Sub-lethal Doses of Polybrominated Diphenyl Ethers, in Vitro, Promote Oxidative Stress and Modulate Molecular Markers Related to Cell Cycle, Antioxidant Balance and Cellular Energy Management

 ,
,  , and
, and
Abstract
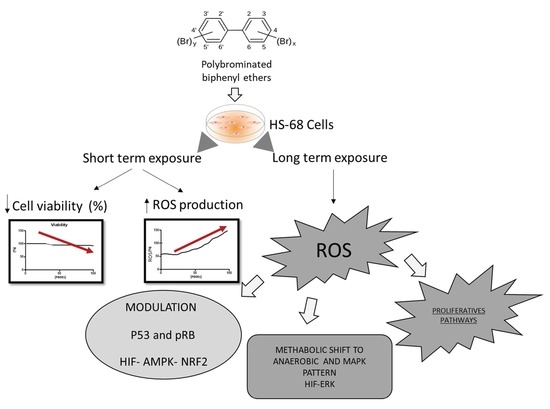
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture Maintenance, Treatment and Cytotoxicity Assay
2.2. Evaluation of Intracellular ROS
2.3. Evaluation of Biomolecular Markers by Immunoblotting
2.4. Image Acquisition
2.5. Statistical Analysis
3. Results
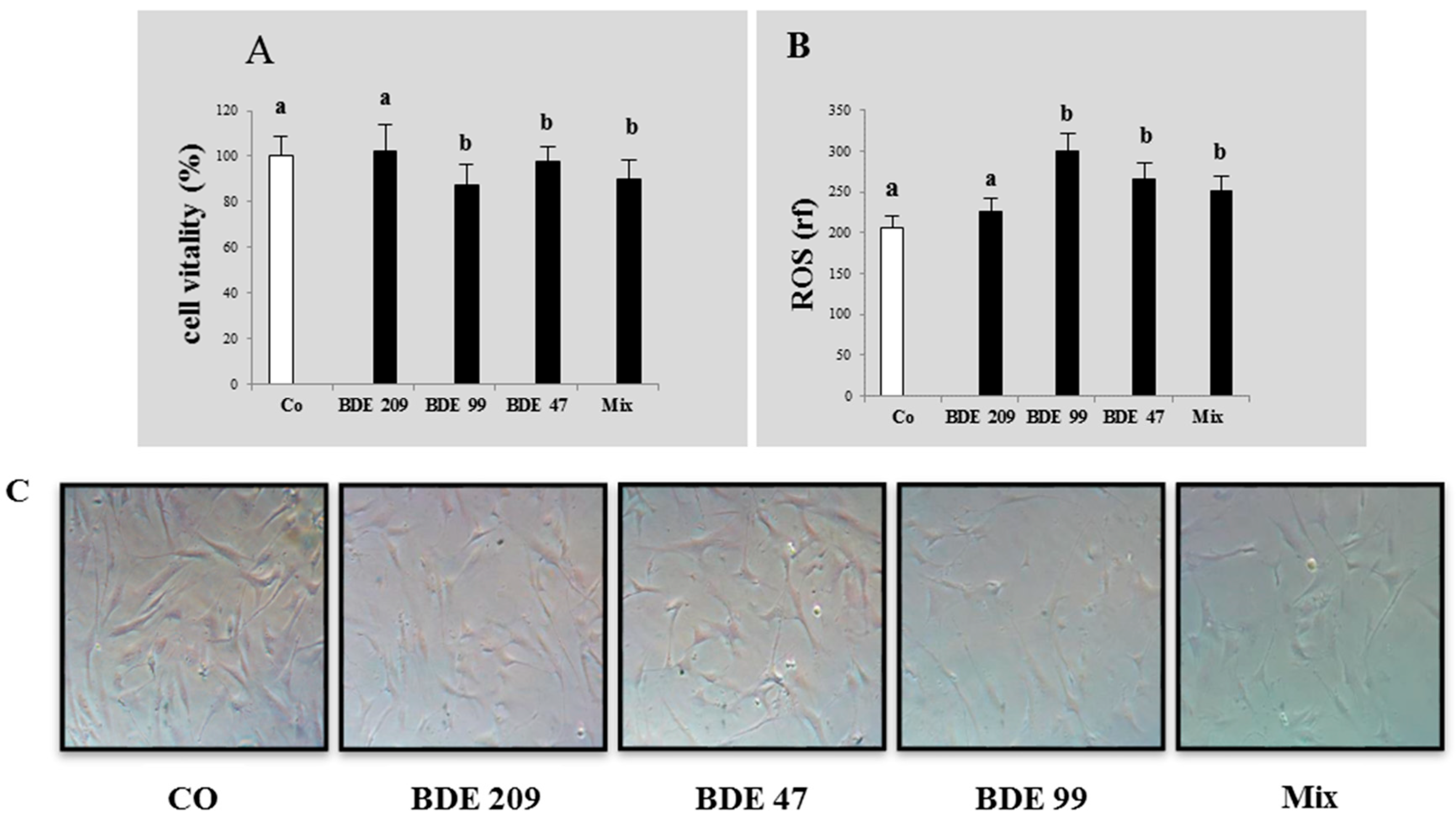
3.1. Effects of PBDE on Cytotoxicity and ROS Production
3.2. Effects of PBDE on Biomolecular Markers: p53, pRB, PARP
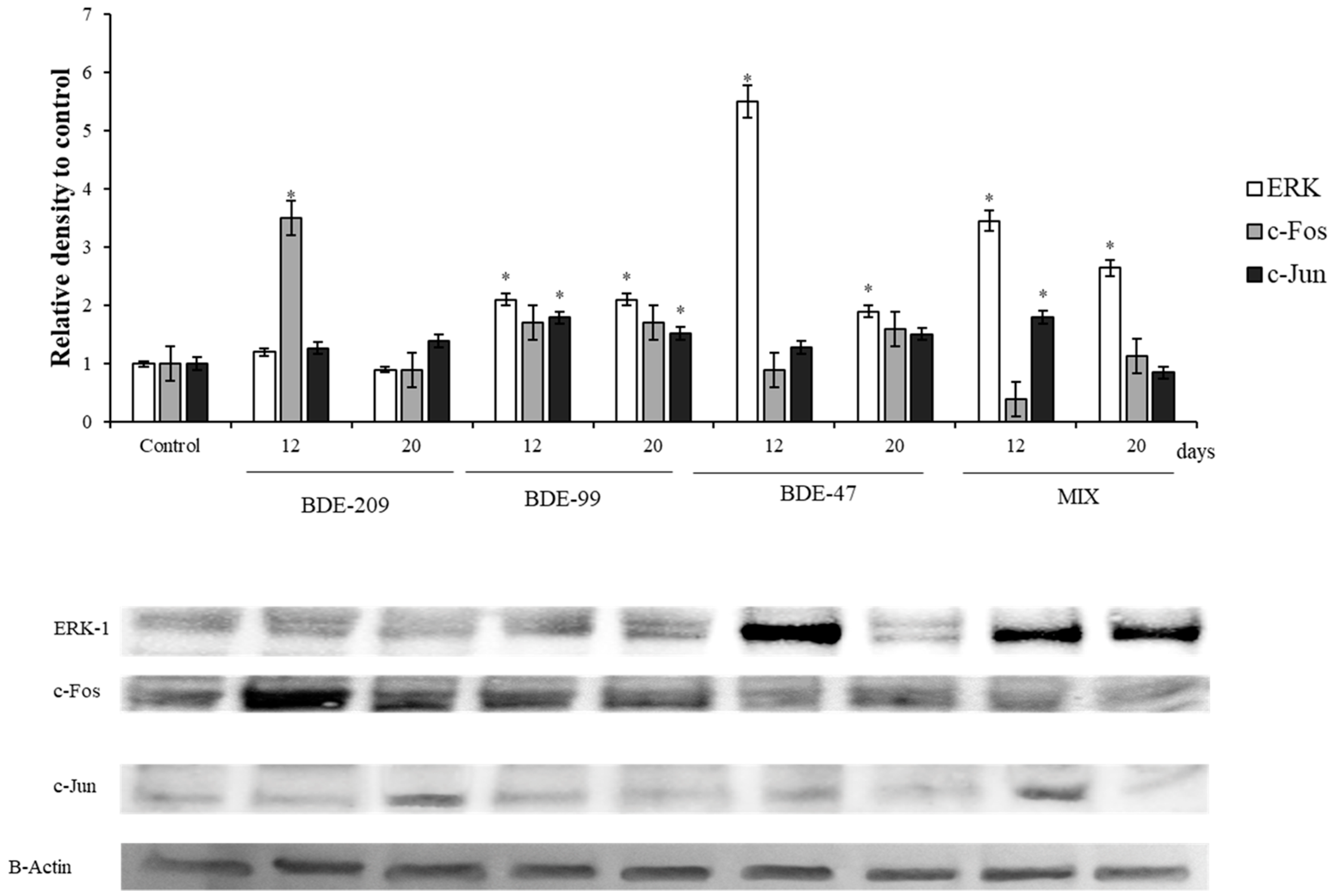
3.3. Effects of PBDEs on ERK, c-Jun and c-Fos
3.4. Effects of PBDEs on NRF2, AMPK, HIF
4. Discussion
4.1. Effects of PBDEs on Cytotoxicity and ROS Production
4.2. Effects of PBDEs on p53, pRB, PARP
4.3. Effects of PBDEs on ERK, c-Jun and c-Fos
4.4. Effects of PBDEs on AMPK, HIF, NRF2
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- He, P.; Wang, A.; Niu, Q.; Guo, L.; Xia, T.; Chen, X. Toxic effect of PBDE-47 on thyroid development, learning, and memory, and the interaction between PBDE-47 and PCB153 that enhances toxicity in rats. Toxicol. Ind. Health 2011, 27, 279–288. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.A. A perspective on the potential health risks of PBDEs. Chemosphere 2002, 46, 745–755. [Google Scholar] [CrossRef]
- Eljarrat, E.; Barceló, D. The Handbook of Environmental Chemistry. Part J. PAHs Relat. Compd. 2011. [Google Scholar] [CrossRef]
- De Boer, J.; Allchin, C.; Law, R.; Zegers, B.; Boon, J.P. Method for the analysis of polybrominated diphenylethers in sediments and biota. Trends Anal. Chem. 2001, 20, 591–599. [Google Scholar] [CrossRef]
- Hong, S.H.; Kannan, N.; Jin, Y.; Won, J.H.; Han, G.M.; Shim, W.J. Temporal trend, spatial distribution, and terrestrial sources of PBDEs and PCBs in Masan Bay, Korea. Mar. Pollut. Bull. 2010, 60, 1836–1841. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Xu, Z.; Dai, J.; Mai, B.; Cao, H.; Wang, J.; Shi, Z.; Xu, M. Distribution of polybrominated diphenyl ethers and decabromodiphenylethane in surface sediments from Fuhe River and Baiyangdian Lake, North China. J. Environ. Sci. (China) 2010, 22, 1833–1839. [Google Scholar] [CrossRef]
- Kierkegaard, A.; Björklund, J.; Fridén, U. Identification of the flame retardant decabromodiphenyl ethane in the environment. Environ. Sci. Technol. 2004, 38, 3247–3253. [Google Scholar] [CrossRef]
- Kim, G.B.; Stapleton, H.M. PBDEs, methoxylated PBDEs and HBCDs in Japanese common squid (Todarodes pacificus) from Korean offshore waters. Mar. Pollut. Bull. 2010, 60, 935–940. [Google Scholar] [CrossRef]
- Oberg, K.; Warman, K.; Oberg, T. Distribution and levels of brominated flame retardants in sewage sludge. Chemosphere 2002, 48, 805–809. [Google Scholar] [CrossRef]
- Sellstrom, U.; Jansson, B. Analysis of tetrabromobisphenol A in a product and environmental samples. Chemosphere 1995, 31, 3085–3092. [Google Scholar] [CrossRef]
- Sjödin, A.; Carlsson, H.; Thuresson, K.; Sjölin, S.; Bergman, A.; Ostman, C. Flame retardants in indoor air at an electronics recycling plant and at other work environments. Environ. Sci. Technol. 2001, 35, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Hites, R.A. Brominated flame retardants in tree bark from North America. Environ. Sci. Technol. 2006, 40, 3711–3716. [Google Scholar] [CrossRef] [PubMed]
- Horri, K.; Alfonso, S.; Cousin, X.; Munschy, C.; Loizeau, V.; Aroua, S.; Bégout, M.-L.; Ernande, B. Fish life-history traits are affected after chronic dietary exposure to an environmentally realistic marine mixture of PCBs and PBDEs. Sci. Total Environ. 2018, 610–611, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Darnerud, P.O.; Eriksen, G.S.; Jóhannesson, T.; Larsen, P.B.; Viluksela, M. Polybrominated diphenyl ethers: Occurrence, dietary exposure, and toxicology. Environ. Health Perspect. 2001, 109, 49–68. [Google Scholar]
- Bi, X.; Thomas, G.O.; Kevin, C.J.; Weiyue, Q.; Guoying, S.; Martin, F.L.; Jiamo, F. Exposure of electronics dismantling workers to polybrominated diphenyl ethers, polychlorinated biphenyls, and organochlorine pesticides in South China. Environ. Sci. Technol. 2007, 41, 5647–5653. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.; Cai, Z.W.; Wong, M.H. Environmental contamination from electronic waste recycling at Guiyu, southeast China. J. Mater. Cycles Waste Manag. 2006, 8, 21–33. [Google Scholar] [CrossRef]
- Wu, N.; Herrmann, T.; Paepke, O.; Tickner, J.; Hale, R.; Harvey, E.; La Guardia, M.; DMcClean Michael FWebster, T. Human exposure to PBDEs: Associations of PBDE body burdens with food consumption and house dust concentrations. Environ. Sci. Technol. 2007, 41, 1584–1589. [Google Scholar] [CrossRef] [PubMed]
- ATSDR; U.S. Department of Health and Human Services; Public Health Service; Agency for Toxic Substances and Disease Registry. Toxicological Profile for Polybrominated Biphenyls and Polybrominated Diphenyl Ethers; Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2004.
- Directive 76/769/EEC. Council Directive of 27 July 1976 on the Approximation of the Laws, Regulations and Administrative Provisions of the Member States Relating to Restrictions on the Marketing and Use of Certain Dangerous Substances and Preparations; The European Parliament and the Council of the European Union: Brussels, Belgium, 1986. [Google Scholar]
- Directive 2002/95/EC of the European Parliament and of the Council of 27 January 2003 on the Restriction of the Use of Certain Hazardous Substance in Electrical and Electric Equipment. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2003:037:0019:0023:en:PDF (accessed on 18 February 2019).
- World Health Organization. Health Risks of Persistent Organic Pollutants from Long-Range Transboundary Air Pollution; World Health Organization: Geneva, Switzerland, 2003. [Google Scholar]
- Chao, S.J.; Huang, C.P.; Chen, P.C.; Huang, C. Teratogenic responses of zebrafish embryos to decabromodiphenyl ether (BDE-209) in the presence of nano-SiO2 particles. Chemosphere 2017, 178, 449–457. [Google Scholar] [CrossRef]
- Mercado-Feliciano, M.; Bigsby, R.M. The Polybrominated diphenyl ether mixture DE-71 ls. Mildly estrogenic. Environ. Health Perspect. 2008, 116, 605–611. [Google Scholar] [CrossRef]
- Tang, S.; Liu, H.; Yin, H.; Liu, X.; Peng, H.; Lu, G.; Dang, Z.; He, C. Effect of 2,2″,4,4″-tetrabromodiphenyl ether (BDE-47) and its metabolites on cell viability, oxidative stress, and apoptosis of HepG2. Chemosphere 2018, 193, 978–988. [Google Scholar] [CrossRef]
- Zhang, L.; Jin, Y.; Han, Z.; Liu, H.; Shi, L.; Hua, X.; Doering, J.A.; Tang, S.; Giesy, J.P.; Yu, H. Integrated in silico and in vivo approaches to investigate effects of BDE-99 mediated by the nuclear receptors on developing zebrafish. Environ. Toxicol. Chem. 2017, 37, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Legler, J. New insights into the endocrine disrupting effects of brominated flame retardants. Chemosphere 2008, 73, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Han, Z.; Liu, C. A review on the effects of PBDEs on thyroid and reproduction systems in fish. Gen. Comp. Endocrinol. 2015, 219, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Mercado-Feliciano, M.; Bigsby, R.M.; Hites, R.A. Measurement of polybrominated diphenyl ethers and metabolites in mouse plasma after exposure to a commercial pentabromodiphenyl ether mixture. Environ. Health Perspect. 2007, 115, 1052–1058. [Google Scholar] [CrossRef]
- Malmberg, T.; Athanasiadou, M.; Marsh, G.; Brandt, I.; Bergman, Å. Identification of hydroxylated polybrominated diphenyl ether metabolites in blood plasma from polybrominated diphenyl ether exposed rats. Environ. Sci. Technol 2005, 39, 5342–5348. [Google Scholar] [CrossRef]
- Meerts, I.A.; van Zanden, J.J.; Luijks, E.A.; van Leeuwen-Bol, I.; Marsh, G.; Jakobsson, E.; Bergman, A.; Brouwer, A. Potent competitive interactions of some brominated flame retardants and related compounds with human transthyretin in vitro. Toxicol. Sci. 2000, 56, 95–104. [Google Scholar] [CrossRef]
- Barber, J.L.; Walsh, M.J.; Hewitt, R.; Jones, K.C.; Martin, F.L. Low-dose treatment with polybrominated diphenyl ethers (PBDEs) induce altered characteristics in MCF-7 cells. Mutagenesis 2006, 21, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Llabjani, V.; Trevisan, J.; Jones, K.C.; Shore, R.F.; Martin, F.L. Binary mixture effects by PBDE and PCB congeners (126 or 153) in MCF-7 cells: Biochemical alterations assessed by IR spectroscopy and multivariate analysis. Environ. Sci. Technol. 2010, 44, 3992–3998. [Google Scholar] [CrossRef]
- Ukpebor, J.; Llabjani, V.; Martin, F.L.; Halsall, C.J. Sublethal genotoxicity and cell alterations by organophosphorus pesticides in MCF-7 cells: Implications for environmentally relevant concentrations. Environ. Toxicol. Chem. 2011, 30, 632–639. [Google Scholar] [CrossRef]
- Li, Z.-H.; Liu, X.-Y.; Wang, N.; Chen, J.-S.; Chen, Y.-H.; Huang, J.-T.; Su, C.-H.; Xie, F.; Yu, B.; Chen, D.-J. Effects of decabrominated diphenyl ether (PBDE-209) in regulation of growth and apoptosis of breast, ovarian, and cervical cancer cells. Environ. Health Perspect. 2012, 120, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tang, X.; Zhou, B.; Xu, N.; Zhou, Z.; Fang, K.; Wang, Y. BDE-47 and BDE-209 inhibit proliferation of Neuro-2a cells via inducing G1-phase arrest. Environ. Toxicol. Pharmacol. 2017, 50, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Singh, S.K. Maternal exposure to polybrominated diphenyl ether (BDE-209) during lactation affects germ cell survival with altered testicular glucose homeostasis and oxidative status through down-regulation of Cx43 and p27Kip1 in prepubertal mice offspring. Toxicology 2017, 386, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, P.; Zhang, S.; Lei, R.; Li, B.; Wu, X.; Jiang, C.; Zhang, X.; Ma, R.; Yang, L.; et al. Oxidative stress-elicited autophagosome accumulation contributes to human neuroblastoma SH-SY5Y cell death induced by PBDE-47. Environ. Toxicol. Pharmacol. 2017, 56, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Rahman, T.; Hosen, I.; Islam, M.M.T.; Shekhar, H.U. Oxidative stress and human health. Adv. Biosci. Biotechnol. 2012, 3, 997–1019. [Google Scholar] [CrossRef]
- Simonelli, V.; Mazzei, F.; Errico, M.D.; Dogliotti, E. Mutation research/fundamental and molecular mechanisms of mutagenesis gene susceptibility to oxidative damage: From single nucleotide polymorphisms to function. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2012, 731, 1–13. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef]
- Barzilai, A.; Yamamoto, K.I. DNA damage responses to oxidative stress. DNA Repair 2004, 3, 1109–1115. [Google Scholar] [CrossRef]
- Oh, S.E.; Mouradian, M.M. Cytoprotective mechanisms of DJ-1 against oxidative stress through modulating ERK1/2 and ASK1 signal transduction. Redox Biol. 2018, 14, 211–217. [Google Scholar] [CrossRef]
- Emanuele, S.; D’Anneo, A.; Calvaruso, G.; Cernigliaro, C.; Giuliano, M.; Lauricella, M. The Double-edged sword profile of redox signaling: Oxidative events as molecular switches in the balance between cell physiology and cancer. Chem. Res. Toxicol. 2018, 31, 201–210. [Google Scholar] [CrossRef]
- Zhang, S.; Kuang, G.; Zhao, G.; Wu, X.; Zhang, C.; Lei, R.; Xia, T.; Chen, J.; Wang, Z.; Ma, R.; et al. Involvement of the mitochondrial p53 pathway in PBDE-47-induced SH-SY5Y cells apoptosis and its underlying activation mechanism. Food Chem. Toxicol. 2013, 62, 699–706. [Google Scholar] [CrossRef]
- Hinds, P.W.; Mittnacht, S.; Dulic, V.; Arnold, A.; Reed, S.I.; Weinberg, R.A. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 1992, 70, 993–1006. [Google Scholar] [CrossRef]
- Pacher, P.; Szabo, C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am. J. Pathol. 2008, 173, 2–13. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Franklin, R.A.; Abrams, S.L.; Chappell, W.H.; Wong, E.W.; Lehmann, B.; Terrian, D.M.; Basecke, J.; Stivala, F.; et al. Targeting the RAF/MEK/ERK, PI3K/AKT and P53 pathways in hematopoietic drug resistance. Adv. Enzym. Regul. 2007, 47, 64–103. [Google Scholar] [CrossRef] [PubMed]
- Turpaev, K.T. Role of transcription factor AP-1 in integration of cellular signaling systems. Mol. Biol. (Mosk) 2006, 40, 945–961. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhang, Q.; Yu, T.; Sun, S.; Wang, W.; Liu, G. Hypoxia promotes drug resistance in osteosarcoma cells via activating AMP-activated protein kinase (AMPK) signaling. J. Bone Oncol. 2016, 5, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Xuan, F.; Fu, H.; Ge, X.; Zhu, J.; Qiao, H.; Jin, S.; Zhang, W. Molecular characterization and mRNA expression of hypoxia inducible factor-1 and cognate inhibiting factor in Macrobrachium nipponense in response to hypoxia. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2016, 196–197, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tang, X.; Zhou, B.; Zhou, Z.; Xu, N.; Wang, Y. A ROS-mediated mitochondrial pathway and Nrf2 pathway activation are involved in BDE-47 induced apoptosis in Neuro-2a cells. Chemosphere 2017, 184, 679–686. [Google Scholar] [CrossRef]
- Espinosa, C.; Manuguerra, S.; Cuesta, A.; Santulli, A.; Messina, C.M. Oxidative Stress, Induced by Sub-Lethal Doses of BDE 209, Promotes Energy Management and Cell Cycle Modulation in the Marine Fish Cell Line SAF-1. Int. J. Environ. Res. Public Health 2019, 16, 474. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Messina, C.M.; Pizzo, F.; Santulli, A.; Bušelić, I.; Boban, M.; Orhanović, S.; Mladineo, I. Anisakis pegreffii (Nematoda: Anisakidae) products modulate oxidative stress and apoptosis-related biomarkers in human cell lines. Parasites Vectors 2016, 9, 607. [Google Scholar] [CrossRef] [PubMed]
- Abbes, M.; Baati, H.; Guermazi, S.; Messina, C.; Santulli, A.; Gharsallah, N.; Ammar, E. Biological properties of carotenoids extracted from Halobacterium halobium isolated from a Tunisian solar saltern. BMC Complement. Altern. Med. 2013, 13, 255. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Lee, K.H.; Chae, S.; Zhang, R.; Jung, M.S.; Kim, S.Y.; Kim, H.S.; Kim, D.H.; Hyun, J.W. Cytoprotective effect of tectorigenin, a metabolite formed by transformation of tectoridin by intestinal microflora, on oxidative stress induced by hydrogen peroxide. Eur. J. Pharmacol. 2005, 519, 16–23. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Chen, S.-J.; Luo, X.-J.; Lin, Z.; Luo, Y.; Li, K.-C.; Peng, X.-Z.; Mai, B.-X.; Ran, Y.; Zeng, E.Y. Time trends of polybrominated diphenyl ethers in sediment cores from the Pearl River Estuary, South China. Environ. Sci. Technol. 2007, 41, 5595–5600. [Google Scholar] [CrossRef] [PubMed]
- Fürst, P. Dioxins, polychlorinated biphenyls and other organohalogen compounds in human milk. Levels, correlations, trends and exposure through breastfeeding. Mol. Nutr. Food Res. 2006, 50, 922–933. [Google Scholar] [CrossRef] [PubMed]
- He, P.; He, W.; Wang, A.; Xia, T.; Xu, B.; Zhang, M.; Chen, X. PBDE-47-induced oxidative stress, DNA damage and apoptosis in primary cultured rat hippocampal neurons. Neurotoxicology 2008, 29, 124–129. [Google Scholar] [CrossRef]
- He, W.; He, P.; Wang, A.; Xia, T.; Xu, B.; Chen, X. Effects of PBDE-47 on cytotoxicity and genotoxicity in human neuroblastoma cells in vitro. Mutat. Res. 2008, 649, 62–70. [Google Scholar] [CrossRef]
- Pellacani, C.; Buschini, A.; Galati, S.; Mussi, F.; Franzoni, S.; Costa, L.G. Evaluation of DNA damage induced by 2 polybrominated diphenyl ether flame retardants (BDE-47 and BDE-209) in SK-N-MC Cells. Int. J. Toxicol. 2012, 31, 372–379. [Google Scholar] [CrossRef]
- Wang, L.; Zou, W.; Zhong, Y.; An, J.; Zhang, X.; Wu, M.; Yu, Z. The hormesis effect of BDE-47 in HepG 2 cells and the potential molecular mechanism. Toxicol. Lett. 2012, 209, 193–201. [Google Scholar] [CrossRef]
- Jiang, Y.; Tang, X.; Zhou, B.; Sun, T.; Chen, H.; Zhao, X.; Wang, Y. The ROS-mediated pathway coupled with the MAPK-p38 signaling pathway and antioxidant system plays roles in the responses of Mytilus edulis haemocytes induced by BDE-47. Aquat. Toxicol. 2017, 187, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011, 51, 327–336. [Google Scholar] [CrossRef]
- Longo, V.; Longo, A.; Di Sano, C.; Cigna, D.; Cibella, F.; Di Felice, G.; Colombo, P. In vitro exposure to 2,2′,4,4′-tetrabromodiphenyl ether (PBDE-47) impairs innate inflammatory response. Chemosphere 2019, 219, 845–854. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Li, S.; Zhong, Y.; Wang, Y.; Zhen, K.; Zhang, X.; Wang, Y.; Wu, M.; Yu, Z.; Sheng, G.; et al. The cytotoxic effects of synthetic 6-hydroxylated and 6-methoxylated polybrominated diphenyl ether 47 (BDE47). Environ. Toxicol. 2011, 26, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, T.A.; Skavicus, S.; Stapleton, H.M.; Seidler, F.J. Brominated and organophosphate flame retardants target different neurodevelopmental stages, characterized with embryonic neural stem cells and neuronotypic PC12 cells. Toxicology 2017, 390, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.; Giordano, G.; Costa, L.G. Comparative cytotoxicity and intracellular accumulation of five polybrominated diphenyl ether congeners in mouse cerebellar granule neurons. Toxicol. Sci. 2010, 114, 124–132. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Dong, W.; Keibler, M.A.; Stephanopoulos, G. Review of metabolic pathways activated in cancer cells as determined through isotopic labeling and network analysis. Metab. Eng. 2017, 43, 113–124. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]
- Mullen, A.R.; DeBerardinis, R.J. Genetically-defined metabolic reprogramming in cancer. Trends Endocrinol. Metab. 2012, 23, 552–559. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Heiden, M.G.V. Altered metabolite levels in cancer: Implications for tumour biology and cancer therapy. Nat. Rev. Cancer 2016, 16, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Böhlig, L.; Rother, K. One function--multiple mechanisms: The manifold activities of p53 as a transcriptional repressor. J. Biomed. Biotechnol. 2011, 464916, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53, growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.; Rotter, V. p53-dependent cell cycle control: Response to genotoxic stress. Semin. Cancer Biol. Med. 1998, 8, 325–336. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 497–501. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Huang, S.; Wang, J.; Cui, Y. 2,2′,4,4′-Tetrabromodiphenyl ether injures cell viability and mitochondrial function of mouse spermatocytes by decreasing mitochondrial proteins Atp5b and Uqcrc1. Environ. Toxicol. Pharmacol. 2016, 46, 301–310. [Google Scholar] [CrossRef]
- Blagosklonny, M.V.; Pardee, A.B. The restriction point of the cell cycle. Cell Cycle 2002, 1, 103–110. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef]
- Luo, R.X.; Postigo, A.A.; Dean, D.C. Rb interacts with histone deacetylase to repress transcription. Cell 1998, 92, 463–473. [Google Scholar] [CrossRef]
- Zheng, L.; Lee, W.H. The retinoblastoma gene: A prototypic and multifunctional tumor suppressor. Exp. Cell Res. 2001, 264, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Almasan, A.; Yin, Y.; Kelly, R.E.; Lee, E.Y.; Bradley, A.; Li, W.; Bertino, J.R.; Wahl, G.M. Deficiency of retinoblastoma protein leads to inappropriate S-phase entry, activation of E2F-responsive genes, and apoptosis. Proc. Natl. Acad. Sci. USA 1995, 92, 5436–5440. [Google Scholar] [CrossRef] [PubMed]
- Sanmartín-Salinas, P.; Del Lobo, M.V.T.; Noguerales-Fraguas, F.; Londoño, M.T.; Jiménez-Ruiz, A.; Guijarro, L.G. Insulin receptor substrate-4 is overexpressed in colorectal cancer and promotes retinoblastoma–cyclin-dependent kinase activation. J. Gastroenterol. 2018, 53, 932–944. [Google Scholar] [CrossRef]
- Kraus, W.L.; Hottiger, M.O. PARP-1 and gene regulation: Progress and puzzles. Mol. Asp. Med. 2013, 34, 1109–1123. [Google Scholar] [CrossRef]
- Bai, P.; Cantó, C. The Role of PARP-1 and PARP-2 Enzymes in metabolic regulation and disease. Cell Metab. 2012, 16, 290–295. [Google Scholar] [CrossRef]
- Fu, T.; Wang, L.; Jin, X.; Sui, H.; Liu, Z.; Jin, Y. Hyperoside induces both autophagy and apoptosis in non-small cell lung cancer cells in vitro. Acta Pharmacol. Sin. 2016, 37, 505–518. [Google Scholar] [CrossRef]
- Sun, W.; Du, L.; Tang, W.; Kuang, L.; Du, P.; Chen, J.; Chen, D. PBDE-209 exposure damages learning and memory ability in rats potentially through increased autophagy and apoptosis in the hippocampus neuron. Environ. Toxicol. Pharmacol. 2017, 50, 151–158. [Google Scholar] [CrossRef]
- Souza, A.O.; Pereira, L.C.; Oliveira, D.P.; Dorta, D.J. BDE-99 congener induces cell death by apoptosis of human hepatoblastoma cell line—HepG2. Toxicol. In Vitro 2013, 27, 580–587. [Google Scholar] [CrossRef]
- Li, P.; Liu, L.; Zhou, G.; Tian, Z.; Luo, C.; Xia, T.; Chen, J.; Niu, Q.; Dong, L.; Zhao, Q.; et al. Perigestational exposure to low doses of PBDE-47 induces excessive ER stress, defective autophagy and the resultant apoptosis contributing to maternal thyroid toxicity. Sci. Total Environ. 2018, 645, 363–371. [Google Scholar] [CrossRef]
- Miller, C.R.; Oliver, K.E.; Farley, J.H. MEK1/2 inhibitors in the treatment of gynecologic malignancies. Gynecol. Oncol. 2014, 133, 128–137. [Google Scholar] [CrossRef]
- El-Baba, C.; Mahadevan, V.; Fahlbusch, F.B.; Suma, M.S.; Rau, T.T.; Gali-Muhtasib, H.; Schneider-Stock, R. Thymoquinone-induced conformational changes of PAK1 interrupt prosurvival MEK-ERK signaling in colorectal cancer. Mol. Cancer 2014, 13, 201. [Google Scholar] [CrossRef]
- Lu, Z.; Ding, L.; Hong, H.; Hoggard, J.; Lu, Q.; Chen, Y.-H. Claudin-7 inhibits human lung cancer cell migration and invasion through ERK/MAPK signaling pathway. Exp. Cell Res. 2011, 317, 1935–1946. [Google Scholar] [CrossRef]
- Pan, F.; Hong, L.-Q. Insulin promotes proliferation and migration of breast cancer cells through the extracellular regulated kinase pathway. Asian Pac. J. Cancer Prev. 2014, 15, 6349–6352. [Google Scholar] [CrossRef]
- Song, X.; Wang, Y.; Du, H.; Fan, Y.; Yang, X.; Wang, X.; Wu, X.; Luo, C. Overexpression of HepaCAM inhibits cell viability and motility through suppressing nucleus translocation of androgen receptor and ERK signaling in prostate cancer. Prostate 2014, 74, 1023–1033. [Google Scholar] [CrossRef]
- Manimala, N.J.; Frost, C.D.; Lane, M.L.; Higuera, M.; Beg, R.; Vesely, D.L. Cardiac hormones target nuclear oncogenes c-Fos and c-Jun in carcinoma cells. Eur. J. Clin. Investig. 2013, 43, 1156–1162. [Google Scholar] [CrossRef]
- Qu, B.-L.; Yu, W.; Huang, Y.-R.; Cai, B.-N.; Du, L.-H.; Liu, F. 6-OH-BDE-47 promotes human lung cancer cells epithelial mesenchymal transition via the AKT/Snail signal pathway. Environ. Toxicol. Pharmacol. 2015, 39, 271–279. [Google Scholar] [CrossRef]
- Lee, M.-C.; Puthumana, J.; Lee, S.-H.; Kang, H.-M.; Park, J.C.; Jeong, C.-B.; Han, J.; Hwang, D.-S.; Seo, J.S.; Park, H.G.; et al. BDE-47 induces oxidative stress, activates MAPK signaling pathway, and elevates de novo lipogenesis in the copepod Paracyclopina nana. Aquat. Toxicol. 2016, 181, 104–112. [Google Scholar] [CrossRef]
- Chambard, J.-C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1299–1310. [Google Scholar] [CrossRef]
- Rodríguez, J.; Calvo, F.; González, J.M.; Casar, B.; Andrés, V.; Crespo, P.; Crespo, P. ERK1/2 MAP kinases promote cell cycle entry by rapid, kinase-independent disruption of retinoblastoma-lamin A complexes. J. Cell Biol. 2010, 191, 967–979. [Google Scholar] [CrossRef]
- Dasgupta, B.; Milbrandt, J. AMP-Activated protein kinase phosphorylates retinoblastoma protein to control mammalian brain development. Dev. Cell 2009, 16, 256–270. [Google Scholar] [CrossRef]
- Shen, Z.; Liang, X.; Rogers, C.Q.; Rideout, D.; You, M. Involvement of adiponectin-SIRT1-AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, 364–374. [Google Scholar] [CrossRef]
- O’Shea, R.S.; Dasarathy, S.; McCullough, A.J. Alcoholic liver disease. Am. J. Gastroenterol. 2010, 105, 14–32. [Google Scholar] [CrossRef]
- Dagon, Y.; Mantzoros, C.S.; Kim, Y.-B. AMPK↔Sirt1, From a signaling network to a combination drug. Metabolism 2016, 65, 1692–1694. [Google Scholar] [CrossRef]
- Qi, L.; Zhu, F.; Li, S.; Si, L.; Hu, L.; Tian, H. Retinoblastoma binding protein 2 (RBP2) promotes HIF-1α–VEGF-induced angiogenesis of non-small cell lung cancer via the AKT pathway. PLoS ONE 2014, 9, e106032. [Google Scholar] [CrossRef]
- Kitajima, S.; Lee, K.L.; Hikasa, H.; Sun, W.; Huang, R.Y.-J.; Yang, H.; Matsunaga, S.; Yamaguchi, T.; Araki, M.; Kato, H.; et al. Hypoxia-inducible factor-1a; promotes cell survival during ammonia stress response in ovarian cancer stem-like cells. Oncotarget 2017, 8, 114481–114494. [Google Scholar] [CrossRef]
- Giudice, A.; Arra, C.; Turco, M.C. Review of molecular mechanisms involved in the activation of the Nrf2-ARE signaling pathway by chemopreventive agents. Methods Mol. Biol. 2010, 647, 37–74. [Google Scholar] [CrossRef]
- Hu, X.Z.; Xu, Y.; Hu, D.C.; Hui, Y.; Yang, F.X. Apoptosis induction on human hepatoma cells HepG2 of decabrominated diphenyl ether (PBDE-209). Toxicol. Lett. 2007, 171, 19–28. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manuguerra, S.; Espinosa Ruiz, C.; Santulli, A.; Messina, C.M. Sub-lethal Doses of Polybrominated Diphenyl Ethers, in Vitro, Promote Oxidative Stress and Modulate Molecular Markers Related to Cell Cycle, Antioxidant Balance and Cellular Energy Management. Int. J. Environ. Res. Public Health 2019, 16, 588. https://doi.org/10.3390/ijerph16040588
Manuguerra S, Espinosa Ruiz C, Santulli A, Messina CM. Sub-lethal Doses of Polybrominated Diphenyl Ethers, in Vitro, Promote Oxidative Stress and Modulate Molecular Markers Related to Cell Cycle, Antioxidant Balance and Cellular Energy Management. International Journal of Environmental Research and Public Health. 2019; 16(4):588. https://doi.org/10.3390/ijerph16040588
Chicago/Turabian StyleManuguerra, Simona, Cristóbal Espinosa Ruiz, Andrea Santulli, and Concetta Maria Messina. 2019. "Sub-lethal Doses of Polybrominated Diphenyl Ethers, in Vitro, Promote Oxidative Stress and Modulate Molecular Markers Related to Cell Cycle, Antioxidant Balance and Cellular Energy Management" International Journal of Environmental Research and Public Health 16, no. 4: 588. https://doi.org/10.3390/ijerph16040588
APA StyleManuguerra, S., Espinosa Ruiz, C., Santulli, A., & Messina, C. M. (2019). Sub-lethal Doses of Polybrominated Diphenyl Ethers, in Vitro, Promote Oxidative Stress and Modulate Molecular Markers Related to Cell Cycle, Antioxidant Balance and Cellular Energy Management. International Journal of Environmental Research and Public Health, 16(4), 588. https://doi.org/10.3390/ijerph16040588