL) | PTV, SVV | 0.025–0.400 µg/L | GC-QMS (SIM)EI | [54] | |
| 32 EDCs and pesticides | water | SPE (LiChrolut EN/RP-18, Strata X) | splitless | 5.3–95.9 ng/L | GC-MS/MS (MRM), EI, quad., | [55] | |
| 58 potential EDCs and PPCPs (18 pesticides) | drinking water, surface, ground, waste water (raw and treated) | SPE (HLB), LLE | 1. splitless2. valve | 1–10 ng/L | 1. GC-MS/MS, EI, IT; 2. LC-MS/MS, ESI+, ESI–, APCI, triplequad. (MRM) | [56]] | |
| 6 EDC herbicides and 3 degrade. products | natural surface water | SPE (Bond Elut-ENV) | splitless | 2.3–115 ng/L | GC-QMS (SIM)EI | [57] | |
| OPPs, OCPs, herbicides, PAHs, PCBs, phenols, organotins | estuarine and coastal water, sediments | SPE (Supelclean ENVI-18) | LVI-PTV, SVV | 10–250 µg/L | GC-QMS (SIM, FS) | [15] | |
| EI | |||||||
| 1. 5 OCPs | wastewaters, surface and ground waters | SPE | 1. PTV, SVV;2. valve | 0.2 and 88.9 ng/L | 1. GC-QMS | [58] | |
| 2. 33 multi-class pollutants | 2. LC-MS/MS | ||||||
| EDCs (1 pesticide), carbamazepine, pharmaceuticals | wastewater irrigated soil | ASE, isolation SPE (Oasis HLB) | splitless | 0.25–2.5 ng/g | GC-QMS (SIM, FS) | [59] | |
| EI | |||||||
| PBDEs, PCBs, insecticides, phthalates | indoor dust from vacuum cleaner | Soxhlet extraction, alumina cleaning | n. r. | 3–10 ng/g | GC-QMS (SIM) | [60] | |
| EI | |||||||
| 18 OCPs | placenta samples from woman | SLE (Alumine), purification - preparative LC | n.r. | n.r. | GC-ECD | [61] | |
| GC-MS, IT | |||||||
| dicofol, DDTs | human milk | LLE, GPC | splitless | 0.1–0.2 ng/g | GC-MS (SIM) | [62] | |
| EI | |||||||
| OCPs, PCBs | Serum samples | SPE (C18), silica gel/florisil SPE cleaning | splitless | 0.4–12.0 ng/g | GC-HRMS (dual focusing sector field MS) | [63] | |
| (SIM), EI | |||||||
| PCBs, 6 DDT metabolites, HCHs, HCB, heptachlor, chlordanes, nanochlors, mirex | blood from delivering woman | SPE (Oasis HLB), florisil SPE cleaning | splitless | n.r. | GC-MS (SIM) | [64] | |
| EI | |||||||
| 1. multiple class of pesticides | meconium | 1. SPE | splitless | 0.01–4.15 μg/g | GC-MS (SIM) | [65] | |
| 2. metabolites | 2. derivatization , LLE | EI | |||||
3.1.2. GC Operating Conditions
3.1.3. Analytical Methods Overview—Analytes vs. Samples
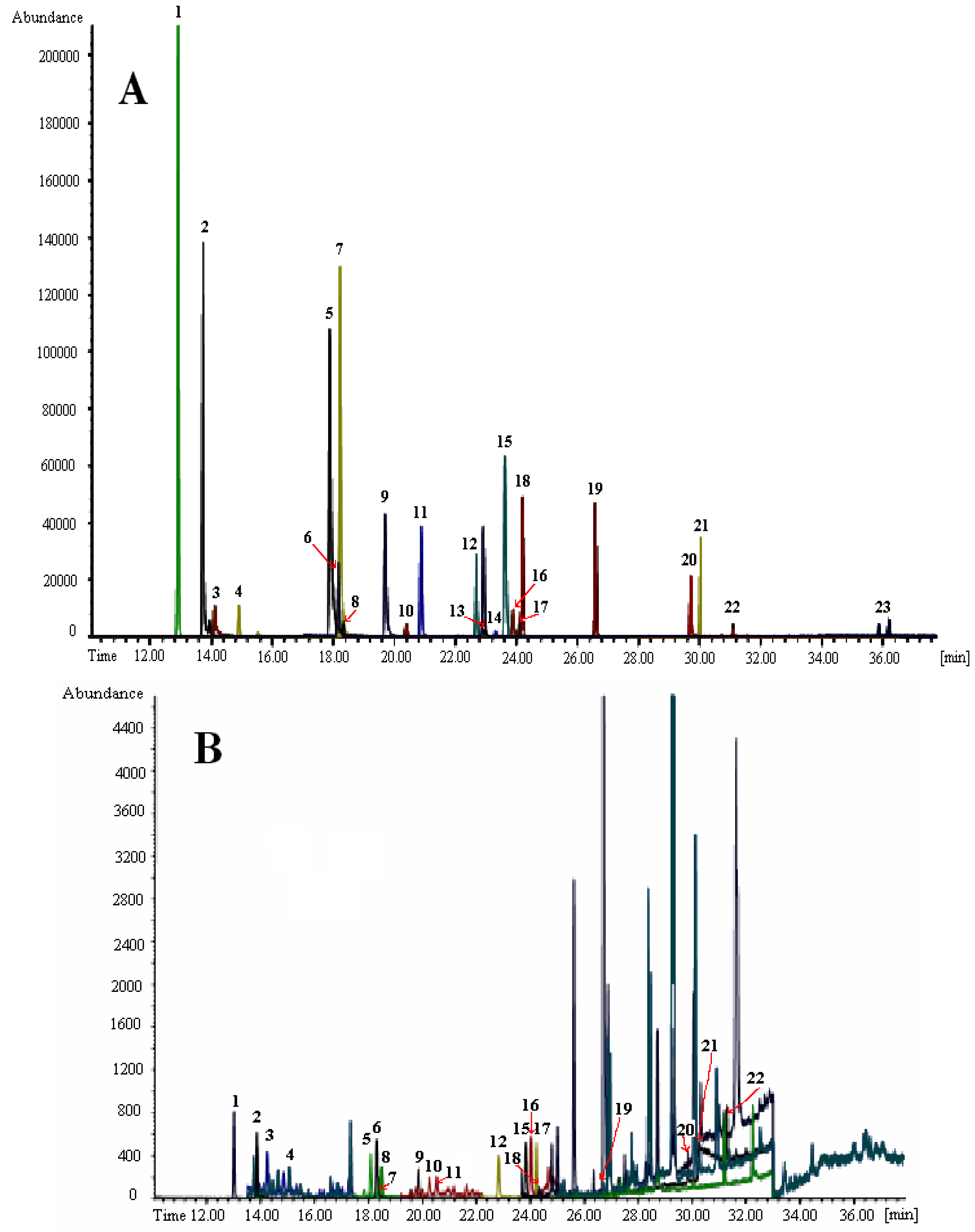
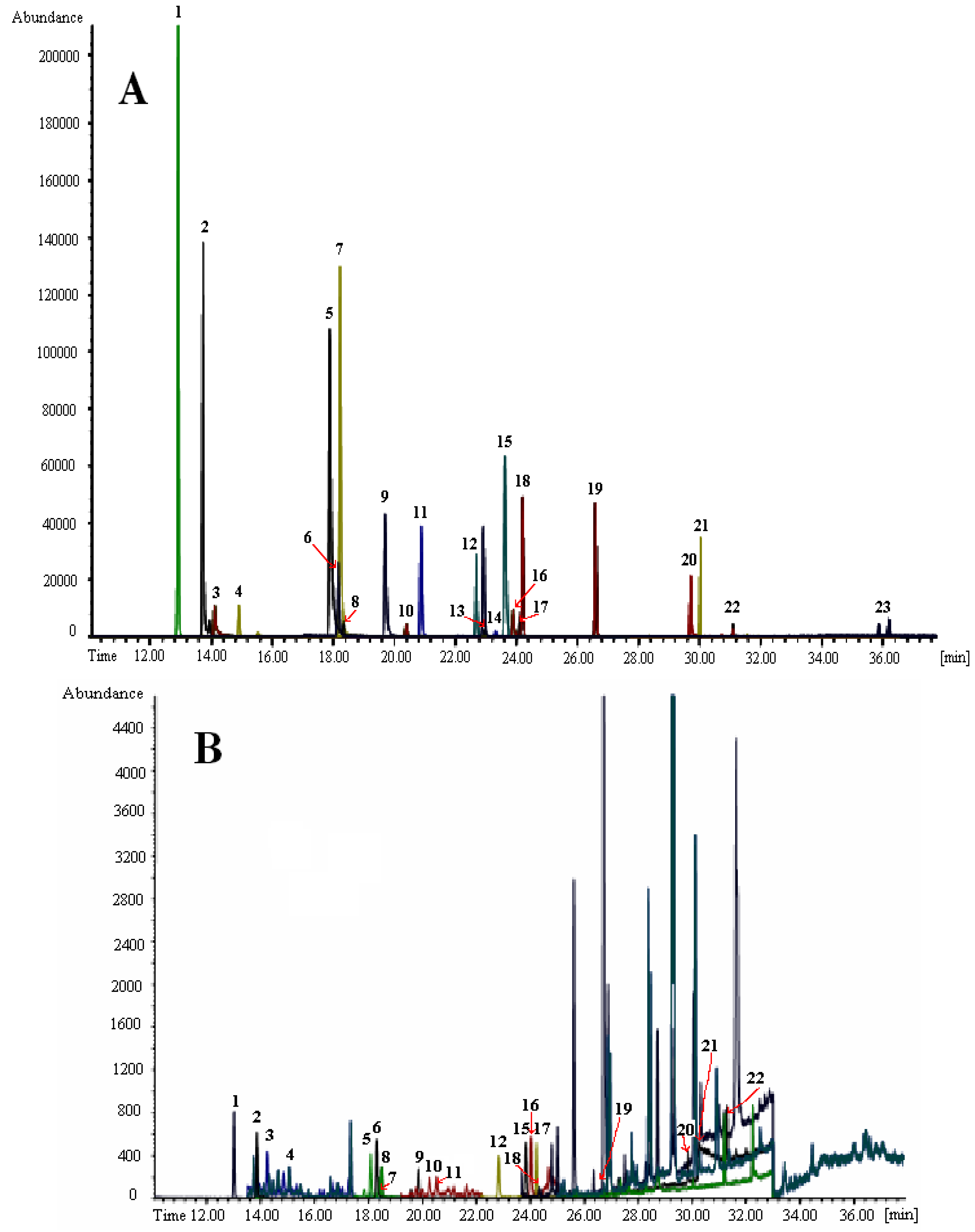
3.2. Fast Capillary GC with MS Detection
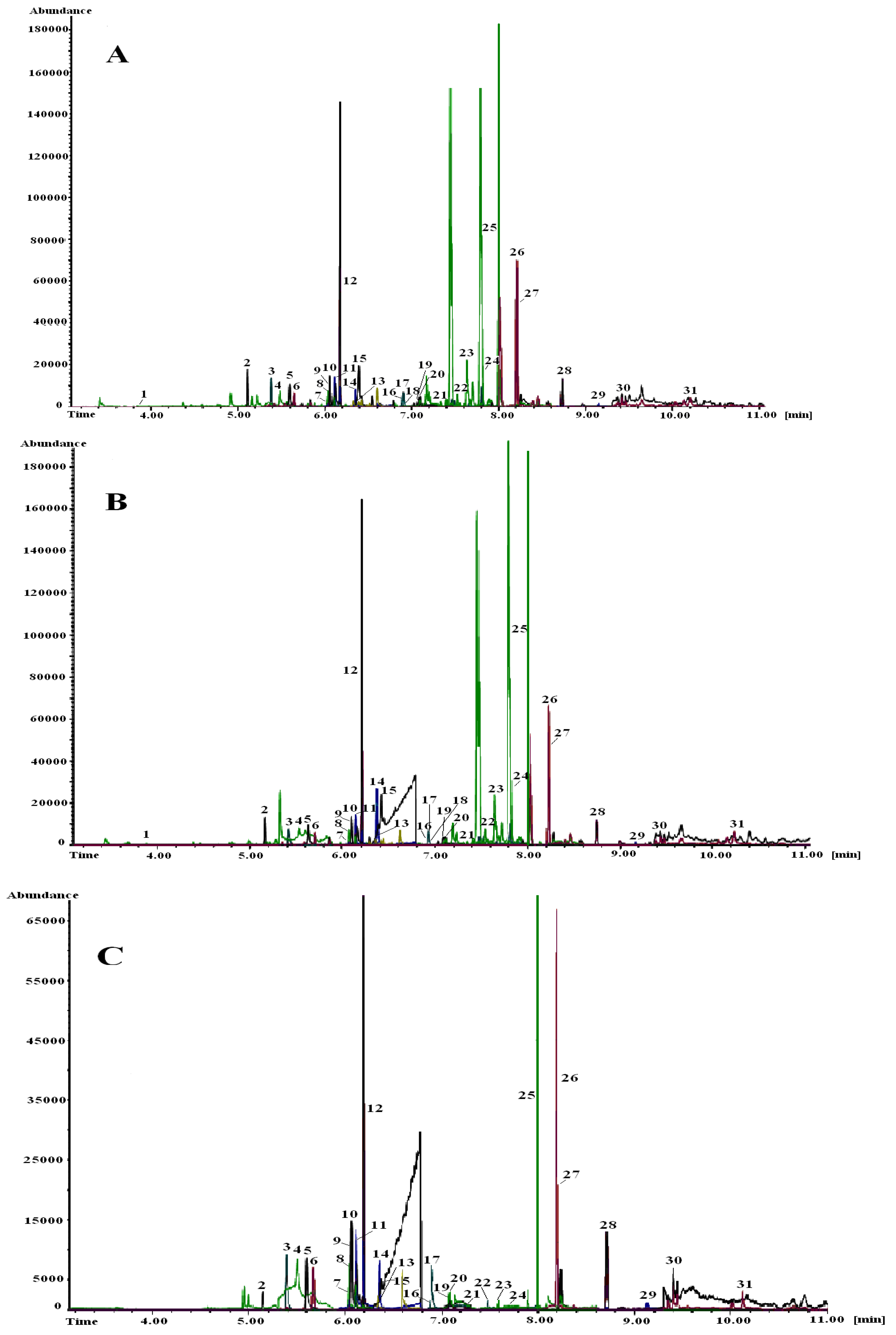
| Method/Results | GC-NCI-MS | GC-EI-MS |
|---|---|---|
| LCLs | 0.01, 0.05 µg/kg | 1 µg/kg |
| R2 | 0.9936–1.0000 | 0.9882–0.9999 |
| LODs | 0.15–88.82 ng/kg | 0.01–6.32 µg/kg |
| LOQs | 0.52–291.35 ng/kg | 0.04–21.07 µg/kg |
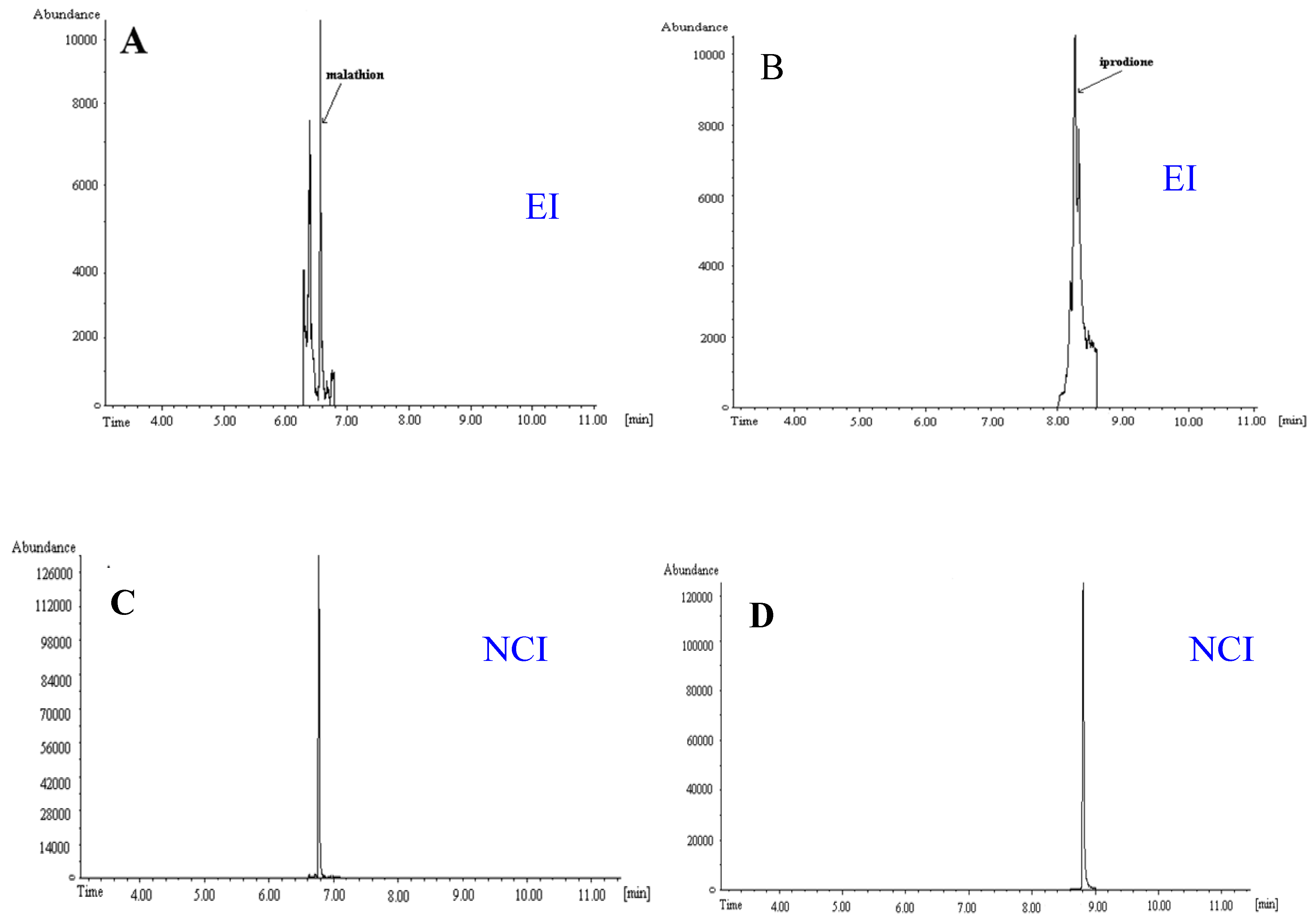
4. Real-Life Samples Analysis
| Matrix | Pesticide | NCI | EI | ||
|---|---|---|---|---|---|
| ci (µg/kg) | RSD (%) | ci (µg/kg) | RSD (%) | ||
| orange | malathion | 50.1 | 0.52 | 52.5 | 3.5 |
| lettuce | iprodione | 40.1 | 1.2 | 42.0 | 3.0 |
| pear A | iprodione | 40.1 | 1.2 | 41.3 | 3.4 |
| pear B | bifenthrin | 69.0 | 5.2 | 64.8 | 4.1 |
| myclobutanil | 0.07 | 6.8 | n.d. | - | |
| kohlrabi | metribuzin | 0.06 | 3.0 | n.d. | - |
| vinclozolin | 0.15 | 2.1 | n.d | - | |
| myclobutanil | 0.25 | 3.6 | n.d. | - | |
| plum | iprodione | 234.3 | 0.31 | 241.1 | 2.8 |
| strawberry | iprodione | 40.9 | 1.2 | 41.3 | 3.4 |
| pepper | myclobutanil | 24.3 | 6.3 | 30.4 | 4.2 |
| cypermethrin | 47.2 | 2.2 | 54.9 | 6.0 | |
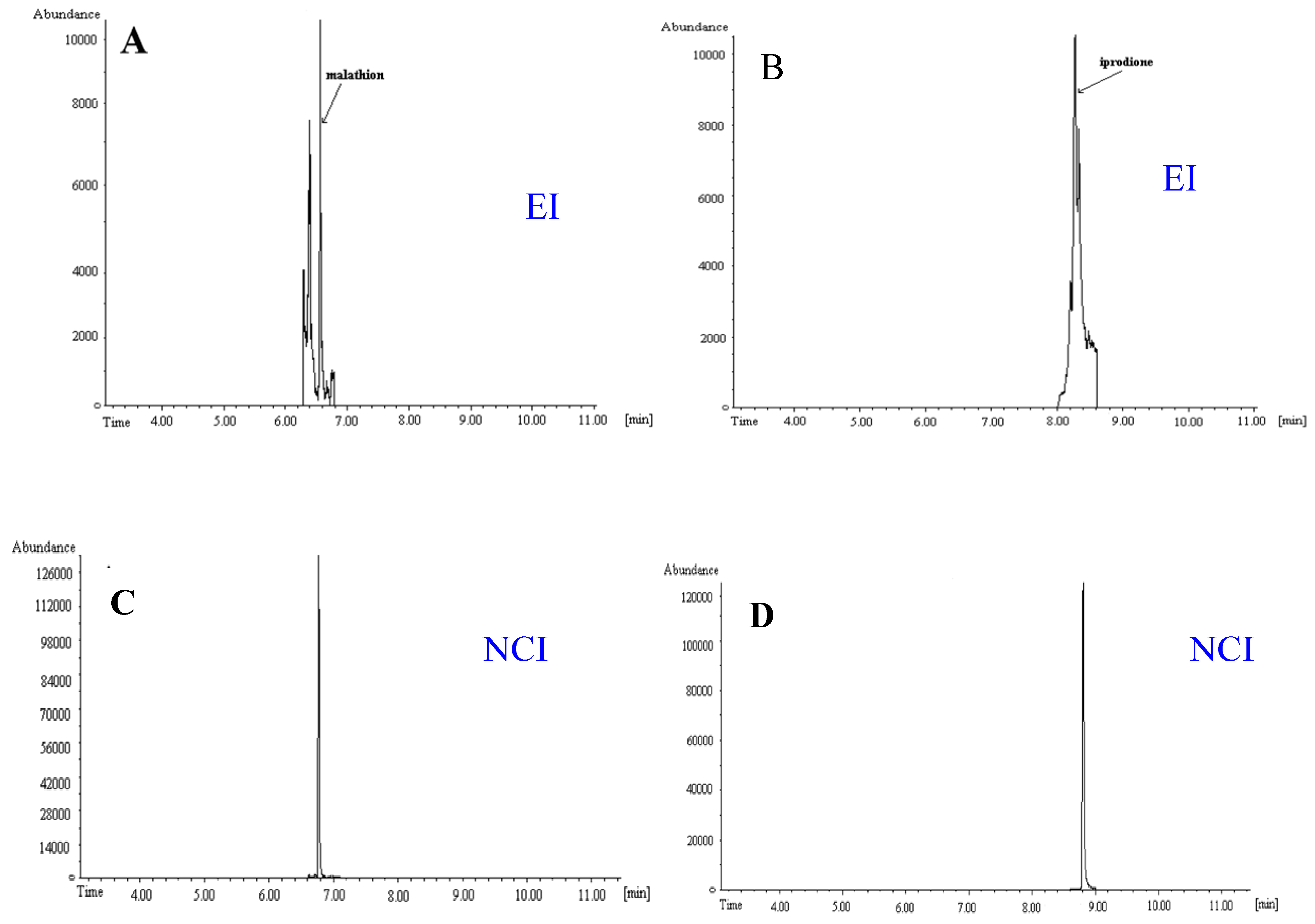
5. Conclusions
Acknowledgments
Conflict of Interest
References
- National Pesticides Strategy: Part 1: The Background and the Need for Change. Available online: http://www.pesticides.gov.uk/environment.asp?id=1523 (accessed on 10 April 2012).
- Mnif, W.; Hassine, A.I.H.; Bouaziz, A.; Bartegi, A.; Thomas, O.; Roig, B. Effect of endocrine disruptor pesticides: A review. Int. J. Environ. Res. Public Health 2011, 8, 2265–2303. [Google Scholar]
- Bhadekar, R.; Swanandi, P.; Tale, V.; Nirichan, B. Developments in analytical methods for detection of pesticides in environmental samples. Am. J. Anal. Chem. 2011, 2, 1–15. [Google Scholar]
- Dömötörová, M.; Matisová, E. Fast gas chromatography for pesticide residues analysis. J. Chromatogr. A 1207, 281–294. [Google Scholar]
- Proposed PAHO/WHO Plan of Action for Technical cooperation in Food Safety, 2006-2007. Available online: ftp://ftp.fao.org/docrep/fao/meeting/010/af272e.pdf (accessed on 10 April 2012).
- Maximum Residue Levels. Available online: http://www.pesticides.gov.uk/guidance/industries/pesticides/advisory-groups/PRiF/PRC+%28Pesticides+Residues+Commitee%29/About+pesticides+in+food/maximum-residue-levels (accessed on 22 August 2012).
- UK Pesticides Strategy: A Strategy for the Sustainable Use of Plant Protection Products, Department of Environment, Food and Rural Affairs, London, United Kingdom. Available online: http://www.pesticides.gov.uk/Resources/CRD/Migrated-Resources/Documents/U/Updated_National_Strategy.pdf (accessed on 22 August 2012).
- Status of Active Substances under EU Review (doc. 3010). Available online: http://ec.europa.eu/food/plant/protection/evaluation/stat_active_subs_3010_en.xls (accessed on 10 April 2012).
- Commission Directive 2003/13/EC of 10 February 2003 amending Directive 96/5/EC on Processed Cereal-Based Foods and Baby Foods for Infants and Young Children. 2003. Available online: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2003:041:0033:0036:EN:PDF (accessed on 10 April 2012).
- Commission Directive 2003/14/EC of 10 February 2003 amending Directive 91/321/EEC on Infant Formulae and Follow-On Formulae. 2003. Available online: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2003:041:0037:0040:EN:PDF (accessed on 10 April 2012).
- Council Directive 98/83/EC of 3 November 1998 on the Quality of Water Intended for Human Consumption. Available online: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:1998:330:0032:0054:EN:PDF (accessed on 10 April 2012).
- Colborn, T.; vom Saal, F.S.; Soto, A.M. Developmental effects of endocrine-disrupting chemicals in wildlife and humans. Environ. Health Perspect. 1993, 101, 378–384. [Google Scholar]
- Lintelmann, J.; Katayama, A.; Kurihara, N.; Shore, L.; Wenzel, A. Endocrine disruptors in the environment. Pure Appl. Chem. 2003, 75, 631–681. [Google Scholar]
- Jobling, S. Endocrine disruption in wild fish. Pure Appl. Chem. 2004, 75, 2219–2234. [Google Scholar] [CrossRef]
- Almeida, C.; Serôdio, P.; Florencio, M.H.; Nogueira, J.M.F. New strategies to screen for endocrine-disrupting chemicals in the Portuguese marine environment utilizing large volume injection-capillary gas chromatography-mass spectrometry combined with retention time locking libraries (LVI-GC-MS-RTL). Anal. Bioanal. Chem. 2007, 87, 2569–2583. [Google Scholar]
- Van Dyk, J.S.; Pletschke, B. Review on the use of enzymes for the detection of organochlorine, organophosphate and carbamate pesticides in the environment. Chemosphere 2011, 82, 291–307. [Google Scholar] [CrossRef]
- Comerton, A.M.; Andrews, R.C.; Bagley, D.M. Practical overview of analytical methods for endocrine-disrupting compounds, pharmaceuticals and personal care products in water and wastewater. Philos. Trans. R. Soc. A 2009, 367, 3923–3939. [Google Scholar]
- Chang, H.S.; Choo, K.H.; Lee, B.; Choi, S.J. The methods of identification, analysis, and removal of endocrine disrupting compounds (EDCs) in water. J. Hazard. Mater. 2009, 172, 1–12. [Google Scholar]
- Endocrine Disruptors Website. Available online: http://ec.europa.eu/environment/endocrine/definitions/endodis_en.htm (accessed on 8 February 2011).
- Diamanti-Kandarakis, E.; Bourguignon, J.P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-disrupting chemicals: An endocrine society scientific statement. Endocr.Rev. 2009, 30, 293–342. [Google Scholar]
- Hrouzková, S.; Matisová, E. Endocrine Disrupting Pesticides. In Pesticide-Advances in Chemical and Botanical Pesticides, 1st; Soundararajan, R.P., Ed.; InTech: Rijeka, Croatia, 2012; pp. 99–126. [Google Scholar]
- Commission of the European Communities: Commission Staff Working Document on the Implementation of the “Community Strategy for Endocrine Disrupters” SEC 2007. Available online: http://ec.europa.eu/environment/endocrine/documents/sec_2007_1635_en.pdf (accessed on 10 April 2012).
- European Commission: Community Strategy for Endocrine Disrupters. Available online: http://ec.europa.eu/environment/docum/99706sm.htm (accessed on 10 March 2011).
- European Commission. Commission Staff Working Document on the Implementation of the “Community Strategy for Endocrine Disrupters”-A Range of Substances Suspected of Interfering with the Hormone Systems of Humans and Wildlife (COM (1999) 706), (COM (2001) 262) and (SEC (2004) 1372). Available online: http://ec.europa.eu/environment/endocrine/documents/sec_2007_1635_en.pdf (accessed on 10 March 2011).
- Final List of Initial Pesticide Active Ingredients and Pesticide Inert Ingredients to be Screened under the Federal Food, Drug, and Cosmetic Act. Available online: http://www.epa.gov/scipoly/oscpendo/pubs/final_list_frn_041509.pdf (accessed on 10 April 2012).
- US Environmental Protection Agency. Endocrine disruptor screening program; Second list of chemicals for tier 1 screening. Fed.Regist. 2010, 75, 70248–70254.
- Snyder, S.A.; Benotti, M.J. Endocrine disruptors and pharmaceuticals: Implications for water sustainability. Water Sci. Technol. 2010, 61, 145–154. [Google Scholar] [CrossRef]
- Mnif, W.; Pillon, A.; Balaguer, P.; Bartegi, A. Endocrine xenoestrogenics disrupters, molecular methods and detection methods. Therapie 2007, 62, 369–386. [Google Scholar] [CrossRef]
- Birnbaum, L.S.; Fenton, S.E. Cancer and developmental exposure to endocrine disruptors. Environ. Health Perspect. 2003, 111, 389–394. [Google Scholar]
- Sharpe, R.R.M. Pathways of endocrine disruption during male sexual differentiation and masculinisation. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 91–110. [Google Scholar] [CrossRef]
- Eskenazi, B.; Marks, A.R.; Bradman, A.; Fenster, L.; Johnson, C.; Barr, D.B. In utero exposure to dichlorodiphenyltrichloroethane (DDT) and dichlorodiphenyldichloroethylene (DDE) and neurodevelopment among young Mexican American children. Pediatrics 2006, 118, 233–241. [Google Scholar]
- Ribas-Fito, N.; Cardo, E.; Sala, M.; Eulalia de Muga, M.; Mazon, C.; Verdu, A.; Kogevinas, M.; Grimalt, J.O.; Sunyer, J. Breastfeeding exposure to organochlorine compounds and neurodevelopment in infants. Pediatrics 2003, 111, 580–585. [Google Scholar] [CrossRef]
- Lagana, A.; Bacaloni, A.; de Leva, I.; Faberi, A.; Fago, G.; Marino, A. Analytical methodologies for determining the occurrence of endocrine disrupting chemicals in sewage treatment plants and natural waters. Anal.Chim. Acta 2004, 501, 79–88. [Google Scholar] [CrossRef]
- Fang, H.; Tong, W.; Shi, L.M.; Blair, R.; Perkins, R.; Branham, W.; Hass, B.S.; Xie, Q.; Dial, S.L.; Moland, C.L.; Sheehan, D.M. Structure-activity relationships for a large diverse set of natural, synthetic, and environmental estrogens. Chem. Res. Toxicol. 2001, 14, 280–294. [Google Scholar] [CrossRef]
- Colborn, T. TEDX-The Endocrine Disruption Exchange,Endocrine Disruption Fact Sheet. Available online: http://www.endocrinedisruption.com/files/EDFactSheet11-7-11.pdf (accessed on 12 April 2012).
- Alder, L.; Greulich, K.; Günther, K.; Kempe, G.; Bärbel, V.; Vieth, B. Residue analysis of 500 high priority pesticides: Better by GC-MS or LC-MS/MS? Mass Spectrom. Rev. 2006, 25, 838–865. [Google Scholar]
- Bezbaruah, A.N.; Kalita, H. Sensors and Biosensors for Endocrine Disrupting Chemicals: State-of-the-Art and Future Trends. In Treatment of Micropollutants in Water and Wastewater; Virkutyte, J., Jegatheesan, V., Varma, R.S., Eds.; IWA Publishing: London, UK, 2010; pp. 93–127. [Google Scholar]
- Dostálek, J.; Přibyl, J.; Homola, J.; Skládal, P. Multichannel SPR biosensor for detection of endocrine disrupting compounds. Anal.Bioanal. Chem. 2007, 389, 1841–1847. [Google Scholar]
- Holland, P.T. Analysis of endocrine active substances in food and the environment. Pure Appl. Chem. 2003, 75, 1843–1857. [Google Scholar] [CrossRef]
- Petrovič, M.; Eljarrat, E.; López de Alda, M.J.; Barceló, D. Recent advances in the mass spectrometric analysis related to endocrine disrupting compounds in aquatic environmental samples. J. Chromatogr. A 2002, 974, 23–51. [Google Scholar]
- LaFleur, A.D.; Schug, K.A. A review of separation methods for the determination of estrogens and plastics-derived estrogen mimics from aqueous systems. Anal.Chim. Acta 2011, 696, 6–26. [Google Scholar] [CrossRef]
- Wille, K.; de Brabander, H.F.; Vanhaecke, L.; de Wulf, E.; van Caeter, P.; Janssen, C.R. Coupled chromatographic and mass-spectrometric techniques for the analysis of emerging pollutants in the aquatic environment. Trends Anal. Chem. 2012, 35, 87–108. [Google Scholar] [CrossRef]
- Kuster, M.; López de Alda, M.; Barceló, D. Liquid chromatography-tandem mass spectrometric analysis and regulatory issues of polar pesticides in natural and treated waters. J. Chromatogr. A 1216, 520–529. [Google Scholar]
- Matisová, E.; Dömötörová, M. Fast gas chromatography and its use in trace analysis. J. Chromatogr. A 1000, 199–221. [Google Scholar]
- Húšková, R.; Matisová, E.; Švorc, Ľ.; Mocák, J.; Kirchner, M. Comparison of negative chemical ionization and electron impact ionization in gas chromatography-mass spectrometry of endocrine disrupting pesticides. J. Chromatogr. A 2009, 1216, 4927–4932. [Google Scholar] [CrossRef]
- Húšková, R.; Matisová, E.; Hrouzková, S.; Švorc, L. Analysis of pesticide residues by fast GC in combination with negative chemical ionization mass spectrometry. J. Chromatogr. A 1216, 6326–6334. [Google Scholar]
- Barriada-Pereira, M.; Serodio, P.; Gonzalez-Castro, M.J.; Nogueira, J.M.F. Determination of organochlorine pesticides in vegetable matrices by stir bar sorptive extraction with liquid desorption and large volume injection-gas chromatography-mass spectrometry towards compliance with European Union directives. J. Chromatogr. A 1217, 119–126. [Google Scholar]
- Húšková, R.; Matisová, E.; Ondreková, S. Fast GC-MS of endocrine disrupting chemicals. Int. J. Environ. Anal. Chem. 2010, 90, 173–187. [Google Scholar] [CrossRef]
- Hercegová, A.; Húšková, R.; Matisová, E. Evaluation of different calibration approaches in pesticide residues analysis in non-fatty food using fast GC-MS. Int. J. Environ. Anal. Chem. 2010, 90, 188–204. [Google Scholar] [CrossRef]
- Hrouzková, S.; Matisová, E.; Andraščíková, M.; Horváth, M.; Húšková, R.; Ďurčanská, J. Survey of low-level endocrine disrupting pesticides in food matrices in Slovakia. Int. J. Environ. Anal. Chem. 2012. [Google Scholar] [CrossRef]
- Brossa, L.; Marcé, R.M.; Borrull, E.; Pocurull, E. Application of on-line solid-phase extraction-gas chromatography-mass spectrometry to the determination of endocrine disruptors in water samples. J. Chromatogr. A 2002, 963, 287–294. [Google Scholar] [CrossRef]
- Brossa, L.; Marcé, R.M.; Borrull, F.; Pocurull, E. Determination of endocrine-disrupting compounds in water samples by on-line solid-phase extraction-programmed-temperature vaporisation-gas chromatography-mass spectrometry. J. Chromatogr. A 2003, 998, 41–50. [Google Scholar] [CrossRef]
- Peñalver, A.; Garcia, V.; Pocurull, E.; Borrull, F.; Marcé, R.M. Stir bar sorptive extraction and large volume injection gas chromatography to determine a group of endocrine disrupters in water samples. J. Chromatogr. A 1007, 1–9. [Google Scholar]
- Serôdio, P.; Nogueira, J.M.F. Multi-residue screening of endocrine disrupters chemicals in water samples by stir bar sorptive extraction-liquid desorption-capillary gas chromatography-mass spectrometry detection. Anal.Chim. Acta 2004, 517, 21–32. [Google Scholar] [CrossRef]
- Mansilha, C.; Melo, A.; Rebelo, H.; Ferreira, I.M.; Pinho, O.; Domingues, V.; Pinho, C.; Gameiro, P. Quantification of endocrine disruptors and pesticides in water by gas chromatography-tandem mass spectrometry: Method validation using weighted linear regression schemes. J. Chromatogr. A 1217, 6681–6691. [Google Scholar]
- Trenholm, R.A.; Vanderford, B.J.; Holady, J.C.; Rexing, D.J.; Snyder, S.A. Broad range analysis of endocrine disruptors and pharmaceuticals using gas chromatography and liquid chromatography tandem mass spectrometry. Chemosphere 2006, 65, 1990–1998. [Google Scholar] [CrossRef]
- Nevado, J.J.B.; Cabanillas, C.G.; Llerena, M.J.V.; Rodriguez Robledo, V. Sensitive SPE GC-MS-SIM screening of endocrine-disrupting herbicides and related degradation products in natural surface waters and robustness study. Microchem.J. 2007, 87, 62–71. [Google Scholar] [CrossRef]
- Baugros, J.B.; Giroud, B.; Dessalces, G.; Grenier-Loustalot, M.F.; Cren-Olivé, C. Multiresidue analytical methods for the ultra-trace quantification of 33 priority substances present in the list of REACH in real water samples. Anal.Chim. Acta 2008, 607, 191–203. [Google Scholar] [CrossRef]
- Durán-Alvarez, J.C.; Becerill-Bravo, E.; Castro, V.S.; Jiménez, B.; Gibson, R. The analysis of a group of acidic pharmaceuticals, carbamazepine, and potential endocrine disrupting compounds in wastewater irrigated soils by gas chromatography-mass spectrometry. Talanta 2009, 78, 1159–1166. [Google Scholar] [CrossRef]
- Hwang, H.M.; Park, E.K.; Young, T.M.; Hammock, B.D. Occurrence of endocrine-disrupting chemicals in indoor dust. Sci. Total Environ. 2008, 404, 26–35. [Google Scholar] [CrossRef]
- Lopez-Espinosa, M.J.; Granada, A.; Carreno, J.; Salvatierra, M.; Olea-Serrano, F.; Olea, N. Organochlorine pesticides in placentas from Southern Spain and some related factors. Placenta 2007, 28, 631–638. [Google Scholar] [CrossRef]
- Fujii, Y.; Haraguchi, K.; Harada, K.H.; Hitomi, T.; Inoue, K.; Itoh, Y.; Watanabe, T.; Takenaka, K.; Uehara, S.; Yang, H.R.; et al. Detection of dicofol and related pesticides in human breast milk from China, Korea and Japan. Chemosphere 2011, 82, 25–31. [Google Scholar] [CrossRef]
- Kang, J.H.; Park, H.; Chang, Y.S.; Choi, J.W. Distribution of organochlorine pesticides (OCPs) and polychlorinated biphenyls (PCBs) in human serum from urban areas in Korea. Chemosphere 2008, 73, 1625–1631. [Google Scholar]
- Röllin, H.B.; Sandanger, T.M.; Hansen, L.; Channa, K.; Odland, J.Ø. Concentration of selected persistent organic pollutants in blood from delivering women in South Africa. Sci. Total Environ. 2009, 408, 146–152. [Google Scholar] [CrossRef]
- Bielawski, D.; Ostrea, E., Jr.; Posecion, N., Jr.; Corrion, M.; Seagraves, J. Detection of several classes of pesticides and metabolites in meconium by gas chromatography-mass spectrometry. Chromatographia 2005, 62, 623–629. [Google Scholar]
- Hajšlová, J.; Zrostlíková, J. Matrix effects in(ultra) trace analysis of pesticide residues in food and biotic matrices. J. Chromatogr. A 1000, 181–197. [Google Scholar]
- Poole, C.F. Matrix-induced response enhancement in pesticide residue analysis by gas chromatography. J. Chromatogr. A 1158, 241–250. [Google Scholar]
- Húšková, R.; Kirchner, M.; Matisová, E. Matrix effects and its elimination in analysis of pesticide residues in food by gas chromatography. Chem. Listy 2007, 101, 1020–1027. [Google Scholar]
- Hercegová, A.; Dömötörová, M.; Kružlicová, D.; Matisová, E. Comparison of sample preparation methods combined with fast gas chromatography-mass spectrometry for ultratrace analysis of pesticide residues in baby food. J. Sep. Sci. 2006, 29, 1102–1109. [Google Scholar] [CrossRef]
- Anastassiades, M.; Lehotay, S.J.; Štajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar]
- Farre, M.; Brix, R.; Kuster, M.; Rubio, F.; Goda, Y.; Lopez de Alda, M.; Barceló, D. Evaluation of commercial immunoassays for the detection of estrogens in water by comparison with high-performance liquid chromatography tandem mass spectrometry HPLC-MS/MS (QqQ). Anal. Bioanal. Chem. 2006, 385, 1001–1011. [Google Scholar] [CrossRef]
- Hercegová, A.; Dömötörová, M.; Matisová, E. Sample preparation methods in the analysis of pesticide residues in baby food with subsequent chromatographic determination. J. Chromatogr. A 1153, 54–73. [Google Scholar]
- Rodriguez-Mozas, S.; Lopez de Alda, M.; Barceló, D. Advantages and limitations of on-line solid phase extraction coupled to liquid chromatography-mass spectrometry technologies versus biosensors for monitoring of emerging contaminants in water. J. Chromatogr. A 1152, 97–115. [Google Scholar]
- Pawliszyn, J.; Lord, H. Reference. In Handbook of Sample Preparation; John Wiley and Sons, Inc.: New Jersey, NJ, USA, 2010. [Google Scholar]
- Fatoki, O.S.; Awofolu, R.O. Methods for selective determination of persistent organochlorine pesticide residues in water and sediments by capillary gas chromatography and electron-capture detection. J. Chromatogr. A 2003, 983, 225–236. [Google Scholar] [CrossRef]
- Xue, N.; Xu, X.; Jin, Z. Screening 31 endocrine-disrupting pesticides in water and surface sediment samples from Beijing Guanting reservoir. Chemosphere 2005, 61, 1594–1606. [Google Scholar] [CrossRef]
- Landrigan, P.J.; Claudio, L.; Markowitz, S.B.; Berkowitz, G.S.; Brenner, B.L.; Romero, H.; Wetmur, J.G.; Matte, T.D.; Gore, A.C.; Godbold, J.H.; Wolff, M.S. Pesticides and inner-city children: Exposures, risks, and prevention. Environ. Health Perspect. 1999, 107, 431–437. [Google Scholar]
- Húšková, R.; Matisová, E.; Hrouzková, S. Mass spectrometry with negative chemical ionization and its use in GC-MS analysis of organic pollutants. Chem. Listy 2010, 104, 913–920. [Google Scholar]
- Tuija Pihlström. Method Validation and Quality Control Procedures for Pesticide Residues Analysis in Food and Feed; Document No. SANCO/10684/2009. National Food Administration: Uppsala, Sweden, 2009.
- Kotretsou, S.I.; Koutsodimou, A. Overview of the applications of tandem mass spectrometry (MS/MS) in food analysis of nutritionally harmful compounds. Food Rev. Int. 2006, 22, 125–172. [Google Scholar] [CrossRef]
- Čajka, T.; Hajšlová, J. Gas chromatography-high-resolution time-of-flight mass spectrometry in pesticide residue analysis: Advantages and limitations. J. Chromatogr.A 1058, 251–261. [Google Scholar]
- Van Hoeck, E.; Canale, F.; Cordero, C.; Compernolle, S.; Bicchi, C.; Sandra, P. Multiresidue screening of endocrine-disrupting chemicals and pharmaceuticals in aqueous samples by multi-stir bar sorptive extraction-single desorption-capillary gas chromatography/mass spectrometry. Anal. Bioanal. Chem. 2008, 393, 907–919. [Google Scholar]
- Ostrea, E.M.; Bielawski, D.M.; Posecion, N.S.; Corrion, M.; Villanueva-Uy, E.; Bernardo, R.C.; Jin, Y.; Janisse, J.J.; Ager, J.W. Combined analysis of prenatal (maternal hair and blood) and neonatal (infant hair, cord blood and meconium) matrices to detect fetal exposure to environmental pesticide. Environ. Res. 2009, 109, 116–122. [Google Scholar] [CrossRef]
- Maštovská, K.; Lehotay, S.J. Practical approaches to fast gas chromatography-mass spectrometry. J. Chromatogr. A 1000, 153–180. [Google Scholar]
- Hrouzková, S.; Matisová, E. Fast Gas Chromatography and Its Use in Pesticide Residues Analysis. In Pesticides-Strategies for Pesticides Analysis, 1st; Stoytcheva, M., Ed.; InTech: Rijeka, Croatia, 2011; pp. 131–154. [Google Scholar]
- Donato, P.; Tranchida, P.Q.; Dugo, P.; Dugo, G.; Mondello, L. Rapid analysis of food products by means of high speed gas chromatography. J. Sep. Sci. 2007, 30, 508–526. [Google Scholar]
- Klee, M.S.; Blumberg, L.M. Theoretical and practical aspects of fast gas chromatography and method translation. J. Chromatogr. Sci. 2002, 40, 234–247. [Google Scholar]
- Dömötörová, M.; Kirchner, M.; Matisová, E.; de Zeeuw, J. Possibilities and limitations of fast GC with narrow-bore columns. J. Sep. Sci. 2006, 29, 1051–1063. [Google Scholar]
- Kirchner, M.; Matisová, E.; Otrekal, R.; Hercegová, A.; de Zeeuw, J. Search on ruggedness of fast gas chromatography-mass spectrometry in pesticide residues analysis. J. Chromatogr. A 1084, 63–70. [Google Scholar]
- Cramers, C.A.; Leclercq, P.A. Strategies for speed optimisation in gas chromatography: An overview. J. Chromatogr. A 1999, 842, 3–13. [Google Scholar] [CrossRef]
- De Zeeuw, J.; Peene, J.; Jansen, H.G.; Lou, X.W. A simple way to speed up separations by GC-MS using short 0.53 mm columns and vacuum outlet conditions. J. High Res. Chromatogr 2000, 23, 677–680. [Google Scholar] [CrossRef]
- Kirchner, M.; Matisová, E. Present state and perspectives of fast GC-MS application. Chem. Listy 2005, 99, 789–801. [Google Scholar]
- Kirchner, M.; Matisová, E.; Hrouzková, S.; de Zeeuw, J. Possibilities and limitations of quadrupole mass spectrometric detector in fast gas chromatography. J. Chromatogr. A 1090, 126–132. [Google Scholar]
- Anastassiades, M.; Maštovská, K.; Lehotay, S.J. Evaluation of analyte protectants to improve gas chromatographic analysis of pesticides. J. Chromatogr. A 1015, 163–184. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Matisová, E.; Hrouzková, S. Analysis of Endocrine Disrupting Pesticides by Capillary GC with Mass Spectrometric Detection. Int. J. Environ. Res. Public Health 2012, 9, 3166-3196. https://doi.org/10.3390/ijerph9093166
Matisová E, Hrouzková S. Analysis of Endocrine Disrupting Pesticides by Capillary GC with Mass Spectrometric Detection. International Journal of Environmental Research and Public Health. 2012; 9(9):3166-3196. https://doi.org/10.3390/ijerph9093166
Chicago/Turabian StyleMatisová, Eva, and Svetlana Hrouzková. 2012. "Analysis of Endocrine Disrupting Pesticides by Capillary GC with Mass Spectrometric Detection" International Journal of Environmental Research and Public Health 9, no. 9: 3166-3196. https://doi.org/10.3390/ijerph9093166
APA StyleMatisová, E., & Hrouzková, S. (2012). Analysis of Endocrine Disrupting Pesticides by Capillary GC with Mass Spectrometric Detection. International Journal of Environmental Research and Public Health, 9(9), 3166-3196. https://doi.org/10.3390/ijerph9093166