The Gut Microbiota of the Egyptian Mongoose as an Early Warning Indicator of Ecosystem Health in Portugal

 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Area
2.2. Animal and Faeces Samples Collection
2.3. Bacterial Isolation, Biochemical and Molecular Identification, Serotyping and Virulence Genes
2.4. Antimicrobial Susceptibility Testing
2.5. Statistical Analysis of Data
3. Results
3.1. Dominant Cultivable Microbiota
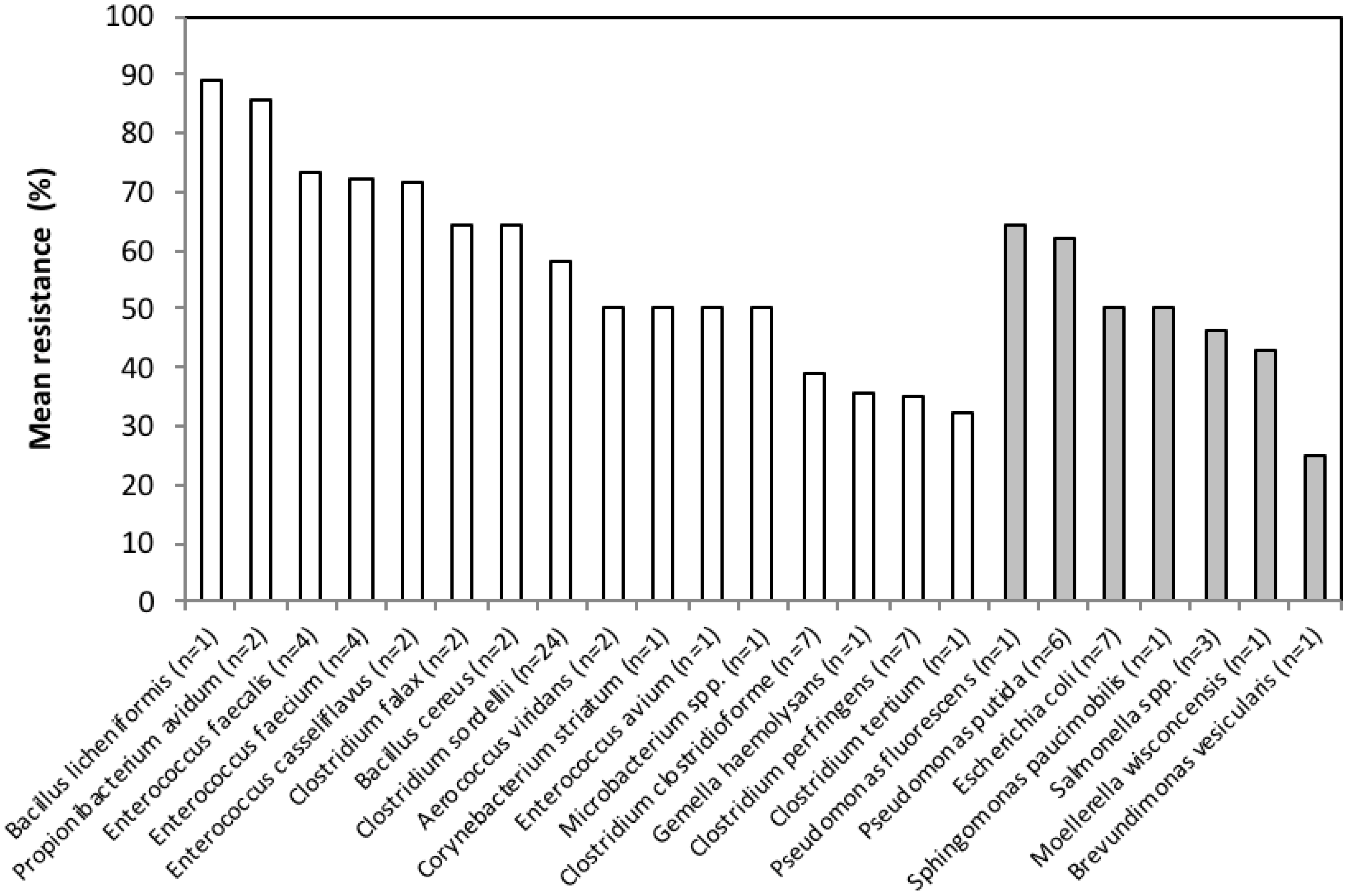
3.2. Antimicrobial Resistance Phenotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ley, R.E.; Lozupone, C.A.; Hamady, M.; Knight, R.; Gordon, J. Worlds within worlds: Evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 2008, 6, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; Cristescu, B.; Northrup, J.M.; Stenhouse, G.B.; Ganzle, M. Diet and environment shape fecal bacterial microbiota composition and enteric pathogen load of grizzly bears. PLoS ONE 2011, 6, e27905. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, S.; Yeoman, C.J.; Sipos, M.; Torralba, M.; Wilson, B.A.; Goldberg, T.L.; Stumpf, R.M.; Leigh, S.R.; White, B.A.; Nelson, K.E. Characterization of the fecal microbiome from non-Human wild primates reveals species specific microbial communities. PLoS ONE 2010, 5, e13963. [Google Scholar] [CrossRef] [PubMed]
- Yeoman, C.J.; Chia, N.; Yildirim, S.; Miller, M.E.B.; Kent, A.; Stumpf, R.; Leigh, S.R.; Nelson, K.E.; White, B.A.; Wilson, B.A. Towards an evolutionary model of animal-associated microbiomes. Entropy 2011, 13, 570–594. [Google Scholar] [CrossRef]
- Cebra, J.J. Influences of microbiota on intestinal immune system development. Am. J. Clin. Nutr. 1999, 69, 1046S–1051S. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.L. The role of natural environments in the evolution of resistance traits in pathogenic bacteria. Proc. R. Soc. B 2009, 276, 2521–2530. [Google Scholar] [CrossRef]
- Baffy, G. Gut Microbiota and Cancer of the Host: Colliding Interests. Adv. Exp. Med. Biol. 2020, 1219, 93–107. [Google Scholar]
- Vemuri, R.; Shankar, E.M.; Chieppa, M.; Eri, R.; Kavanagh, K. Beyond Just Bacteria: Functional Biomes in the Gut Ecosystem Including Virome, Mycobiome, Archaeome and Helminths. Microorganisms 2020, 8, 483. [Google Scholar] [CrossRef]
- Oh, P.L.; Benson, A.K.; Peterson, D.A.; Patil, P.B.; Moriyama, E.N.; Roos, S.; Walter, J. Diversification of the gut symbiont Lactobacillus reuteri as a result of host-driven evolution. ISME J. 2010, 4, 377–387. [Google Scholar] [CrossRef]
- Gill, N.; Finlay, B.B. The gut microbiota: Challenging immunology. Nat. Rev. Immunol. 2011, 11, 636–637. [Google Scholar] [CrossRef]
- Amato, K.; Righini, N. The howler monkey as a model for exploring host-gut microbiota interactions in primates. In Howler Monkeys; Kowalewski, M.M., Garber, P.A., Cortés-Ortiz, L., Urbani, B., Youlatos, D., Eds.; Springer: New York, NY, USA, 2015; pp. 229–258. [Google Scholar]
- Waite, D.W.; Deines, P.; Taylor, M.W. Gut microbiome of the Critically Endangered New Zealand parrot, the Kakapo (Strigops habroptilus). PLoS ONE 2012, 7, e35803. [Google Scholar] [CrossRef] [PubMed]
- Rwego, I.B.; Isabirye-Basuta, G.; Gillespie, T.R.; Goldberg, T.L. Gastrointestinal bacterial transmission among humans, mountain gorillas, and livestock in Bwindi Impenetrable National Park, Uganda. Conserv. Biol. 2008, 22, 1600–1607. [Google Scholar] [CrossRef] [PubMed]
- Barros, T.; Fonseca, C. Expansão do sacarrabos Herpestes ichneumon (Linnaeus, 1758) em Portugal. Galemys 2011, 23, 9–15. [Google Scholar]
- Bandeira, V.; Virgós, E.; Barros, T.; Cunha, M.V.; Fonseca, C. Geographic variation and sexual dimorphism in body size of Egyptian mongoose (Herpestes ichneumon, Linnaeus, 1758) (Mammalia, Carnivora, Herpestidae) in the western limit of its European distribution. Zool. Anz. 2016, 264, 1–10. [Google Scholar] [CrossRef]
- Rosalino, L.M.; Santos, M.J.; Pereira, I.; Santos-Reis, M. Sex-driven differences in Egyptian mongoose’s (Herpestes ichneumon) diet in its northwestern European range. Eur. J. Wildl. Res. 2009, 55, 293–299. [Google Scholar] [CrossRef]
- Pyšek, P.; Richardson, D.M. Invasive species, environmental change and management, and health. Ann. Rev. Environ. Resour. 2010, 35, 25–55. [Google Scholar] [CrossRef]
- Nowak, R.M. Walker’s Carnivores of the World; The Johns Hopkins University Press: Baltimore, MD, USA, 2005. [Google Scholar]
- Bandeira, V.; Virgós, E.; Carvalho, J.; Barros, T.; Cunha, M.V.; Fonseca, C. Diet footprint of Egyptian mongoose along ecological gradients: Effects of primary productivity and life history traits. Mamm. Biol. 2018, 88, 16–25. [Google Scholar] [CrossRef]
- Rosalino, L.M.; Chambel, I. Sacarrabos (Herpestes ichneumon): O emigrante africano. In Um Olhar Sobre os Carnívoros Portugueses; Loureiro, F., Pedroso, N.M., Santos, M.J., Rosalino, L.M., Eds.; Carnivora: Lisboa, Portugal, 2012; pp. 181–191. [Google Scholar]
- Domingos, S.A. O Sacarrabos na Região do Paul do Boquilobo: Um Estudo de Rádio-Rastreio. Bachelor’s Thesis, (level 3). Faculdade de Ciências da Universidade de Lisboa, Lisboa, Portugal, 1999. [Google Scholar]
- Cabral, M.J.; Almeida, J.; Almeida, P.R.; Dellinger, T.; Ferrand de Almeida, N.; Oliveira, M.E.; Palmeirim, J.M.; Queiroz, A.I.; Rogado, L.; Santos-Reis, M. Livro Vermelho dos Vertebrados de Portugal; Instituto da Conservação da Natureza: Lisboa, Portugal, 2006; pp. 517–527. [Google Scholar]
- Palomo, L.J.; Gisbert, J.; Blanco, J.C. Atlas y Libro Rojo de los Mamíferos Terrestres de España; Dirección General para la Biodiversidad-SECEM-SECEMU: Madrid, Spain, 2007. [Google Scholar]
- Mesquita, S.; Sousa, A.J. Bioclimatic mapping using geostatistical approaches: Application to mainland Portugal. Int. J. Climatol. 2009, 29, 2156–2170. [Google Scholar] [CrossRef]
- Grimont, P.A.; Weill, F.X. Antigenic Formulae of the Salmonella Serovars, 9th ed.; WHO Collaborating Centre for Reference and Research on Salmonella, Institute Pasteur: Paris, France, 2007. [Google Scholar]
- Marchesi, J.R.; Sato, T.; Weightman, A.J.; Martin, T.A.; Fry, J.C.; Hiom, S.J.; Dymock, D.; Wade, W.G. Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl. Environ. Microbiol. 1998, 64, 795–799. [Google Scholar] [CrossRef]
- Jerse, A.E.; Yu, J.; Kaper, J.B. A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7839–7843. [Google Scholar] [CrossRef]
- Read, S.C.; Clarke, R.C.; Martin, A.; De Grandis, S.A.; Hii, J.; McEwen, S.; Gyles, C.L. Polymerase chain reaction for detection of verocytotoxigenic Escherichia coli isolated from animal and food sources. Mol. Cell Probes 1992, 6, 153–161. [Google Scholar] [CrossRef]
- Woodward, M.J.; Carroll, P.J.; Wray, C. Detection of entero-and verocyto- toxin genes in Escherichia coli from diarrhoeal in animals using the polymerase chain reaction. Vet. Microbiol. 1992, 31, 251–261. [Google Scholar] [CrossRef]
- O’Brien, A.D.; Karmali, M.A.; Scotland, S.M. A proposal for rationalization of the Escherichia coli cytotoxins. In Recent Advances in Verocytotoxin-Producing Escherichia coli Infections; Karmali, M.A., Goglio, A.G., Eds.; Elsevier Science, B.V.: Amsterdam, The Netherlands, 1994; pp. 147–149. [Google Scholar]
- Paton, A.W.; Paton, J.C. Detection and characterization of Shiga toxigenic Escherichia coli by using multiplex PCR assays for stx1, stx2, eaeA, enterohemorrhagic E. coli hlyA, rfbO111, and rfbO157. J. Clin. Microbiol. 1998, 36, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Posthaus, H.; Breitenmoser-Wursten, C.; Posthaus, H.L.; Bacciarini, L.; Breitenmoser, U. Causes of mortality in reintroduced Eurasian lynx in Switzerland. J. Wildl. Dis. 2002, 38, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Morabito, S.; Dell’Omo, G.; Agrimi, U.; Schmidt, H.; Karch, H.; Cheasty, T.; Caprioli, A. Detection and characterization of Shiga toxin-producing Escherichia coli in feral pigeons. Vet. Microbiol. 2001, 82, 275–283. [Google Scholar] [CrossRef]
- Perna, N.T.; Plunkett, G., 3rd; Burland, V.; Mau, B.; Glasner, J.D.; Rose, D.J.; Mayhew, G.F.; Evans, P.S.; Gregor, J.; Kirkpatrick, H.A.; et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 2001, 409, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, A.W.; Bielaszewská, M.; Zhang, W.L.; Pulz, M.; Kuczius, T.; Ammon, A.; Karch, H. Escherichia coli harboring Shiga toxin 2 gene variants: Frequency and association with clinical symptoms. J. Infect. Dis. 2002, 185, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Persson, S.; Olsen, K.E.P.; Ethelberg, S.; Scheutz, F. Subtyping typing method for Escherichia coli Shiga toxin (Verocytotoxin) 2 variants and correlations to clinical manifestations. J. Clin. Microbiol. 2007, 45, 2020–2024. [Google Scholar] [CrossRef]
- Scheutz, F.; Teel, L.D.; Beutin, L.; Piérard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S.; et al. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef] [PubMed]
- Southwood, T.R.E. Ecological Methods, 2nd ed.; Chapman and Hall Ltd.: New York, NY, USA, 1978; pp. 420–455. [Google Scholar]
- Diniz-Filho, J.A.F.; Bini, L.M.; Hawkins, B.A. Spatial autocorrelation and red herrings in geographical ecology. Glob. Ecol. Biogeogr. 2003, 12, 53–64. [Google Scholar] [CrossRef]
- Zuur, A.F.; Ieno, E.N.; Smith, G.M. Analysing Ecological Data; Springer: New York, NY, USA, 2007. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014; Available online: http://www.R-project.org/ (accessed on 1 February 2016).
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package. R Package Version 2.3-0. 2015. Available online: http://CRAN.R-project.org/package=vegan) (accessed on 1 February 2016).
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Zar, J.H. Biostatistical Analysis; Pearson Prentice Hall: Upper Saddle River, NJ, USA, 2010. [Google Scholar]
- Livermore, D.M.; Winstanley, T.G.; Shannon, K.P. Interpretative reading: Recognizing the unusual and inferring resistance mechanisms from resistance phenotypes. J. Antimicrob. Chemother. 2001, 48, 87–102. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Goatcher, L.J.; Barrett, M.W.; Coleman, R.N.; Hawley, A.W.; Qureshi, A.A. A study of predominant aerobic microflora of black bears (Ursus americanus) and grizzly bears (Ursus arctos) in northwestern Alberta. Can. J. Microbiol. 1987, 33, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Suchodolski, J.S.; Camacho, J.; Steiner, J.M. Analysis of bacterial diversity in the canine duodenum, jejunum, ileum, and colon by comparative 16S rRNA gene analysis. FEMS Microbiol. Ecol. 2008, 66, 567–578. [Google Scholar] [CrossRef]
- Oliveira, M.; Sales-Luís, T.; Duarte, A.; Nunes, S.F.; Carneiro, C.; Tenreiro, T.; Tenreiro, R.; Santos-Reis, M.; Tavares, L.; Vilela, C.L. First assessment of microbial diversity in faecal microflora of Eurasian otter (Lutra lutra Linnaeus, 1758) in Portugal. Eur. J. Wildl. Res. 2008, 54, 245–252. [Google Scholar] [CrossRef]
- Zentek, J.; Marquart, B.; Pietrzak, T.; Ballèvre, O.; Rochat, F. Dietary effects on bifidobacteria and Clostridium perfringens in the canine intestinal tract. J. Anim. Physiol. Anim. Nutr. 2003, 87, 397–407. [Google Scholar] [CrossRef]
- Zentek, J.; Fricke, S.; Hewicker-Trautwein, M.; Ehinger, B.; Amtsberg, G.; Baums, C. Dietary protein source and manufacturing processes affect macronutrient digestibility, fecal consistency, and presence of fecal Clostridium perfringens in adult dogs. J. Nutr. 2004, 134, 2158S–2161S. [Google Scholar] [CrossRef]
- Poeta, P.; Costa, D.; Sáenz, Y.; Klibi, N.; Ruiz-Larrea, F.; Rodrigues, J.; Torres, C. Characterization of antibiotic resistance genes and virulence factors in faecal Enterococci of wild animals in Portugal. J. Vet. Med. Ser. B 2005, 52, 396–402. [Google Scholar] [CrossRef]
- Rosalino, L.M.; Basto, M.; Sales-Luís, T.; Pedroso, N.; Tavares, L.; Vilela, C.L.; Oliveira, M. Bacterial diversity in faecal microbiota of badgers (Meles meles Linnaeus, 1758) in Portugal. In Animal Diversity, Natural History and Conservation; Gupta, V.K., Verma, A.K., Eds.; Daya Publishing House: New Delhi, India, 2013; pp. 1–17. [Google Scholar]
- Semedo-Lemsaddek, T.; Nóbrega, C.S.; Ribeiro, T.; Pedroso, N.M.; Sales-Luís, T.; Lemsaddek, A.; Tenreiro, R.; Tavares, L.; Vilela, C.L.; Oliveira, M. Virulence traits and antibiotic resistance among enterococci isolated from Eurasian otter (Lutra lutra). Vet. Microbiol. 2013, 163, 378–382. [Google Scholar] [CrossRef]
- Millán, J.; Candela, M.G.; Palomares, F.; Cubero, M.J.; Rodríguez, A.; Barral, M.; de la Fuente, J.; Almería, S.; León-Vizcaíno, L. Disease threats to the endangered Iberian lynx (Lynx pardinus). Vet. J. 2009, 182, 114–124. [Google Scholar] [CrossRef]
- Chen, L.; Liu, M.; Zhu, J.; Gao, Y.; Sha, W.; Ding, H.; Jiang, W.; Shenping, W. Age, Gender, and Feeding Environment Influence Fecal Microbial Diversity in Spotted Hyenas (Crocuta crocuta). Curr. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- de la Fe, C.; Rodríguez, J.M.; Ramírez, G.A.; Hervás, J.; Gil, J.; Poveda, J.B. Sudden death associated with Clostridium sordellii in captive lions (Panthera leo). Vet. Pathol. 2006, 43, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Robert, N.; Walzer, C. Pathological disorders in captive cheetahs. In Iberian Lynx Ex-Situ Conservation: An Interdisciplinary Approach; Vargas, A., Breitenmoser, C., Breitenmoser, U., Eds.; Fundación Biodiversidad: Madrid, Spain, 2009; pp. 265–272. [Google Scholar]
- Hoover, J.P.; Tyler, R.D. Renal function and fractional clearances of American river otters (Lutra canadensis). J. Wildl. Dis. 1986, 22, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Hancox, M. Parasites and infectious diseases of the Eurasian badger (Meles meles L.): A review. Mammal Rev. 1980, 10, 151–162. [Google Scholar] [CrossRef]
- Aarestrup, F.M.; Wegener, H.C.; Collignon, P. Resistance in bacteria of the food chain: Epidemiology and control strategies. Expert Rev. Anti-Infect. Ther. 2008, 6, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, A.; Igrejas, G.; Radhouani, H.; Estepa, V.; Pacheco, R.; Monteiro, R.; Brito, F.; Guerra, A.; Petrucci-Fonseca, F.; Torres, C.; et al. Iberian wolf as a reservoir of extended-spectrum beta- Lactamase- Producing Escherichia coli of the TEM, SHV, and CTX-M Groups. Microb. Drug Resist. 2012, 18, 215–219. [Google Scholar]
- Simões, R.; Ferreira, C.; Gonçalves, J.; Álvares, F.; Rio-Maior, H.; Roque, S.; Brandão, R.; Costa, P.M. Occurrence of virulence genes in multidrug-resistant Escherichia coli isolates from Iberian wolves (Canis lupus signatus) in Portugal. Eur. J. Wildl. Res. 2012, 58, 677–684. [Google Scholar]
- Osterblad, M.; Norrdahl, K.; Korpimaki, E.; Huovinen, P. How wild are wild mammals? Nature 2001, 409, 37–38. [Google Scholar] [CrossRef]
- Wright, G.D. Antibiotic resistance in the environment: A link to the clinic? Curr. Opin. Microbiol. 2010, 13, 589–594. [Google Scholar] [CrossRef]
- Thaller, M.C.; Migliore, L.; Marquez, C.; Tapia, W.; Cedeño, V.; Rossolini, G.M.; Gentile, G. Tracking acquired antibiotic resistance in commensal bacteria of Galápagos land iguanas: No man, no resistance. PLoS ONE 2010, 5, e8989. [Google Scholar] [CrossRef]
- Brownstein, D.; Miller, M.A.; Oates, S.C.; Byrne, B.A.; Jang, S.; Murray, M.J.; Jessup, D.A.; Gill, V.A. Antimicrobial susceptibility of bacterial isolates from sea otters (Enhydra lutris). J. Wildl. Dis. 2011, 47, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Azcón, J.M.B.; Duperón, E.B. Carnívoros Ibéricos; Colegio Oficial de Biólogos de Andalucía: Granada, Spain, 1999. [Google Scholar]
- Palomares, F. Site fidelity and effects of body mass on home-range size of Egyptian mongooses. Can. J. Zool. 1994, 72, 465–469. [Google Scholar] [CrossRef]
- Palomares, F. Herpestes ichneumon (Linnaeus, 1758). In Atlas y Libro Rojo de los Mamíferos Terrestres de España; Palomo, L.J., Gisbert, J., Blanco, J.C., Eds.; Dirección General para la Biodiversidad-SECEM-SECEMU: Madrid, Spain, 2007; pp. 327–329. [Google Scholar]
- Duarte, M.D.; Henriques, A.M.; Barros, S.C.; Fagulha, T.; Mendonça, P.; Carvalho, P.; Monteiro, M.; Fevereiro, M.; Basto, M.P.; Rosalino, L.M.; et al. Snapshot of viral infections in wild carnivores reveals ubiquity of parvovirus and susceptibility of Egyptian mongoose to Feline Panleukopenia Virus. PLoS ONE 2013, 8, e59399. [Google Scholar] [CrossRef] [PubMed]
- Cunha, M.V.; Rosalino, L.M.; Leão, C.; Bandeira, V.; Fonseca, C.; Botelho, A.; Reis, A.C. Ecological drivers of Mycobacterium avium subsp. paratuberculosis detection in mongoose (Herpestes ichneumon) using IS900 as proxy. Sci. Rep. 2020, 10, 860. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Phylum | Bacterial Phenotypic Species (no. Isolates) | PEN | AMO | AMC | OXA | CFT | CFP | STR | SPE | KAN | GEN | APR | CMP | TET | DOT |
| Firmicutes | Aerococcus viridans (n = 2) | 100 | 100 | 100 | 100 | 0 | 0 | 100 | 0 | 100 | 0 | 0 | 0 | 100 | 0 |
| Firmicutes | Bacillus cereus (n = 2) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 50 | 50 | 50 | 50 | 0 | 100 | 50 |
| Firmicutes | Bacillus licheniformis (n = 1) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 100 | 100 |
| Firmicutes | Clostridium clostridioforme (n = 7) | 100 | 0 | 0 | 0 | 0 | 0 | 100 | 0 | 100 | 100 | 0 | 0 | 0 | 0 |
| Firmicutes | Clostridium falax (n = 1) | 100 | 0 | 0 | 0 | 0 | 0 | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 |
| Firmicutes | Clostridium perfringens (n = 7) | 43 | 0 | 0 | 0 | 29 | 0 | 100 | 71 | 100 | 100 | 100 | 0 | 29 | 0 |
| Firmicutes | Clostridium sordellii (n = 24) | 88 | 0 | 0 | 75 | 0 | 0 | 100 | 96 | 100 | 100 | 92 | 75 | 79 | 0 |
| Firmicutes | Clostridium tertium (n = 1) | 100 | 0 | 0 | 0 | 0 | 0 | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 0 |
| Actinobacteria | Corynebacterium striatum (n = 1) | 100 | 100 | 100 | 100 | 0 | 0 | 100 | 0 | 100 | 0 | 0 | 0 | 100 | 0 |
| Firmicutes | Enterococcus avium (n = 1) | 100 | 0 | 0 | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 0 | 0 | 0 |
| Firmicutes | Enterococcus casseliflavus (n = 2) | 100 | 0 | 0 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 0 |
| Firmicutes | Enterococcus faecalis (n = 4) | 100 | 25 | 25 | 100 | 75 | 75 | 100 | 75 | 100 | 75 | 75 | 75 | 25 | 0 |
| Firmicutes | Enterococcus faecium (n = 4) | 100 | 0 | 0 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 0 |
| Firmicutes | Gemella haemolysans (n = 1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 100 | 100 |
| Actinobacteria | Microbacterium spp. (n = 1) | 100 | 100 | 100 | 100 | 0 | 0 | 100 | 0 | 100 | 0 | 0 | 0 | 100 | 0 |
| Actinobacteria | Propionibacterium avidum (n = 2) | 0 | 0 | 0 | 100 | 0 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Proteobacteria | Brevundimonas vesicularis (n = 1) | 100 | 100 | 100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Proteobacteria | Escherichia coli (n = 7) | 100 | 43 | 0 | 100 | 100 | 0 | 57 | 0 | 57 | 57 | 57 | 57 | 0 | 0 |
| Proteobacteria | Moellerella wisconcensis (n = 1) | 100 | 100 | 0 | 100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Proteobacteria | Pseudomonas fluorescens (n = 1) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 0 | 0 | 100 | 0 | 0 |
| Proteobacteria | Pseudomonas putida (n = 6) | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 0 | 0 | 0 | 83 | 0 | 0 |
| Proteobacteria | Salmonella spp. (n = 3) | 100 | 100 | 100 | 100 | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Proteobacteria | Sphingomonas paucimobilis (n = 1) | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Phylum | Bacterial Phenotypic Species (no. Isolates) | ERY | LIN | PRI | TYL | COL | TSU | SUL | FLU | OXO | ENR | FUR | FUC | RFA | MTR |
| Firmicutes | Aerococcus viridans (n = 2) | 0 | 100 | 0 | 0 | 100 | 0 | 100 | 100 | 100 | 100 | 0 | 0 | 0 | 100 |
| Firmicutes | Bacillus cereus (n = 2) | 0 | 100 | 0 | 0 | 100 | 100 | 100 | 50 | 50 | 50 | 0 | 100 | 0 | 100 |
| Firmicutes | Bacillus licheniformis (n = 1) | 0 | 100 | 100 | 100 | 100 | 0 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Firmicutes | Clostridium clostridioforme (n = 7) | 0 | 100 | 0 | 0 | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 0 | 0 |
| Firmicutes | Clostridium falax (n = 1) | 100 | 100 | 0 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 100 | 100 |
| Firmicutes | Clostridium perfringens (n = 7) | 57 | 43 | 0 | 0 | 100 | 14 | 14 | 43 | 43 | 100 | 0 | 0 | 0 | 0 |
| Firmicutes | Clostridium sordellii (n = 24) | 0 | 96 | 0 | 0 | 100 | 83 | 88 | 100 | 100 | 100 | 0 | 75 | 0 | 79 |
| Firmicutes | Clostridium tertium (n = 1) | 100 | 100 | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Actinobacteria | Corynebacterium striatum (n = 1) | 0 | 100 | 0 | 0 | 100 | 0 | 100 | 100 | 100 | 100 | 0 | 0 | 0 | 100 |
| Firmicutes | Enterococcus avium (n = 1) | 0 | 0 | 0 | 0 | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 0 | 100 |
| Firmicutes | Enterococcus casseliflavus (n = 2) | 100 | 100 | 0 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 100 |
| Firmicutes | Enterococcus faecalis (n = 4) | 75 | 100 | 75 | 75 | 100 | 75 | 100 | 100 | 100 | 100 | 75 | 0 | 50 | 100 |
| Firmicutes | Enterococcus faecium (n = 4) | 100 | 100 | 0 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 25 | 0 | 100 | 100 |
| Firmicutes | Gemella haemolysans (n = 1) | 0 | 100 | 0 | 0 | 100 | 0 | 100 | 100 | 100 | 100 | 0 | 0 | 0 | 100 |
| Actinobacteria | Microbacterium spp. (n = 1) | 0 | 100 | 0 | 0 | 100 | 0 | 100 | 100 | 100 | 100 | 0 | 0 | 0 | 100 |
| Actinobacteria | Propionibacterium avidum (n = 2) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Proteobacteria | Brevundimonas vesicularis (n = 1) | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 100 | 100 | 0 | 0 | 0 | 0 | 100 |
| Proteobacteria | Escherichia coli (n = 7) | 100 | 100 | 100 | 100 | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 100 | 100 | 100 |
| Proteobacteria | Moellerella wisconcensis (n = 1) | 100 | 100 | 100 | 100 | 100 | 0 | 100 | 0 | 0 | 0 | 100 | 100 | 0 | 100 |
| Proteobacteria | Pseudomonas fluorescens (n = 1) | 100 | 100 | 100 | 100 | 0 | 0 | 0 | 100 | 100 | 100 | 100 | 100 | 0 | 100 |
| Proteobacteria | Pseudomonas putida (n = 6) | 100 | 100 | 100 | 100 | 0 | 83 | 100 | 83 | 0 | 0 | 100 | 100 | 83 | 100 |
| Proteobacteria | Salmonella spp. (n = 3) | 100 | 100 | 100 | 100 | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 100 | 100 | 100 |
| Proteobacteria | Sphingomonas paucimobilis (n = 1) | 100 | 100 | 100 | 100 | 0 | 0 | 100 | 0 | 0 | 0 | 100 | 100 | 0 | 100 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cunha, M.V.; Albuquerque, T.; Themudo, P.; Fonseca, C.; Bandeira, V.; Rosalino, L.M. The Gut Microbiota of the Egyptian Mongoose as an Early Warning Indicator of Ecosystem Health in Portugal. Int. J. Environ. Res. Public Health 2020, 17, 3104. https://doi.org/10.3390/ijerph17093104
Cunha MV, Albuquerque T, Themudo P, Fonseca C, Bandeira V, Rosalino LM. The Gut Microbiota of the Egyptian Mongoose as an Early Warning Indicator of Ecosystem Health in Portugal. International Journal of Environmental Research and Public Health. 2020; 17(9):3104. https://doi.org/10.3390/ijerph17093104
Chicago/Turabian StyleCunha, Mónica V., Teresa Albuquerque, Patrícia Themudo, Carlos Fonseca, Victor Bandeira, and Luís M. Rosalino. 2020. "The Gut Microbiota of the Egyptian Mongoose as an Early Warning Indicator of Ecosystem Health in Portugal" International Journal of Environmental Research and Public Health 17, no. 9: 3104. https://doi.org/10.3390/ijerph17093104
APA StyleCunha, M. V., Albuquerque, T., Themudo, P., Fonseca, C., Bandeira, V., & Rosalino, L. M. (2020). The Gut Microbiota of the Egyptian Mongoose as an Early Warning Indicator of Ecosystem Health in Portugal. International Journal of Environmental Research and Public Health, 17(9), 3104. https://doi.org/10.3390/ijerph17093104