
In Silico Core Proteomics and Molecular Docking Approaches for the Identification of Novel Inhibitors against Streptococcus pyogenes

 ,
, 
 , ,
, , 
Abstract
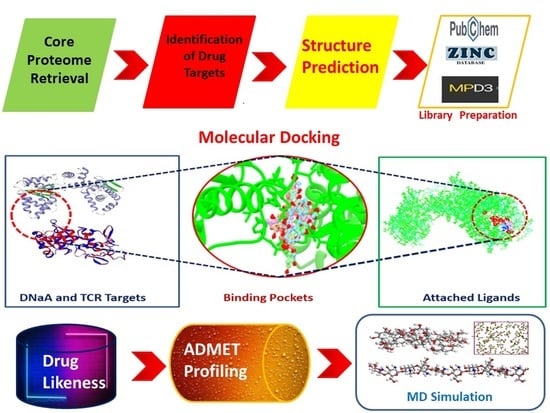
1. Introduction
2. Materials and Methods
2.1. Core Proteome Retrieval
2.2. Identification of Drug Targets
2.3. Structure Prediction
2.4. Structure Evaluation
2.5. Preparation of Target Proteins
2.6. Library Preparation
2.7. Molecular Docking Analysis
2.8. Evaluation of Inhibitors’ Druglikeness
2.9. ADMET Profiling
2.10. Molecular Dynamics Simulation Protocol
2.11. MMPB/GBSA Analysis
3. Results
3.1. Core Proteome Retrieval
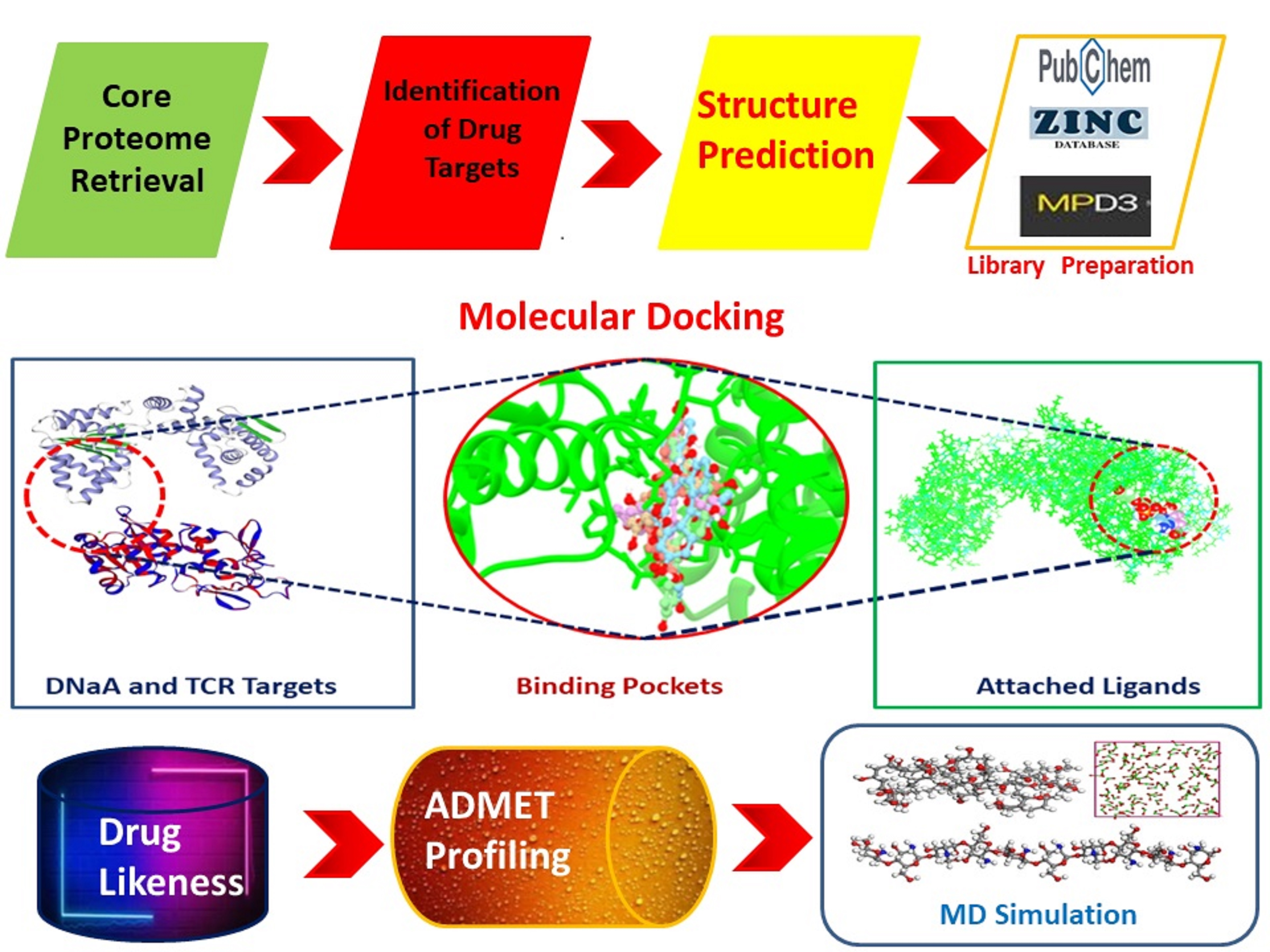
3.2. Identification of Drug Targets
3.3. Structure Prediction
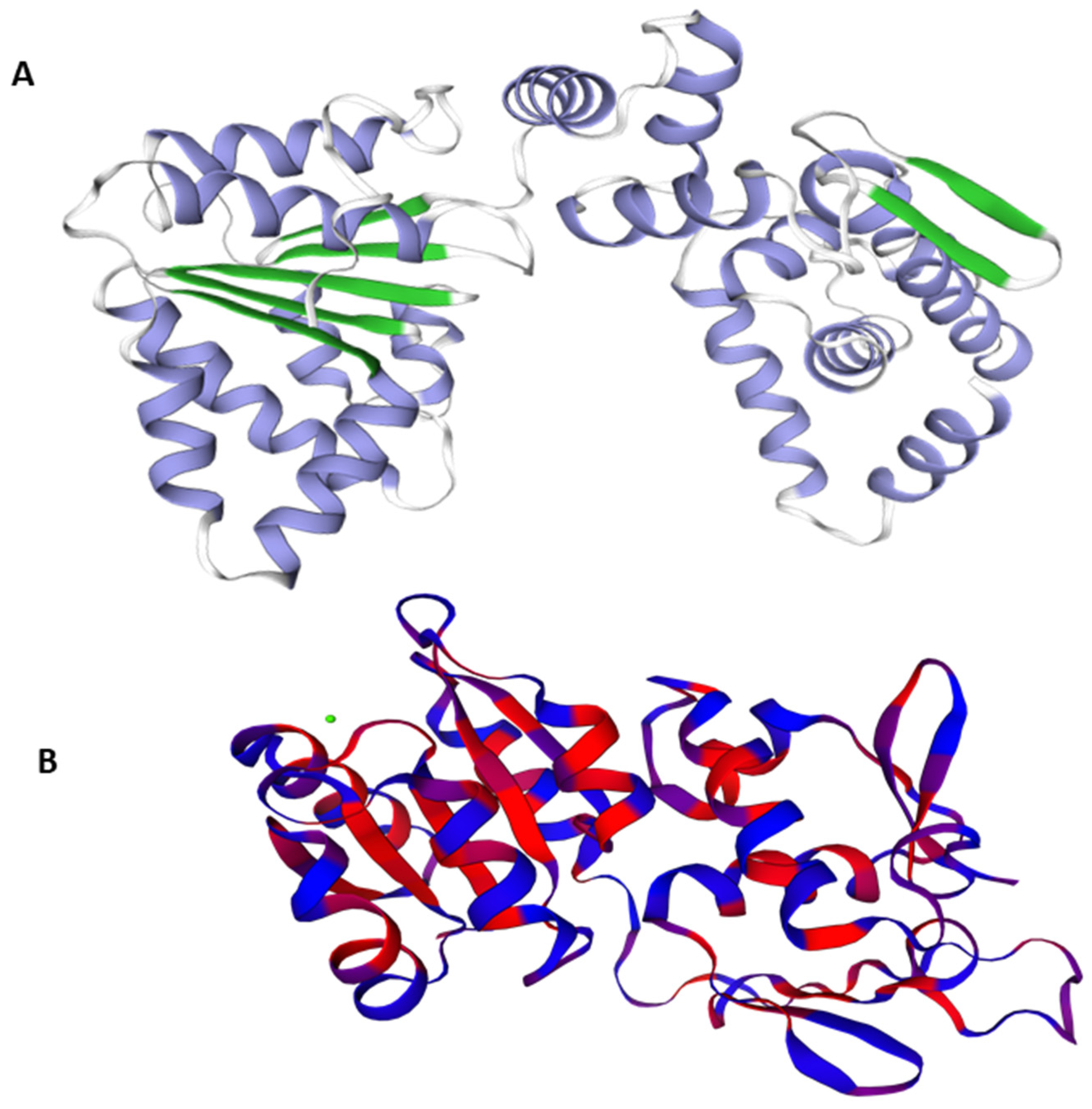
3.4. Model Evaluation
3.5. Molecular Docking Analysis
3.6. Druglikeness Prediction
3.7. ADMET Profiling
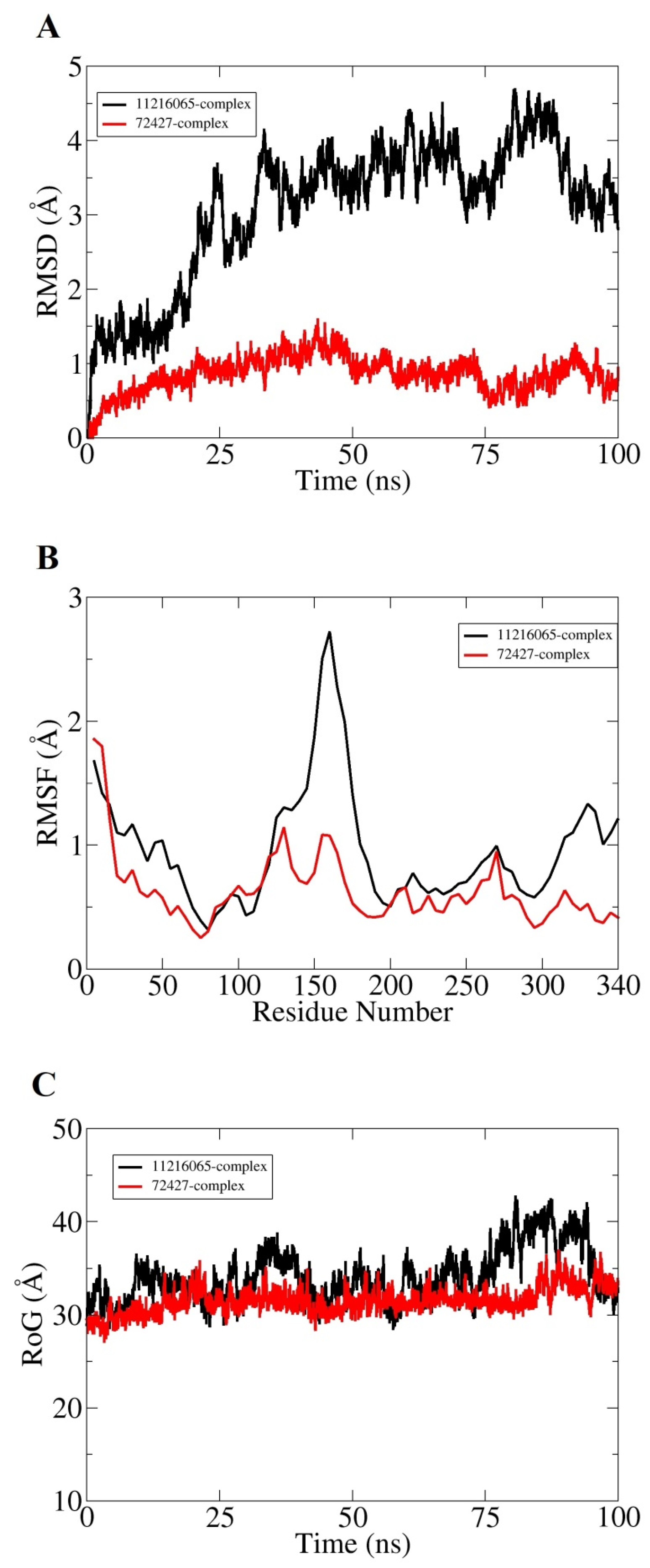
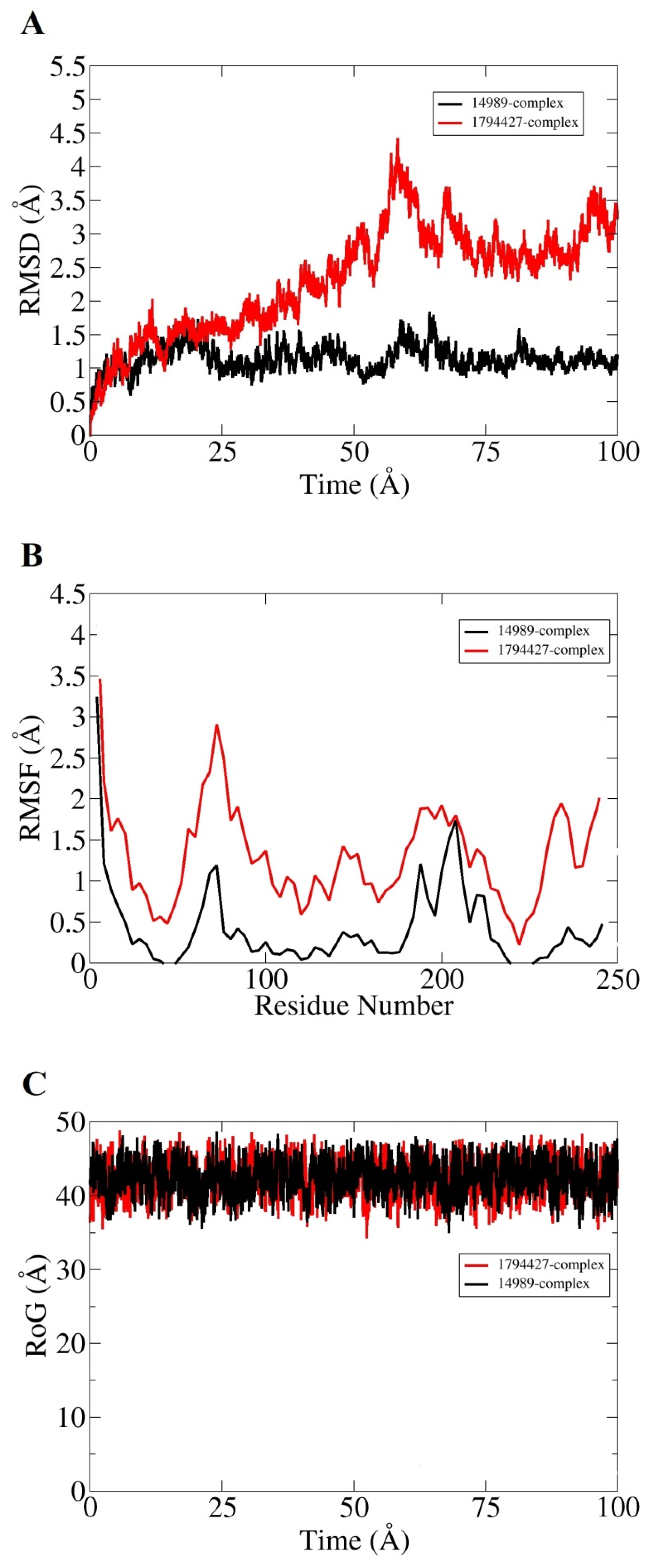
3.8. MD Simulation
3.9. Binding Free Energy Calculations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cunningham, M.W. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 2000, 13, 470–511. [Google Scholar] [CrossRef] [PubMed]
- Sartelli, M.; Malangoni, M.A.; May, K.A.; Viale, P.; Kao, L.S.; Catena, F.; Ansaloni, L.; Moore, E.E.; Moore, F.A.; Peitzman, A.B.; et al. World Society of Emergency Surgery (WSES) guidelines for management of skin and soft tissue infections. World J. Emerg. Surg. 2014, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.L. Invasive group A streptococcus infections. Clin. Infect. Dis. 1992, 14, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Suvorov, N.A.; Ferretti, J.J. Physical and genetic chromosomal map of an M type 1 strain of Streptococcus pyogenes. J. Bacteriol. 1996, 178, 5546–5549. [Google Scholar] [CrossRef][Green Version]
- Bisno, A.; Brito, M.; Collins, C. Molecular basis of group A streptococcal virulence. Lancet Infect. Dis. 2003, 3, 191–200. [Google Scholar] [CrossRef]
- Ikebe, T.; Hirasawa, K.; Suzuki, R.; Isobe, J.; Tanaka, D.; Katsukawa, C.; Kawahara, R.; Tomita, M.; Ogata, K.; Endoh, M.; et al. Antimicrobial susceptibility survey of Streptococcus pyogenes isolated in Japan from patients with severe in-vasive group A streptococcal infections. Antimicrob. Agents Chemother. 2005, 49, 788–790. [Google Scholar] [CrossRef]
- Choby, B.A. Diagnosis and treatment of streptococcal pharyngitis. Am. Fam. Physician 2009, 79, 383–390. [Google Scholar]
- Al-Hamad, A.M. Streptococcal throat: Therapeutic options and macrolide resistance. Saudi Med. J. 2015, 36, 1128. [Google Scholar] [CrossRef]
- Singh, S.; Singh, D.B.; Singh, A.; Gautam, B.; Ram, G.; Dwivedi, S.; Ramteke, P.W. An approach for identification of novel drug targets in Streptococcus pyogenes SF370 through pathway analysis. Interdiscip. Sci. Comput. Life Sci. 2016, 8, 388–394. [Google Scholar] [CrossRef]
- Ghosh, S.; Prava, J.; Samal, H.B.; Suar, M.; Mahapatra, R.K. Comparative genomics study for the identification of drug and vaccine targets in Staphylococcus aureus: MurA ligase enzyme as a proposed candidate. J. Microbiol. Methods 2014, 101, 1–8. [Google Scholar] [CrossRef]
- Kumar, A.; Thotakura, P.L.; Tiwary, B.K.; Krishna, R. Target identification in Fusobacterium nucleatum by subtractive genomics approach and enrichment analysis of host-pathogen protein-protein interactions. BMC Microbiol. 2016, 16, 84. [Google Scholar] [CrossRef]
- Mondal, S.I.; Ferdous, S.; Jewel, N.A.; Akter, A.; Mahmud, Z.; Islam, M.M.; Afrin, T.; Karim, N. Identification of potential drug targets by subtractive genome analysis of Escherichia coli O157:H7: An in silico approach. Adv. Appl. Bioinform. Chem. AABC 2015, 8, 49. [Google Scholar] [CrossRef]
- Shahid, F.; Ashfaq, U.A.; Saeed, S.; Munir, S.; Almatroudi, A.; Khurshid, M. In Silico Subtractive Proteomics Approach for Identification of Potential Drug Targets in Staphylococcus saprophyticus. Int. J. Environ. Res. Public Health 2020, 17, 3644. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Emms, M.D.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves or-thogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed]
- Randow, F.; Seed, B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat. Cell Biol. 2001, 3, 891–896. [Google Scholar] [CrossRef]
- Wei, W.; Ning, L.-W.; Ye, Y.-N.; Guo, F.-B. Geptop: A gene essentiality prediction tool for sequenced bacterial genomes based on orthology and phylogeny. PLoS ONE 2013, 8, e72343. [Google Scholar] [CrossRef] [PubMed]
- Anishetty, S.; Pulimi, M.; Pennathur, G. Potential drug targets in Mycobacterium tuberculosis through metabolic pathway analysis. Comput. Biol. Chem. 2005, 29, 368–378. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Mirza, M.U.; Song, J.-M.; Rao, M.J.; Zhu, X.; Chen, L.-L. Probing the structural basis of Citrus phytochrome B using computational modelling and molecular dynamics simulation approaches. J. Mol. Liq. 2021, 340, 116895. [Google Scholar] [CrossRef]
- Laskowski, R.; MacArthur, M.W.; Moss, D.S.; Thornton, J. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. [20] VERIFY3D: Assessment of protein models with three-dimensional profiles. Heterotrimeric G-Protein Effectors 1997, 277, 396–404. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Mumtaz, A.; Ashfaq, U.A.; Qamar, M.T.U.; Anwar, F.; Gulzar, F.; Ali, M.A.; Saari, N.; Pervez, M.T. MPD3: A useful medicinal plants database for drug designing. Nat. Prod. Res. 2016, 31, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.; Ashfaq, U.A.; Qasim, M.; Yasmeen, E.; Qamar, M.T.U.; Anwar, F. Screening of medicinal plant phytochemicals as natural antagonists of p53–MDM2 interaction to reactivate p53 functioning. Anti-Cancer Drugs 2017, 28, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Christopher, A.; Lipinski, F.; Lombardo, B.; Dominy, W.P.; Feeney, J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Suleman, M.; Qamar, M.T.U.; Saleem, S.; Ahmad, S.; Ali, S.S.; Khan, H.; Akbar, F.; Khan, W.; Alblihy, A.; Alrumaihi, F.; et al. Mutational Landscape of Pirin and Elucidation of the Impact of Most Detrimental Missense Variants That Accelerate the Breast Cancer Pathways: A Computational Modelling Study. Front. Mol. Biosci. 2021, 8, 692835. [Google Scholar] [CrossRef]
- Weiner, P.K.; Kollman, P.A. AMBER: Assisted model building with energy refinement. A general program for modeling molecules and their interactions. J. Comput. Chem. 1981, 2, 287–303. [Google Scholar] [CrossRef]
- Li, C.; Tan, T.; Zhang, H.; Feng, W. Analysis of the conformational stability and activity of Candida antarctica lipase B in organic solvents: Insight from molecular dynamics and quantum mechanics/simulations. J. Biol. Chem. 2010, 285, 28434–28441. [Google Scholar] [CrossRef]
- Hammonds, K.D.; Ryckaert, J.-P. On the convergence of the SHAKE algorithm. Comput. Phys. Commun. 1991, 62, 336–351. [Google Scholar] [CrossRef]
- Abro, A.; Azam, S.S. Binding free energy based analysis of arsenic (+3 oxidation state) methyltransferase with S-adenosylmethionine. J. Mol. Liq. 2016, 220, 375–382. [Google Scholar] [CrossRef]
- Ismail, S.; Shahid, F.; Khan, A.; Bhatti, S.; Ahmad, S.; Naz, A.; Almatroudi, A.; Qamar, M.T.U. Pan-vaccinomics approach towards a universal vaccine candidate against WHO priority pathogens to address growing global antibiotic resistance. Comput. Biol. Med. 2021, 136, 104705. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Ismail, S.; Ahmad, S.; Mirza, M.U.; Abbasi, S.W.; Ashfaq, U.A.; Chen, L.-L. Development of a Novel Multi-Epitope Vaccine Against Crimean-Congo Hemorrhagic Fever Virus: An Integrated Reverse Vaccinology, Vaccine Informatics and Biophysics Approach. Front. Immunol. 2021, 12, 12. [Google Scholar] [CrossRef]
- Alamri, M.A.; Qamar, M.T.U.; Afzal, O.; Alabbas, A.B.; Riadi, Y.; Alqahtani, S.M. Discovery of anti-MERS-CoV small covalent inhibitors through pharmacophore modeling, covalent docking and molecular dynamics simulation. J. Mol. Liq. 2021, 330, 115699. [Google Scholar] [CrossRef]
- Rehman, A.; Ahmad, S.; Shahid, F.; Albutti, A.; Alwashmi, A.; Aljasir, M.; Alhumeed, N.; Qasim, M.; Ashfaq, U.; Qamar, M.T.U. Integrated Core Proteomics, Subtractive Proteomics, and Immunoinformatics Investigation to Unveil a Potential Multi-Epitope Vaccine against Schistosomiasis. Vaccines 2021, 9, 658. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A.; et al. Designing multi-epitope vaccine against Staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef] [PubMed]
- Uddin, R.; Siddiqui, Q.N.; Sufian, M.; Azam, S.S.; Wadood, A. Proteome-wide subtractive approach to prioritize a hypothetical protein of XDR-Mycobacterium tuberculosis as potential drug target. Genes Genom. 2019, 41, 1281–1292. [Google Scholar] [CrossRef]
- Sakharkar, K.R.; Sakharkar, M.K.; Chow, V. A novel genomics approach for the identification of drug targets in pathogens, with special reference to Pseudomonas aeruginosa. Silico Biol. 2004, 4, 355–360. [Google Scholar]
- Rehman, A.; Ashfaq, U.A.; Shahid, F.; Noor, F.; Aslam, S. The Screening of phytochemicals against NS5 Polymerase to treat Zika Virus infection: Integrated computational based approach. Comb. Chem. High Throughput Screen. 2021, 24, 1. [Google Scholar] [CrossRef]
- Butt, A.M.; Nasrullah, I.; Tahir, S.; Tong, Y. Comparative genomics analysis of Mycobacterium ulcerans for the identification of putative essential genes and therapeutic candidates. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Noor, F.; Noor, A.; Ishaq, A.R.; Farzeen, I.; Saleem, M.H.; Ghaffar, K.; Aslam, M.F.; Aslam, S.; Chen, J.-T. Recent Advances in Diagnostic and Therapeutic Approaches for Breast Cancer: A Comprehensive Review. Curr. Pharm. Des. 2021, 27, 2344–2365. [Google Scholar] [CrossRef]
- Tsaioun, K.; Bottlaender, M.; Mabondzo, A. ADDME – Avoiding Drug Development Mistakes Early: Central nervous system drug discovery perspective. BMC neurology 2009, 9, S1. [Google Scholar] [CrossRef]
- Javed, S.; Shoaib, A.; Mahmood, Z.; Nawaz, S.; Khan, K.M. Phytochemical, pharmacological and GC-MS characterization of the lipophilic fraction of Monotheca buxifolia. Asian J. Agric. Biol. 2021. [Google Scholar] [CrossRef]
- Ahmad, S.; Shahid, F.; Qamar, M.T.U.; Rehman, H.; Abbasi, S.; Sajjad, W.; Ismail, S.; Alrumaihi, F.; Allemailem, K.; Almatroudi, A.; et al. Immuno-Informatics Analysis of Pakistan-Based HCV Subtype-3a for Chimeric Polypeptide Vaccine Design. Vaccines 2021, 9, 293. [Google Scholar] [CrossRef]
- Khalid, R.R.; Qamar, M.T.U.; Maryam, A.; Ashique, A.; Anwar, F.; Geesi, M.H.; Siddiqi, A.R. Comparative Studies of the Dynamics Effects of BAY60-2770 and BAY58-2667 Binding with Human and Bacterial H-NOX Domains. Molecules 2018, 23, 2141. [Google Scholar] [CrossRef]
- Piard, J.C.; Hautefort, I.; A Fischetti, V.; Ehrlich, S.D.; Fons, M.; Gruss, A. Cell wall anchoring of the Streptococcus pyogenes M6 protein in various lactic acid bacteria. J. Bacteriol. 1997, 179, 3068–3072. [Google Scholar] [CrossRef]
- Sarangi, A.N.; Aggarwal, R.; Rahman, Q.; Trivedi, N. Subtractive Genomics Approach for in Silico Identification and Characterization of Novel Drug Targets in Neisseria Meningitides Serogroup B. J. Comput. Sci. Syst. Biol. 2009, 2, 255–258. [Google Scholar] [CrossRef]
- Barh, D.; Tiwari, S.; Jain, N.; Ali, A.; Santos, A.R.; Misra, A.N.; Azevedo, V.; Kumar, A. In silico subtractive genomics for target identification in human bacterial pathogens. Drug Dev. Res. 2011, 72, 162–177. [Google Scholar] [CrossRef]
- Goyal, M.; Citu, C.; Singh, N. In silico identification of novel drug targets in acinetobacter baumannii by subtractive genomic approach. Asian J. Pharm. Clin. Res. 2018, 11, 230–236. [Google Scholar] [CrossRef]
- Qureshi, N.A.; Bakhtiar, S.M.; Faheem, M.; Shah, M.; Bari, A.; Mahmood, H.M.; Sohaib, M.; Mothana, R.A.; Ullah, R.; Jamal, S.B. Genome-Based Drug Target Identification in Human Pathogen Streptococcus gallolyticus. Front. Genet. 2021, 12, 303. [Google Scholar] [CrossRef] [PubMed]
- Fatoba, A.; Okpeku, M.; Adeleke, M. Subtractive Genomics Approach for Identification of Novel Therapeutic Drug Targets in Mycoplasma genitalium. Pathogens 2021, 10, 921. [Google Scholar] [CrossRef] [PubMed]
- Amineni, U.; Pradhan, D.; Marisetty, H. In silico identification of common putative drug targets in Leptospira interrogans. J. Chem. Biol. 2010, 3, 165–173. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Maryam, A.; Muneer, I.; Xing, F.; Ashfaq, U.A.; Khan, F.A.; Anwar, F.; Geesi, M.H.; Khalid, R.R.; Rauf, S.A.; et al. Computational screening of medicinal plant phytochemicals to discover potent pan-serotype inhibitors against dengue virus. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Durdagi, S.; Qamar, M.T.U.; Salmas, R.E.; Tariq, Q.; Anwar, F.; Ashfaq, U.A. Investigating the molecular mechanism of staphylococcal DNA gyrase inhibitors: A combined ligand-based and structure-based resources pipeline. J. Mol. Graph. Model. 2018, 85, 122–129. [Google Scholar] [CrossRef]
- Lin, J.; Sahakian, D.C.; De Morais, S.M.F.; Xu, J.J.; Polzer, R.J.; Winter, S.M. The Role of Absorption, Distribution, Metabolism, Excretion and Toxicity in Drug Discovery. Curr. Top. Med. Chem. 2003, 3, 1125–1154. [Google Scholar] [CrossRef]
- Vasanthanathan, P.; Taboureau, O.; Oostenbrink, C.; Vermeulen, N.P.E.; Olsen, L.; Jørgensen, F.S. Classification of Cytochrome P450 1A2 Inhibitors and Noninhibitors by Machine Learning Techniques. Drug Metab. Dispos. 2009, 37, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.; Price, A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Common Pathways | Unique Pathways |
|---|---|---|
| Glucose-6-phosphate isomerase | Metabolic pathways Glycolysis Carbon metabolism Pentose phosphate pathway Amino sugar and nucleotide sugar metabolism Starch and sucrose metabolism | Biosynthesis of secondary metabolites Microbial metabolism in diverse environments |
| UDP-N-acetylenolpyruvoylglucosamine reductase | Metabolic pathways Amino sugar and nucleotide sugar metabolism | Peptidoglycan biosynthesis |
| Riboflavin biosynthesis protein | Biosynthesis of cofactors Metabolic pathways Riboflavin metabolism | Biosynthesis of secondary metabolites |
| Alanine racemase | Metabolic pathways | d-Alanine metabolism Vancomycin resistance |
| Chromosomal replication initiator protein DnaA | Two-component system | |
| Two-component response regulator | Two-component system | |
| Phosphate acyltransferase | Glycerolipid metabolism Metabolic pathways | Biosynthesis of secondary metabolites |
| Fructose-bisphosphate aldolase | Metabolic pathways Glycolysis Carbon metabolism Biosynthesis of amino acids Fructose and mannose metabolism Pentose phosphate pathway Methane metabolism | Biosynthesis of secondary metabolites Microbial metabolism in diverse environments |
| UDP-N-acetylmuramoyl-tripeptide—D-alanyl- D-alanine ligase | Metabolic pathways | Vancomycin resistance Peptidoglycan biosynthesis |
| Acetyl-coenzyme A carboxylase carboxyl transferase subunit alpha | Metabolic pathways | Biosynthesis of secondary metabolites |
| Target Proteins | Ramachandran Plot Statistics (%) | Verify 3D | ERRAT | ProSA | |||
|---|---|---|---|---|---|---|---|
| Core | Allowed | General | Disallowed | Compatibility Score (%) | Quality Factor | z-Score | |
| Chromosomal replication initiator protein DnaA | 88.4% | 10.0% | 1.6% | 0.0% | 74.71% | 90.0602 | −8.48 |
| Two-component response regulator | 89.6% | 9.5% | 0.5% | 0.5% | 85.47% | 92.4779 | −7.86 |
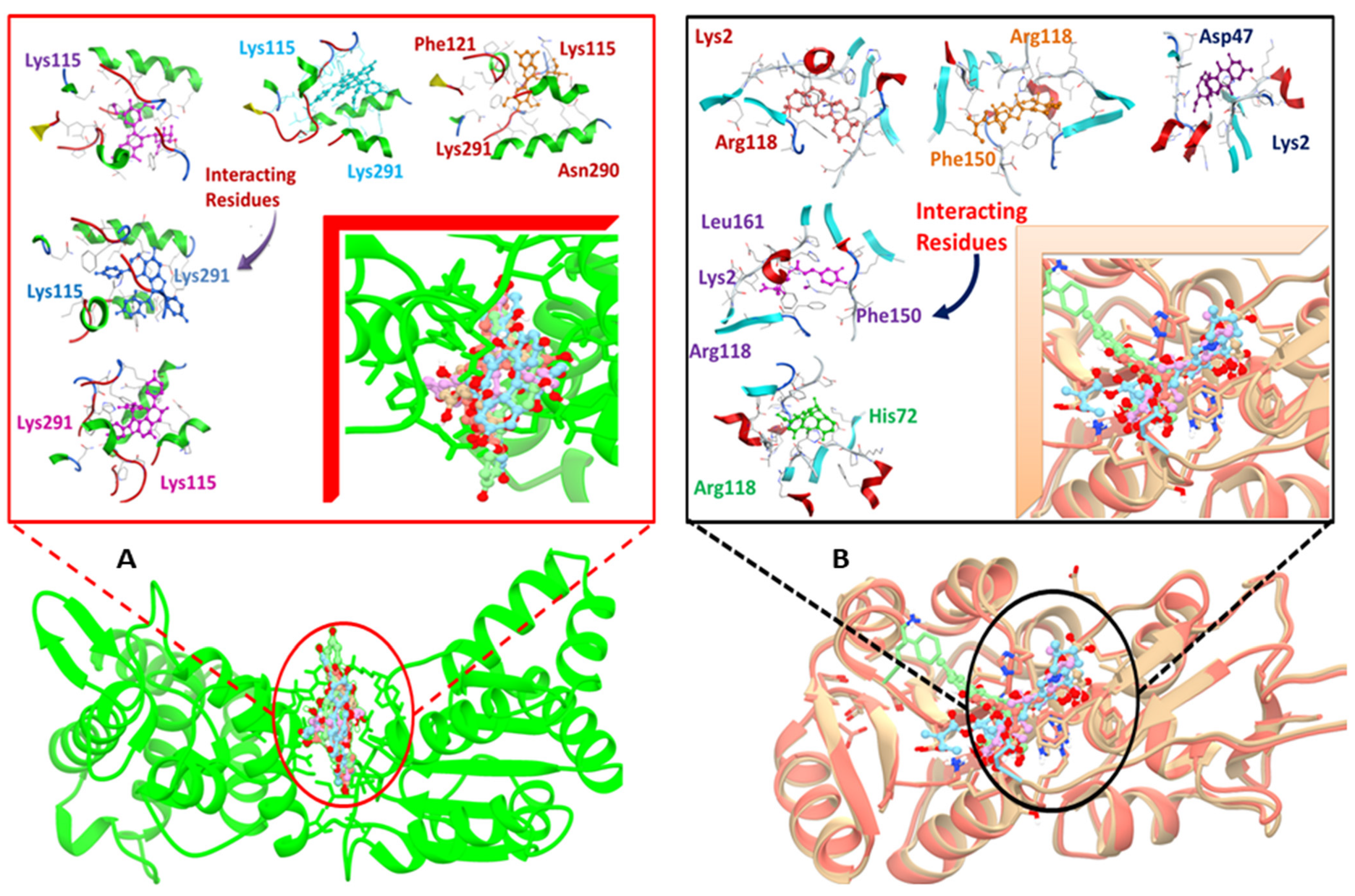
| Target Proteins | Compound ID’s | Compounds Name | Docking Score (kcal/mol) | RMSD | Interacting Residues |
|---|---|---|---|---|---|
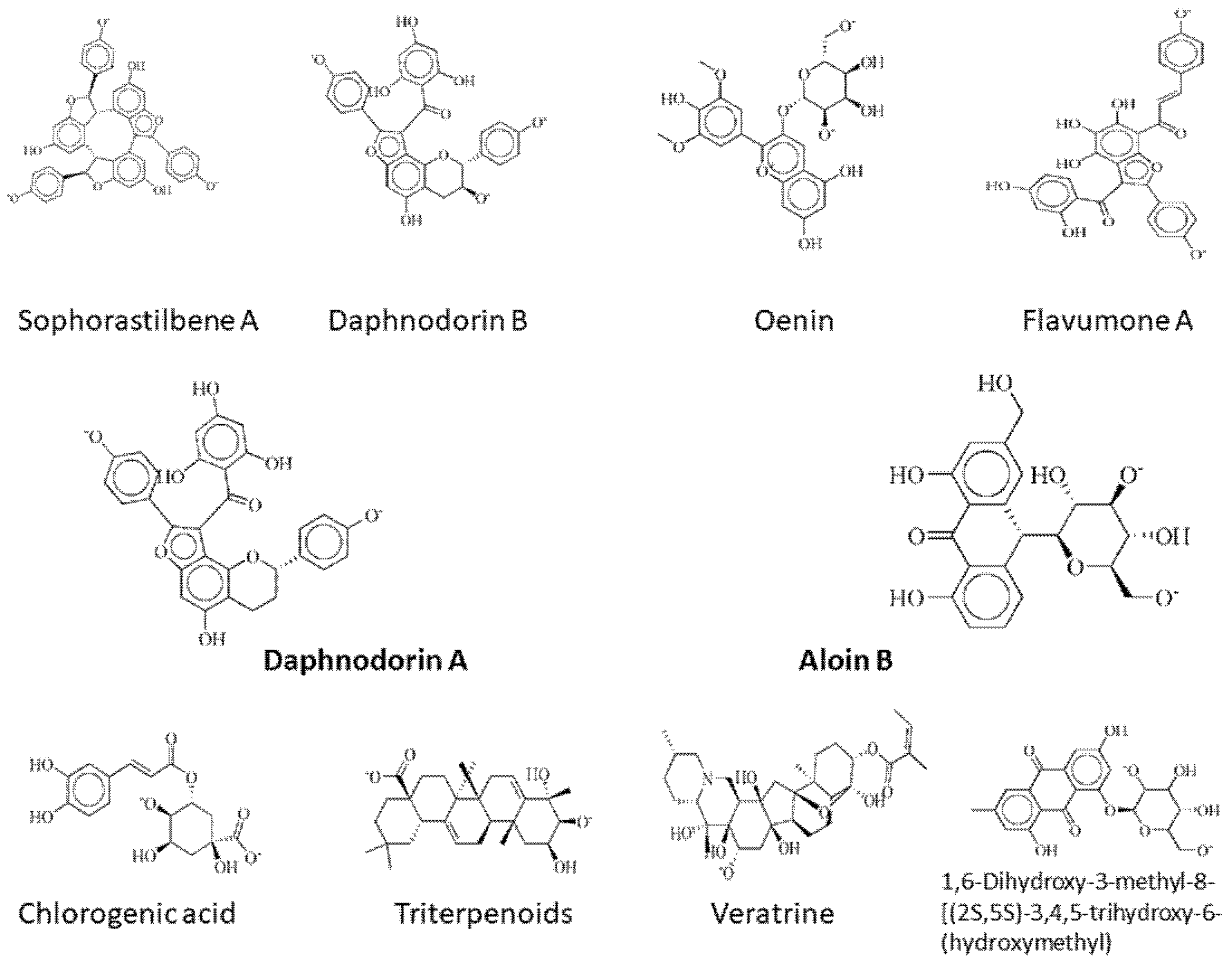
| DNaA Protein | 11216065 | Sophorastilbene A | −21.31 | 3.61 | Phe121 Lys115 Lys291 Asn290 |
| 72427 | Daphnodorin B | −20.77 | 1.70 | Lys115 Lys291 | |
| 443652 | Oenin | −20.18 | 1.96 | Lys115 | |
| 12096478 | Flavumone A | −20.06 | 2.32 | Lys115 Lys291 | |
| 72426 | Daphnodorin A | −16.00 | 3.26 | Lys115 Lys291 | |
| TCR protein | 14989 | Aloin B | −18.02 | 1.55 | Arg118 His72 |
| 1794427 | Chlorogenic acid | −17.47 | 1.23 | Lys2 Arg118 Phe150 Leu161 | |
| 71597391 | Triterpenoids | −17.47 | 1.42 | Lys2 Arg118 | |
| 5380394 | Veratrine | −16.96 | 2.22 | Arg118 Phe150 | |
| 118855584 | 1,6-Dihydroxy-3-methyl-8-[(2S,5S)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyanthracene-9,10-dione | −16.84 | 1.51 | Asp47 Lys2 |
| Target Proteins | Compounds | Molecular Weight (g/mol) | Number of HBA (nON) | Number of HBD (nOHNH) | mi-LogP |
|---|---|---|---|---|---|
| DNaA | 11216065 | 673.65 | 9 | 3 | 4.36 |
| 72427 | 539.47 | 10 | 4 | 2.24 | |
| 443652 | 491.43 | 12 | 5 | −2.39 | |
| 12096478 | 538.46 | 10 | 5 | 2.29 | |
| 72426 | 524.48 | 9 | 4 | 3.25 | |
| TCR | 14989 | 416.38 | 9 | 5 | −3.04 |
| 1794427 | 352.30 | 9 | 4 | −3.24 | |
| 71597391 | 470.65 | 5 | 2 | 1.70 | |
| 5380394 | 590.73 | 10 | 5 | −0.54 | |
| 118855584 | 430.37 | 10 | 4 | −1.82 |
| Standard Parameters | Target | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DNaA | TCR | |||||||||
| 11216065 | 72427 | 443652 | 12096478 | 72426 | 14989 | 1794427 | 71597391 | 5380394 | 118855584 | |
| Absorption | ||||||||||
| Aqueous solubility (LogS) | −3.3170 | −3.2562 | −2.7564 | −3.0485 | −2.9143 | −2.3269 | −2.5951 | −3.9108 | −2.4829 | −2.8081 |
| Human Intestinal Absorption | 0.9727 | + 0.9394 | −0.9165 | +0.9300 | +0.8623 | +0.7201 | −0.8658 | +0.7320 | −0.7652 | −0.8845 |
| Blood Brain Barrier | +0.8635 | +0.7154 | −0.8897 | +0.6248 | +0.7183 | +0.5432 | +0.5612 | +0.7302 | −0.8131 | −0.6852 |
| Caco-2 permeability | −0.6888 | −0.8292 | −0.4982 | −0.0172 | 0.6303 | 0.2248 | −0.5040 | 1.1647 | 0.2244 | −0.4438 |
| Distribution | ||||||||||
| P-gp Substrate | Non-Substrate | Non-Substrate | Substrate | Substrate | Substrate | Substrate | Substrate | Substrate | Substrate | Substrate |
| P-gp Inhibitor | Non-Inhibitor | Non-Inhibitor | Non-Inhibitor | Non-Inhibitor | Non-Inhibitor | Non-Inhibitor | Non-Inhibitor | Non-Inhibitor | Inhibitor | Non-Inhibitor |
| Metabolism | ||||||||||
| CYP450 2D6 Substrate | x | x | x | X | x | x | x | x | x | x |
| CYP450 3A4 Substrate | x | x | √ | X | x | x | x | √ | √ | x |
| CYP450 1A2 Inhibitor | √ | √ | X | √ | √ | x | x | x | x | x |
| CYP450 2C9 Inhibitor | √ | √ | X | √ | √ | x | x | x | x | x |
| CYP450 2D6 Inhibitor | x | x | X | X | x | x | x | x | x | x |
| CYP450 2C19 Inhibitor | √ | √ | X | X | √ | x | x | x | x | x |
| CYP450 3A4 Inhibitor | x | √ | X | X | √ | x | x | x | x | x |
| Toxicity | ||||||||||
| Salmonella typhimurium reverse mutation assay AMES Test | Non-AMES Toxic | Non-AMES Toxic | Non-AMES Toxic | Non-AMES Toxic | Non-AMES Toxic | AMES Toxic | Non-AMES Toxic | Non-AMES Toxic | Non-AMES Toxic | AMES Toxic |
| Human Ether-à-go-go-Related Gene (hERG) Inhibition | Weak inhibitors | Weak inhibitors | Weak inhibitors | Weak inhibitors | Weak inhibitors | Weak inhibitor | Weak inhibitor | Weak inhibitor | Weak inhibitor | Weak inhibitor |
| Carcinogens | Non-Carcinogens | Non-Carcinogens | Non-Carcinogens | Non-Carcinogens | Non-Carcinogens | Non-Carcinogens | Non-Carcinogens | Non-Carcinogens | Non-Carcinogens | Non-Carcinogens |
| Rat Acute Toxicity (LD50, mol/kg) | 2.4083 | 2.5846 | −2.7564 | 2.5248 | 3.0847 | 2.5732 | 2.6020 | 2.8611 | 3.3693 | 2.9432 |
| Energy Parameter | TCR | DNaA | ||
|---|---|---|---|---|
| 14989-Complex | 1794427-Complex | 72427-Complex | 11216065-Complex | |
| MM-GBSA | ||||
| VDWAALS | −22.23 | −21.65 | −18.16 | −22.46 |
| EEL | −10.11 | −9.10 | −11.21 | −10.58 |
| EGB | 13.20 | 12.11 | 16.58 | 15.21 |
| ESURF | −2.00 | −2.54 | −3.39 | −4.00 |
| Delta G gas | −32.34 | −30.75 | −29.37 | −33.04 |
| Delta G solv | 11.20 | 9.57 | 13.13 | 11.21 |
| Delta Total | −21.14 | −21.18 | −16.24 | −21.83 |
| MM-PBSA | ||||
| VDWAALS | −22.23 | −21.65 | −18.16 | −22.46 |
| EEL | −10.11 | −9.10 | −11.21 | −10.58 |
| EPB | 10.28 | 6.28 | 12.07 | 9.41 |
| ENPOLAR | −5.00 | −3.96 | −2.98 | −3.51 |
| Delta G gas | −32.34 | −30.75 | −29.37 | −33.04 |
| Delta G solv | 5.28 | 2.32 | 9.09 | 5.9 |
| Delta Total | −27.06 | −28.43 | −20.28 | −27.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehman, A.; Wang, X.; Ahmad, S.; Shahid, F.; Aslam, S.; Ashfaq, U.A.; Alrumaihi, F.; Qasim, M.; Hashem, A.; Al-Hazzani, A.A.; et al. In Silico Core Proteomics and Molecular Docking Approaches for the Identification of Novel Inhibitors against Streptococcus pyogenes. Int. J. Environ. Res. Public Health 2021, 18, 11355. https://doi.org/10.3390/ijerph182111355
Rehman A, Wang X, Ahmad S, Shahid F, Aslam S, Ashfaq UA, Alrumaihi F, Qasim M, Hashem A, Al-Hazzani AA, et al. In Silico Core Proteomics and Molecular Docking Approaches for the Identification of Novel Inhibitors against Streptococcus pyogenes. International Journal of Environmental Research and Public Health. 2021; 18(21):11355. https://doi.org/10.3390/ijerph182111355
Chicago/Turabian StyleRehman, Abdur, Xiukang Wang, Sajjad Ahmad, Farah Shahid, Sidra Aslam, Usman Ali Ashfaq, Faris Alrumaihi, Muhammad Qasim, Abeer Hashem, Amal A. Al-Hazzani, and et al. 2021. "In Silico Core Proteomics and Molecular Docking Approaches for the Identification of Novel Inhibitors against Streptococcus pyogenes" International Journal of Environmental Research and Public Health 18, no. 21: 11355. https://doi.org/10.3390/ijerph182111355
APA StyleRehman, A., Wang, X., Ahmad, S., Shahid, F., Aslam, S., Ashfaq, U. A., Alrumaihi, F., Qasim, M., Hashem, A., Al-Hazzani, A. A., & Abd_Allah, E. F. (2021). In Silico Core Proteomics and Molecular Docking Approaches for the Identification of Novel Inhibitors against Streptococcus pyogenes. International Journal of Environmental Research and Public Health, 18(21), 11355. https://doi.org/10.3390/ijerph182111355