Fetal Cerebral Artery Mitochondrion as Target of Prenatal Alcohol Exposure


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
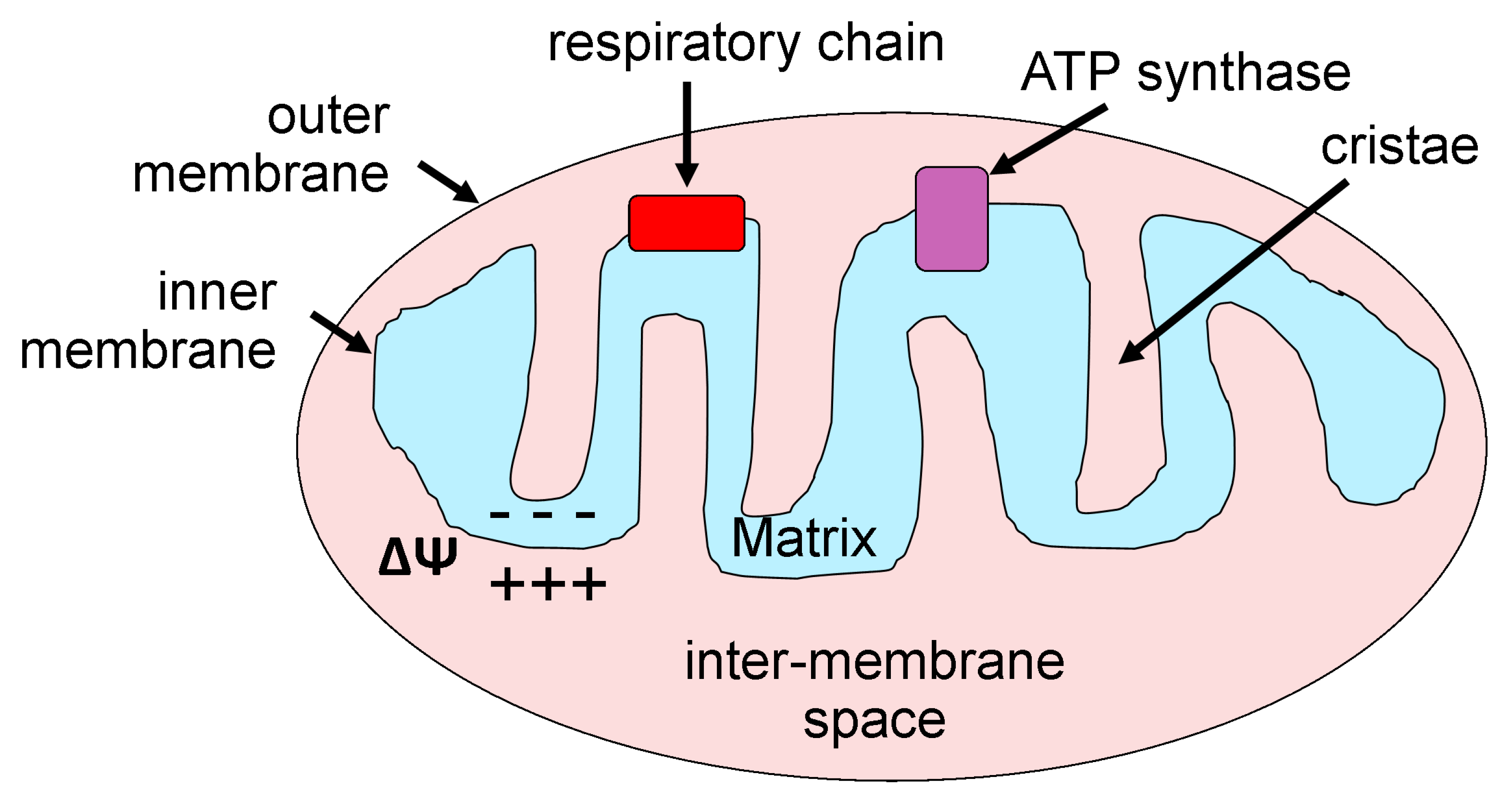
2. Mitochondria: Basic Morphology and Function
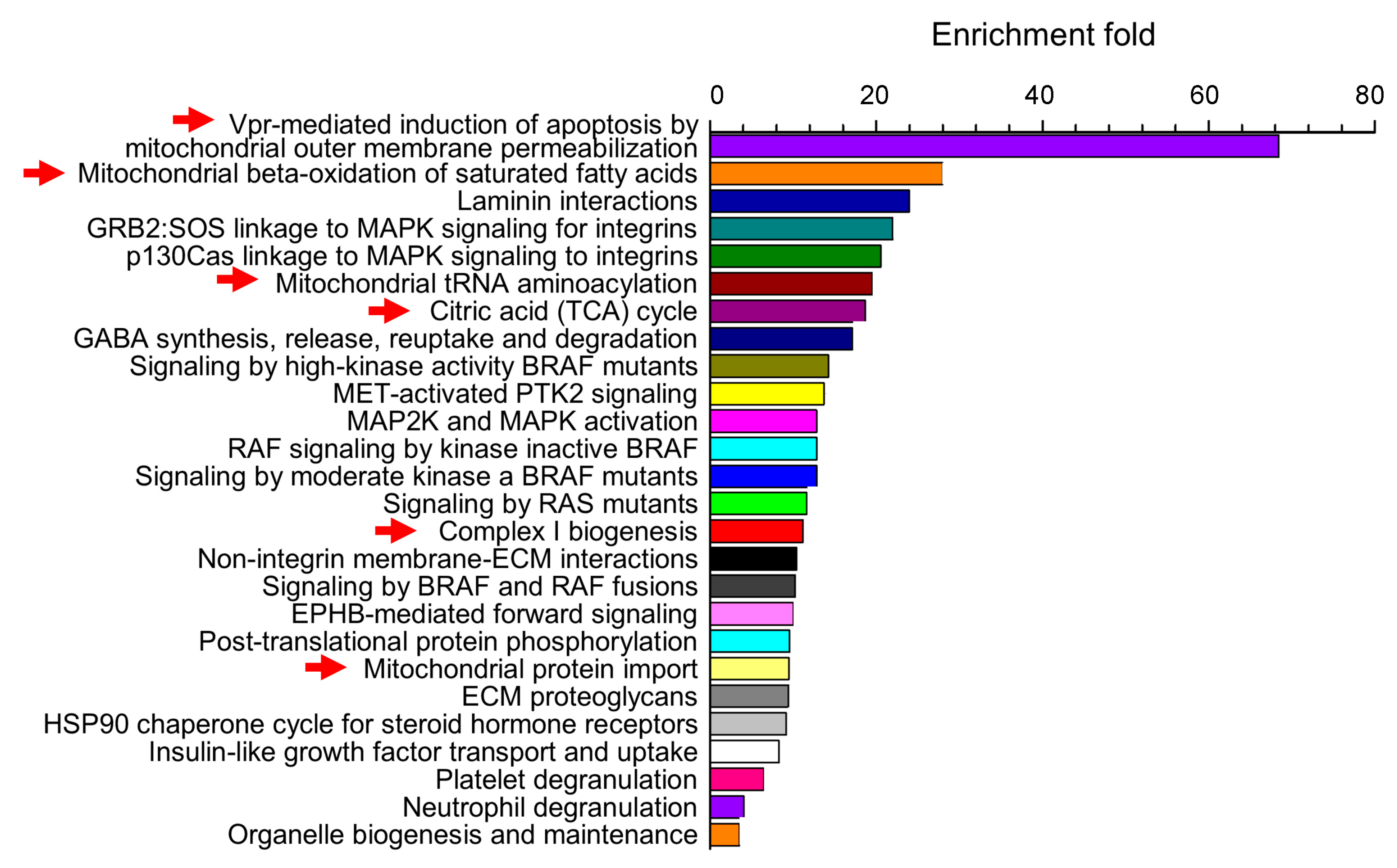
3. Cerebrovascular Mitochondria
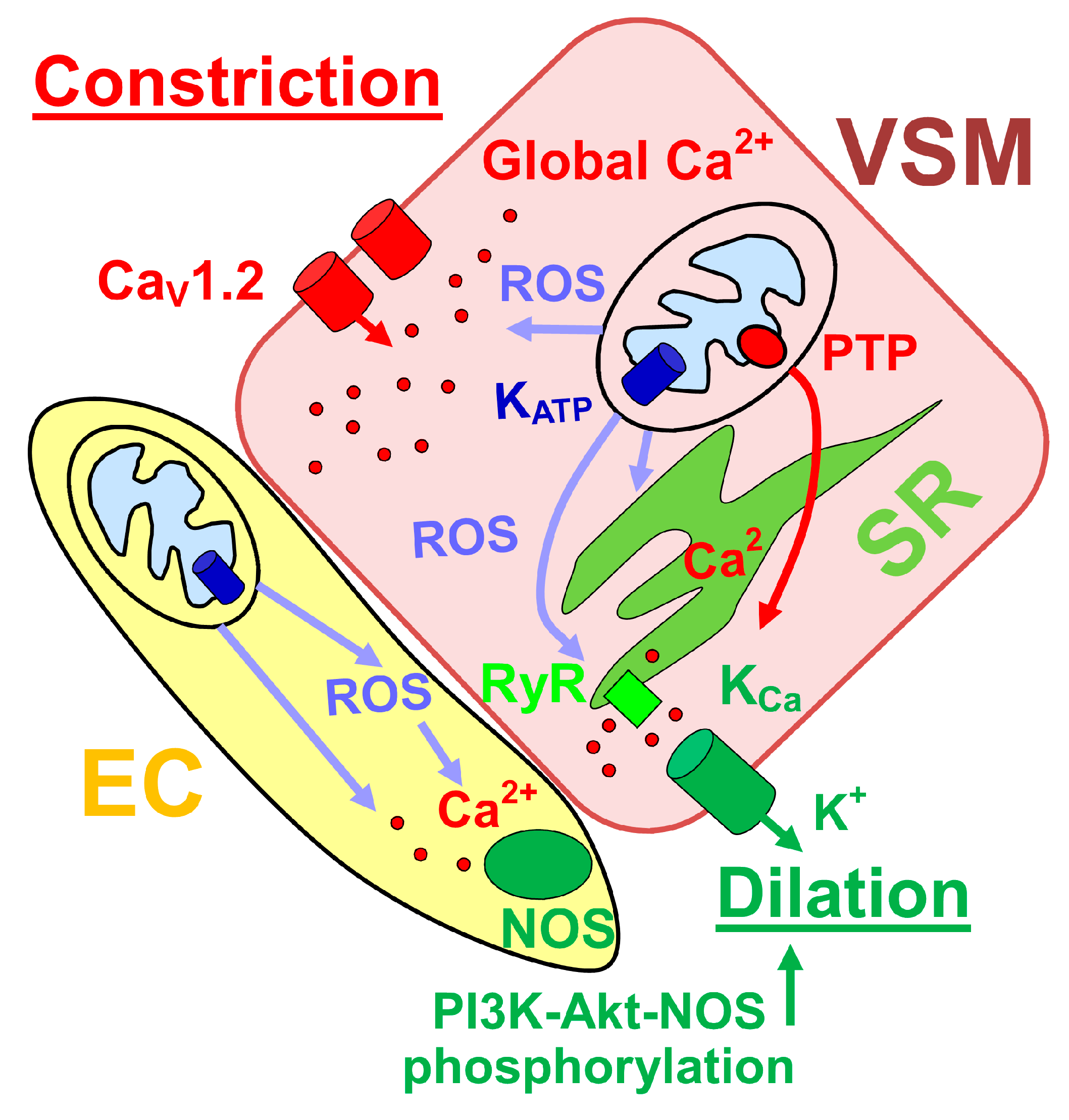
4. Mitochondrial Function and Control of Cerebral Artery Diameter
5. Mitochondria during Fetal Development
6. Alcohol Modifications in Fetal Mitochondria Morphology and Function
7. Alcohol and Fetal Cerebrovascular Mitochondria
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADH | alcohol dehydrogenase |
| ALDH | aldehyde dehydrogenase |
| AngII | angiotensin II |
| ATP | adenosine triphosphate |
| BAL | blood alcohol level |
| BKCa | voltage- and Ca2+-gated potassium channels of large conductance |
| EC | endothelial cell |
| FAS | fetal alcohol syndrome |
| FASDs | fetal alcohol spectrum disorders |
| GPx | glutathione peroxidase |
| HNE | 4-hydroxynonenal |
| MCA | middle cerebral artery |
| MCU | mitochondrial Ca2+ uniporter |
| MnSOD | manganese superoxide dismutase |
| PTP | permeability transition pore |
| NAD+ | nicotinamide adenine dinucleotide |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NOS | nitric oxide synthase |
| SR | sarcoplasmic reticulum |
| PI3K | phophoinositide-3 kinase |
| ROS | reactive oxygen species |
| RyR | ryanodine receptor |
| VSM | vascular smooth muscle |
References
- Ferreira, M.P.; Willoughby, D. Alcohol consumption: The good, the bad, and the indifferent. Appl. Physiol. Nutr. Metab. 2008, 33, 12–20. [Google Scholar] [CrossRef]
- Popova, S.; Lange, S.; Probst, C.; Gmel, G.; Rehm, J. Estimation of national, regional, and global prevalence of alcohol use during pregnancy and fetal alcohol syndrome: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e290–e299. [Google Scholar] [CrossRef]
- Popova, S.; Lange, S.; Probst, C.; Gmel, G.; Rehm, J. Global prevalence of alcohol use and binge drinking during pregnancy, and fetal alcohol spectrum disorder. Biochem. Cell Biol. 2018, 96, 237–240. [Google Scholar] [CrossRef]
- Pierce, D.R.; West, J.R. Blood alcohol concentration: A critical factor for producing fetal alcohol effects. Alcohol 1986, 3, 269–272. [Google Scholar] [CrossRef]
- Maier, S.E.; West, J.R. Drinking patterns and alcohol-related birth defects. Alcohol Res. Health 2001, 25, 168–174. [Google Scholar]
- May, P.A.; Gossage, J.P. Maternal risk factors for fetal alcohol spectrum disorders: Not as simple as it might seem. Alcohol Res. Health 2011, 34, 15–26. [Google Scholar] [PubMed]
- Riley, E.P.; Infante, M.A.; Warren, K.R. Fetal alcohol spectrum disorders: An overview. Neuropsychol. Rev. 2011, 21, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Burd, L. Fetal alcohol spectrum disorder: Complexity from comorbidity. Lancet 2016, 387, 926–927. [Google Scholar] [CrossRef]
- May, P.A.; Gossage, J.P.; Kalberg, W.O.; Robinson, L.K.; Buckley, D.; Manning, M.; Hoyme, H.E. Prevalence and epidemiologic characteristics of FASD from various research methods with an emphasis on recent in-school studies. Dev. Disabil. Res. Rev. 2009, 15, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Olivier, L.; Curfs, L.M.; Viljoen, D.L. Fetal alcohol spectrum disorders: Prevalence rates in South Africa. S. Afr. Med. J. 2016, 106, S103–S106. [Google Scholar] [CrossRef]
- Roozen, S.; Peters, G.J.; Kok, G.; Townend, D.; Nijhuis, J.; Curfs, L. Worldwide Prevalence of Fetal Alcohol Spectrum Disorders: A Systematic Literature Review Including Meta-Analysis. Alcohol. Clin. Exp. Res. 2016, 40, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.; Probst, C.; Gmel, G.; Rehm, J.; Burd, L.; Popova, S. Global Prevalence of Fetal Alcohol Spectrum Disorder Among Children and Youth: A Systematic Review and Meta-analysis. JAMA Pediatr. 2017, 171, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Murawski, N.J.; Moore, E.M.; Thomas, J.D.; Riley, E.P. Advances in Diagnosis and Treatment of Fetal Alcohol Spectrum Disorders: From Animal Models to Human Studies. Alcohol Res. 2015, 37, 97–108. [Google Scholar] [PubMed]
- Wilhoit, L.F.; Scott, D.A.; Simecka, B.A. Fetal Alcohol Spectrum Disorders: Characteristics, Complications, and Treatment. Community Ment. Health J. 2017, 53, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.N.; Anderson, G.D. Pharmacokinetic and pharmacodynamic drug interactions with ethanol (alcohol). Clin. Pharm. 2014, 53, 1115–1136. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Koob, G.F. Titrating Tipsy Targets: The Neurobiology of Low-Dose Alcohol. Trends Pharm. Sci. 2017, 38, 556–568. [Google Scholar] [CrossRef]
- Roberto, M.; Varodayan, F.P. Synaptic targets: Chronic alcohol actions. Neuropharmacology 2017, 122, 85–99. [Google Scholar] [CrossRef]
- Mattson, S.N.; Schoenfeld, A.M.; Riley, E.P. Teratogenic effects of alcohol on brain and behavior. Alcohol Res. Health 2001, 25, 185–191. [Google Scholar]
- Caputo, C.; Wood, E.; Jabbour, L. Impact of fetal alcohol exposure on body systems: A systematic review. Birth Defects Res. C Embryo Today 2016, 108, 174–180. [Google Scholar] [CrossRef]
- Bukiya, A.N.; Dopico, A.M. Fetal Cerebral Circulation as Target of Maternal Alcohol Consumption. Alcohol. Clin. Exp. Res. 2018, 42, 1006–1018. [Google Scholar] [CrossRef]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The neurovascular unit—Concept review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef]
- Aliev, G.; Seyidova, D.; Lamb, B.T.; Obrenovich, M.E.; Siedlak, S.L.; Vinters, H.V.; Friedland, R.P.; LaManna, J.C.; Smith, M.A.; Perry, G. Mitochondria and vascular lesions as a central target for the development of Alzheimer's disease and Alzheimer disease-like pathology in transgenic mice. Neurol. Res. 2003, 25, 665–674. [Google Scholar] [CrossRef]
- Zhu, X.; Smith, M.A.; Honda, K.; Aliev, G.; Moreira, P.I.; Nunomura, A.; Casadesus, G.; Harris, P.L.; Siedlak, S.L.; Perry, G. Vascular oxidative stress in Alzheimer disease. J. Neurol. Sci. 2007, 257, 240–246. [Google Scholar] [CrossRef]
- Dai, D.F.; Rabinovitch, P.S.; Ungvari, Z. Mitochondria and cardiovascular aging. Circ. Res. 2012, 110, 1109–1124. [Google Scholar] [CrossRef]
- Carvalho, C.; Moreira, P.I. Oxidative Stress: A Major Player in Cerebrovascular Alterations Associated to Neurodegenerative Events. Front. Physiol. 2018, 9, 806. [Google Scholar] [CrossRef]
- Bisen, S.; Kakhniashvili, D.; Johnson, D.L.; Bukiya, A.N. Proteomic Analysis of Baboon Cerebral Artery Reveals Potential Pathways of Damage by Prenatal Alcohol Exposure. Mol. Cell. Proteomics 2019, 18, 294–307. [Google Scholar] [CrossRef]
- Henderson, G.I.; Chen, J.J.; Schenker, S. Ethanol, oxidative stress, reactive aldehydes, and the fetus. Front. Biosci. 1999, 4, D541–D550. [Google Scholar]
- Nyquist-Battie, C.; Freter, M. Cardiac mitochondrial abnormalities in a mouse model of the fetal alcohol syndrome. Alcohol. Clin. Exp. Res. 1988, 12, 264–267. [Google Scholar] [CrossRef]
- Busija, D.W.; Katakam, P.V. Mitochondrial mechanisms in cerebral vascular control: Shared signaling pathways with preconditioning. J. Vasc. Res. 2014, 51, 175–189. [Google Scholar] [CrossRef]
- van der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell Biology of the Mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Yoon, Y. Mitochondrial fission and fusion. Biochem. Soc. Trans. 2016, 44, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef]
- Lee, S.; Jeong, S.Y.; Lim, W.C.; Kim, S.; Park, Y.Y.; Sun, X.; Youle, R.J.; Cho, H. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J. Biol. Chem. 2007, 282, 22977–22983. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Kijima, K.; Numakura, C.; Izumino, H.; Umetsu, K.; Nezu, A.; Shiiki, T.; Ogawa, M.; Ishizaki, Y.; Kitamura, T.; Shozawa, Y.; et al. Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum. Genet. 2005, 116, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.N.; Coetzee, W.A. KATP Channels in the Cardiovascular System. Physiol. Rev. 2016, 96, 177–252. [Google Scholar] [CrossRef]
- Krabbendam, I.E.; Honrath, B.; Culmsee, C.; Dolga, A.M. Mitochondrial Ca2+-activated K+ channels and their role in cell life and death pathways. Cell Calcium 2018, 69, 101–111. [Google Scholar] [CrossRef]
- Adebiyi, A.; McNally, E.M.; Jaggar, J.H. Vasodilation induced by oxygen/glucose deprivation is attenuated in cerebral arteries of SUR2 null mice. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1360–H1368. [Google Scholar] [CrossRef]
- Busija, D.W.; Rutkai, I.; Dutta, S.; Katakam, P.V. Role of Mitochondria in Cerebral Vascular Function: Energy Production, Cellular Protection, and Regulation of Vascular Tone. Compr. Physiol. 2016, 6, 1529–1548. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Lambert, A.J.; Brand, M.D. Reactive oxygen species production by mitochondria. Methods Mol. Biol. 2009, 554, 165–181. [Google Scholar]
- Olson, M.L.; Chalmers, S.; McCarron, J.G. Mitochondrial organization and Ca2+ uptake. Biochem. Soc. Trans. 2012, 40, 158–167. [Google Scholar] [CrossRef]
- Blacker, T.S.; Duchen, M.R. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic. Biol. Med. 2016, 100, 53–65. [Google Scholar] [CrossRef]
- Blajszczak, C.; Bonini, M.G. Mitochondria targeting by environmental stressors: Implications for redox cellular signaling. Toxicology 2017, 391, 84–89. [Google Scholar] [CrossRef]
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar]
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef]
- Szymański, J.; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M.; Ziółkowski, W.; Duszyński, J.; Pinton, P.; Dobrzyń, A.; Więckowski, M.R. Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. Int. J. Mol. Sci. 2017, 18, 1576. [Google Scholar] [CrossRef]
- Bernardi, P. Mitochondria in muscle cell death. Ital. J. Neurol. Sci. 1999, 20, 395–400. [Google Scholar] [CrossRef]
- Williams, G.S.; Boyman, L.; Lederer, W.J. Mitochondrial calcium and the regulation of metabolism in the heart. J. Mol. Cell. Cardiol. 2015, 78, 35–45. [Google Scholar] [CrossRef]
- Quintana-Cabrera, R.; Bolaños, J.P. Glutathione and γ-glutamylcysteine in the antioxidant and survival functions of mitochondria. Biochem. Soc. Trans. 2013, 41, 106–110. [Google Scholar] [CrossRef]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharm. 2014, 5, 151. [Google Scholar] [CrossRef]
- Calabrese, G.; Morgan, B.; Riemer, J. Mitochondrial Glutathione: Regulation and Functions. Antioxid. Redox Signal. 2017, 27, 1162–1177. [Google Scholar] [CrossRef]
- Wiesenfeld, M.; Schimpfessel, L.; Crokaert, R. Multiple forms of mitochondrial alcohol dehydrogenase in Saccharomyces cerevisiae. Biochim. Biophys. Acta 1975, 405, 500–512. [Google Scholar] [CrossRef]
- DiFabio, J.; Ji, Y.; Vasiliou, V.; Thatcher, G.R.; Bennett, B.M. Role of mitochondrial aldehyde dehydrogenase in nitrate tolerance. Mol. Pharm. 2003, 64, 1109–1116. [Google Scholar] [CrossRef]
- Crichton, P.G.; Affourtit, C.; Moore, A.L. Identification of a mitochondrial alcohol dehydrogenase in Schizosaccharomyces pombe: New insights into energy metabolism. Biochem. J. 2007, 401, 459–464. [Google Scholar] [CrossRef]
- Chen, C.H.; Sun, L.; Mochly-Rosen, D. Mitochondrial aldehyde dehydrogenase and cardiac diseases. Cardiovasc. Res. 2010, 88, 51–57. [Google Scholar] [CrossRef]
- Manzo-Avalos, S.; Saavedra-Molina, A. Cellular and mitochondrial effects of alcohol consumption. Int. J. Environ. Res. Public Health 2010, 7, 4281–4304. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Du, Y.H.; Tang, Y.B.; Lv, X.F.; Liu, J.; Zhou, J.G.; Guan, Y.Y. ClC-3 chloride channel prevents apoptosis induced by hydrogen peroxide in basilar artery smooth muscle cells through mitochondria dependent pathway. Apoptosis 2011, 16, 468–477. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, Y.; Ma, M.M.; Tang, Y.B.; Zhou, J.G.; Guan, Y.Y. Mitochondria dependent pathway is involved in the protective effect of bestrophin-3 on hydrogen peroxide-induced apoptosis in basilar artery smooth muscle cells. Apoptosis 2013, 18, 556–565. [Google Scholar] [CrossRef]
- Zhang, R.; Ran, H.H.; Cai, L.L.; Zhu, L.; Sun, J.F.; Peng, L.; Liu, X.J.; Zhang, L.N.; Fang, Z.; Fan, Y.Y.; et al. Simulated microgravity-induced mitochondrial dysfunction in rat cerebral arteries. FASEB J. 2014, 28, 2715–2724. [Google Scholar] [CrossRef]
- Ungvari, Z.; Orosz, Z.; Labinskyy, N.; Rivera, A.; Xiangmin, Z.; Smith, K.; Csiszar, A. Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H37–H47. [Google Scholar] [CrossRef] [PubMed]
- Springo, Z.; Tarantini, S.; Toth, P.; Tucsek, Z.; Koller, A.; Sonntag, W.E.; Csiszar, A.; Ungvari, Z. Aging Exacerbates Pressure-Induced Mitochondrial Oxidative Stress in Mouse Cerebral Arteries. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, S.; Saunter, C.D.; Girkin, J.M.; McCarron, J.G. Age decreases mitochondrial motility and increases mitochondrial size in vascular smooth muscle. J. Physiol. 2016, 594, 4283–4295. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, S.; Saunter, C.; Wilson, C.; Coats, P.; Girkin, J.M.; McCarron, J.G. Mitochondrial motility and vascular smooth muscle proliferation. Arter. Thromb. Vasc. Biol. 2012, 32, 3000–3011. [Google Scholar] [CrossRef] [PubMed]
- Rutkai, I.; Dutta, S.; Katakam, P.V.; Busija, D.W. Dynamics of enhanced mitochondrial respiration in female compared with male rat cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1490–H1500. [Google Scholar] [CrossRef]
- Merdzo, I.; Rutkai, I.; Tokes, T.; Sure, V.N.; Katakam, P.V.; Busija, D.W. The mitochondrial function of the cerebral vasculature in insulin-resistant Zucker obese rats. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H830–H838. [Google Scholar] [CrossRef]
- Katakam, P.V.; Domoki, F.; Snipes, J.A.; Busija, A.R.; Jarajapu, Y.P.; Busija, D.W. Impaired mitochondria-dependent vasodilation in cerebral arteries of Zucker obese rats with insulin resistance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R289–R298. [Google Scholar] [CrossRef] [PubMed]
- Merdzo, I.; Rutkai, I.; Sure, V.N.; McNulty, C.A.; Katakam, P.V.; Busija, D.W. Impaired Mitochondrial Respiration in Large Cerebral Arteries of Rats with Type 2 Diabetes. J. Vasc. Res. 2017, 54, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rutkai, I.; Katakam, P.V.; Dutta, S.; Busija, D.W. Sustained mitochondrial functioning in cerebral arteries after transient ischemic stress in the rat: A potential target for therapies. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H958–H966. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rutkai, I.; Merdzo, I.; Wunnava, S.V.; Curtin, G.T.; Katakam, P.V.; Busija, D.W. Cerebrovascular function and mitochondrial bioenergetics after ischemia-reperfusion in male rats. J. Cereb. Blood Flow Metab. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cheranov, S.Y.; Jaggar, J.H. Mitochondrial modulation of Ca2+ sparks and transient KCa currents in smooth muscle cells of rat cerebral arteries. J. Physiol. 2004, 556, 755–771. [Google Scholar] [CrossRef]
- Chaplin, N.L.; Nieves-Cintrón, M.; Fresquez, A.M.; Navedo, M.F.; Amberg, G.C. Arterial Smooth Muscle Mitochondria Amplify Hydrogen Peroxide Microdomains Functionally Coupled to L-Type Calcium Channels. Circ. Res. 2015, 117, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, D.; Xi, Q.; Pfeffer, L.M.; Jaggar, J.H. Mitochondria control functional CaV1.2 expression in smooth muscle cells of cerebral arteries. Circ. Res. 2010, 107, 631–641. [Google Scholar] [CrossRef]
- Jaggar, J.H.; Wellman, G.C.; Heppner, T.J.; Porter, V.A.; Perez, G.J.; Gollasch, M.; Kleppisch, T.; Rubart, M.; Stevenson, A.S.; Lederer, W.J.; et al. Ca2+ channels, ryanodine receptors and Ca(2+)-activated K+ channels: A functional unit for regulating arterial tone. Acta Physiol. Scand. 1998, 164, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, P.; Li, Y. Impaired development of mitochondria plays a role in the central nervous system defects of fetal alcohol syndrome. Birth Defects Res. A Clin. Mol. Teratol. 2005, 73, 83–91. [Google Scholar] [CrossRef]
- Katakam, P.V.; Gordon, A.O.; Sure, V.N.; Rutkai, I.; Busija, D.W. Diversity of mitochondria-dependent dilator mechanisms in vascular smooth muscle of cerebral arteries from normal and insulin-resistant rats. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H493–H503. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, W.A. Multiplicity of effectors of the cardioprotective agent, diazoxide. Pharmacol. Ther. 2013, 140, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Katakam, P.V.; Wappler, E.A.; Katz, P.S.; Rutkai, I.; Institoris, A.; Domoki, F.; Gáspár, T.; Grovenburg, S.M.; Snipes, J.A.; Busija, D.W. Depolarization of mitochondria in endothelial cells promotes cerebral artery vasodilation by activation of nitric oxide synthase. Arter. Thromb. Vasc. Biol. 2013, 33, 752–759. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef]
- Budohoski, K.P.; Czosnyka, M.; Kirkpatrick, P.J.; Smielewski, P.; Steiner, L.A.; Pickard, J.D. Clinical relevance of cerebral autoregulation following subarachnoid haemorrhage. Nat. Rev. Neurol. 2013, 9, 152–163. [Google Scholar] [CrossRef]
- Meng, L.; Hou, W.; Chui, J.; Han, R.; Gelb, A.W. Cardiac Output and Cerebral Blood Flow: The Integrated Regulation of Brain Perfusion in Adult Humans. Anesthesiology 2015, 123, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Szarka, N.; Pabbidi, M.R.; Amrein, K.; Czeiter, E.; Berta, G.; Pohoczky, K.; Helyes, Z.; Ungvari, Z.; Koller, A.; Buki, A.; et al. Traumatic Brain Injury Impairs Myogenic Constriction of Cerebral Arteries: Role of Mitochondria-Derived H2O2 and TRPV4-Dependent Activation of BKca Channels. J. Neurotrauma 2018. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.M.; Kelley, S.W.; Meehan, J.M. Ventricular mitochondrial gene expression during development and following embryonic ethanol exposure. J. Mol. Cell. Cardiol. 1993, 25, 117–131. [Google Scholar] [CrossRef]
- Goyal, R.; Longo, L.D. Metabolic Profiles in Ovine Carotid Arteries with Developmental Maturation and Long-Term Hypoxia. PLoS ONE 2015, 10, e0130739. [Google Scholar] [CrossRef]
- Horton, A.A.; Mills, D.J. Developmental patterns of alcohol dehydrogenase and aldehyde dehydrogenases in homogenates and subcellular fractions of rat liver. Mech. Ageing Dev. 1979, 11, 363–370. [Google Scholar] [CrossRef]
- Semple, B.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sanchis, R.; Guerri, C. Alcohol-metabolizing enzymes in placenta and fetal liver: Effect of chronic ethanol intake. Alcohol. Clin. Exp. Res. 1986, 10, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Devi, B.G.; Henderson, G.I.; Frosto, T.A.; Schenker, S. Effect of acute ethanol exposure on cultured fetal rat hepatocytes: Relation to mitochondrial function. Alcohol. Clin. Exp. Res. 1994, 18, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Rømert, P.; Matthiessen, M.E. Alcohol-induced injury of mitochondria in hepatocytes of mini-pig fetuses. Virchows Arch. A Pathol. Anat. Histopathol. 1983, 399, 299–305. [Google Scholar] [CrossRef]
- Amankwah, K.S.; Weberg, A.D.; Kaufmann, R.C. Ultrastructural changes in preputial neural tissues: Effects of maternal drinking. Early Hum. Dev. 1982, 6, 375–380. [Google Scholar] [CrossRef]
- Ramachandran, V.; Watts, L.T.; Maffi, S.K.; Chen, J.; Schenker, S.; Henderson, G. Ethanol-induced oxidative stress precedes mitochondrially mediated apoptotic death of cultured fetal cortical neurons. J. Neurosci. Res. 2003, 74, 577–588. [Google Scholar] [CrossRef]
- Ramachandran, V.; Perez, A.; Chen, J.; Senthil, D.; Schenker, S.; Henderson, G.I. In utero ethanol exposure causes mitochondrial dysfunction, which can result in apoptotic cell death in fetal brain: A potential role for 4-hydroxynonenal. Alcohol. Clin. Exp. Res. 2001, 25, 862–871. [Google Scholar] [CrossRef]
- Chu, J.; Tong, M.; de la Monte, S.M. Chronic ethanol exposure causes mitochondrial dysfunction and oxidative stress in immature central nervous system neurons. Acta Neuropathol. 2007, 113, 659–673. [Google Scholar] [CrossRef] [PubMed]
- de La Monte, S.M.; Wands, J.R. Mitochondrial DNA damage and impaired mitochondrial function contribute to apoptosis of insulin-stimulated ethanol-exposed neuronal cells. Alcohol. Clin. Exp. Res. 2001, 25, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Xi, Q.; Cheranov, S.Y.; Jaggar, J.H. Mitochondria-derived reactive oxygen species dilate cerebral arteries by activating Ca2+ sparks. Circ. Res. 2005, 97, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Green, C.R.; Watts, L.T.; Kobus, S.M.; Henderson, G.I.; Reynolds, J.N.; Brien, J.F. Effects of chronic prenatal ethanol exposure on mitochondrial glutathione and 8-iso-prostaglandin F2alpha concentrations in the hippocampus of the perinatal guinea pig. Reprod. Fertil. Dev. 2006, 18, 517–524. [Google Scholar] [CrossRef]
- Chen, J.J.; Schenker, S.; Henderson, G.I. 4-hydroxynonenal levels are enhanced in fetal liver mitochondria by in utero ethanol exposure. Hepatology 1997, 25, 142–147. [Google Scholar]
- Nakai, A.; Taniuchi, Y.; Asakura, H.; Oya, A.; Yokota, A.; Koshino, T.; Araki, T. Developmental changes in tolerance to transient intrauterine ischemia in rat cerebral mitochondria. Am. J. Obs. Gynecol. 2001, 184, 731–735. [Google Scholar] [CrossRef]
- Marin-Garcia, J.; Ananthakrishnan, R.; Goldenthal, M.J. Mitochondrial dysfunction after fetal alcohol exposure. Alcohol. Clin. Exp. Res. 1996, 20, 1029–1032. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Wands, J.R. Chronic gestational exposure to ethanol impairs insulin-stimulated survival and mitochondrial function in cerebellar neurons. Cell. Mol. Life Sci. 2002, 59, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Seleverstov, O.; Tobiasz, A.; Jackson, J.S.; Sullivan, R.; Ma, D.; Sullivan, J.P.; Davison, S.; Akkhawattanangkul, Y.; Tate, D.L.; Costello, T.; et al. Maternal alcohol exposure during mid-pregnancy dilates fetal cerebral arteries via endocannabinoid receptors. Alcohol 2017, 61, 51–61. [Google Scholar] [CrossRef]
- Tobiasz, A.M.; Duncan, J.R.; Bursac, Z.; Sullivan, R.D.; Tate, D.L.; Dopico, A.M.; Bukiya, A.N.; Mari, G. The Effect of Prenatal Alcohol Exposure on Fetal Growth and Cardiovascular Parameters in a Baboon Model of Pregnancy. Reprod. Sci. 2018, 25, 1116–1123. [Google Scholar] [CrossRef]
- Simakova, M.; Tobiasz, A.; Sullivan, R.D.; Bisen, S.; Duncan, J.; Sullivan, J.P.; Davison, S.; Tate, D.L.; Barnett, S.; Mari, G.; et al. Gestational age-dependent interplay between endocannabinoid receptors and alcohol in fetal cerebral arteries. J. Drug Alcohol Res. 2019, 8, 1–11. [Google Scholar] [CrossRef]
- Bauer, C. The baboon (Papio sp.) as a model for female reproduction studies. Contraception 2015, 92, 120–123. [Google Scholar] [CrossRef]
- Parnell, S.E.; Ramadoss, J.; Delp, M.D.; Ramsey, M.W.; Chen, W.J.; West, J.R.; Cudd, T.A. Chronic ethanol increases fetal cerebral blood flow specific to the ethanol-sensitive cerebellum under normoxaemic, hypercapnic and acidaemic conditions: Ovine model. Exp. Physiol. 2007, 92, 933–943. [Google Scholar] [CrossRef]
- Coleman, W.B.; Cunningham, C.C. Effect of chronic ethanol consumption on hepatic mitochondrial transcription and translation. Biochim. Biophys. Acta 1991, 1058, 178–186. [Google Scholar] [CrossRef]
- Venkatraman, A.; Landar, A.; Davis, A.J.; Chamlee, L.; Sanderson, T.; Kim, H.; Page, G.; Pompilius, M.; Ballinger, S.; Darley-Usmar, V.; et al. Modification of the mitochondrial proteome in response to the stress of ethanol-dependent hepatotoxicity. J. Biol. Chem. 2004, 279, 22092–22101. [Google Scholar] [CrossRef] [PubMed]
- Rømert, P.; Matthiessen, M.E. Ultrastructural and morphometric study of hepatocytes from near-term minipig fetuses exposed to ethanol in vivo. Acta Anat. 1992, 143, 301–305. [Google Scholar] [PubMed]
- Bake, S.; Gardner, R.; Tingling, J.D.; Miranda, R.C.; Sohrabji, F. Fetal Alcohol Exposure Alters Blood Flow and Neurological Responses to Transient Cerebral Ischemia in Adult Mice. Alcohol. Clin. Exp. Res. 2017, 41, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Cananzi, S.G.; Mayhan, W.G. In Utero Exposure to Alcohol Impairs Reactivity of Cerebral Arterioles and Increases Susceptibility of the Brain to Damage Following Ischemia/Reperfusion in Adulthood. Alcohol. Clin. Exp. Res. 2019. [Google Scholar] [CrossRef]
- Shibazaki, Y.; Nakai, A.; Koshino, T.; Yokoyama, K. Effect of the immunosuppressant drug FK506 on neonatal cerebral mitochondrial function and energy metabolism after transient intrauterine ischemia in rats. Brain Res. 2001, 892, 351–358. [Google Scholar] [CrossRef]
- Watanabe, K.; Wakatsuki, A.; Shinohara, K.; Ikenoue, N.; Yokota, K.; Fukaya, T. Maternally administered melatonin protects against ischemia and reperfusion-induced oxidative mitochondrial damage in premature fetal rat brain. J. Pineal Res. 2004, 37, 276–280. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, D.; Li, G.; Liu, J.; Tian, J.; Fu, F.; Liu, K. Neuroprotective effects of safflor yellow B on brain ischemic injury. Exp. Brain Res. 2007, 177, 533–539. [Google Scholar] [CrossRef]
- Bakken, T.E.; Miller, J.A.; Ding, S.L.; Sunkin, S.M.; Smith, K.A.; Ng, L.; Szafer, A.; Dalley, R.A.; Royall, J.J.; Lemon, T.; et al. A comprehensive transcriptional map of primate brain development. Nature 2016, 535, 367–375. [Google Scholar] [CrossRef]
- Zhu, Y.; Sousa, A.M.M.; Gao, T.; Skarica, M.; Li, M.; Santpere, G.; Esteller-Cucala, P.; Juan, D.; Ferrández-Peral, L.; Gulden, F.O.; et al. Spatiotemporal transcriptomic divergence across human and macaque brain development. Science 2018, 362. [Google Scholar] [CrossRef]
- Nakai, A.; Shibazaki, Y.; Taniuchi, Y.; Oya, A.; Asakura, H.; Koshino, T.; Araki, T. Vitamins ameliorate secondary mitochondrial failure in neonatal rat brain. Pediatr. Neurol. 2002, 27, 30–35. [Google Scholar] [CrossRef]



© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bukiya, A.N. Fetal Cerebral Artery Mitochondrion as Target of Prenatal Alcohol Exposure. Int. J. Environ. Res. Public Health 2019, 16, 1586. https://doi.org/10.3390/ijerph16091586
Bukiya AN. Fetal Cerebral Artery Mitochondrion as Target of Prenatal Alcohol Exposure. International Journal of Environmental Research and Public Health. 2019; 16(9):1586. https://doi.org/10.3390/ijerph16091586
Chicago/Turabian StyleBukiya, Anna N. 2019. "Fetal Cerebral Artery Mitochondrion as Target of Prenatal Alcohol Exposure" International Journal of Environmental Research and Public Health 16, no. 9: 1586. https://doi.org/10.3390/ijerph16091586
APA StyleBukiya, A. N. (2019). Fetal Cerebral Artery Mitochondrion as Target of Prenatal Alcohol Exposure. International Journal of Environmental Research and Public Health, 16(9), 1586. https://doi.org/10.3390/ijerph16091586