Spatial-Temporal Variation of Bacterial Communities in Sediments in Lake Chaohu, a Large, Shallow Eutrophic Lake in China


Abstract
1. Introduction
2. Materials and Methods
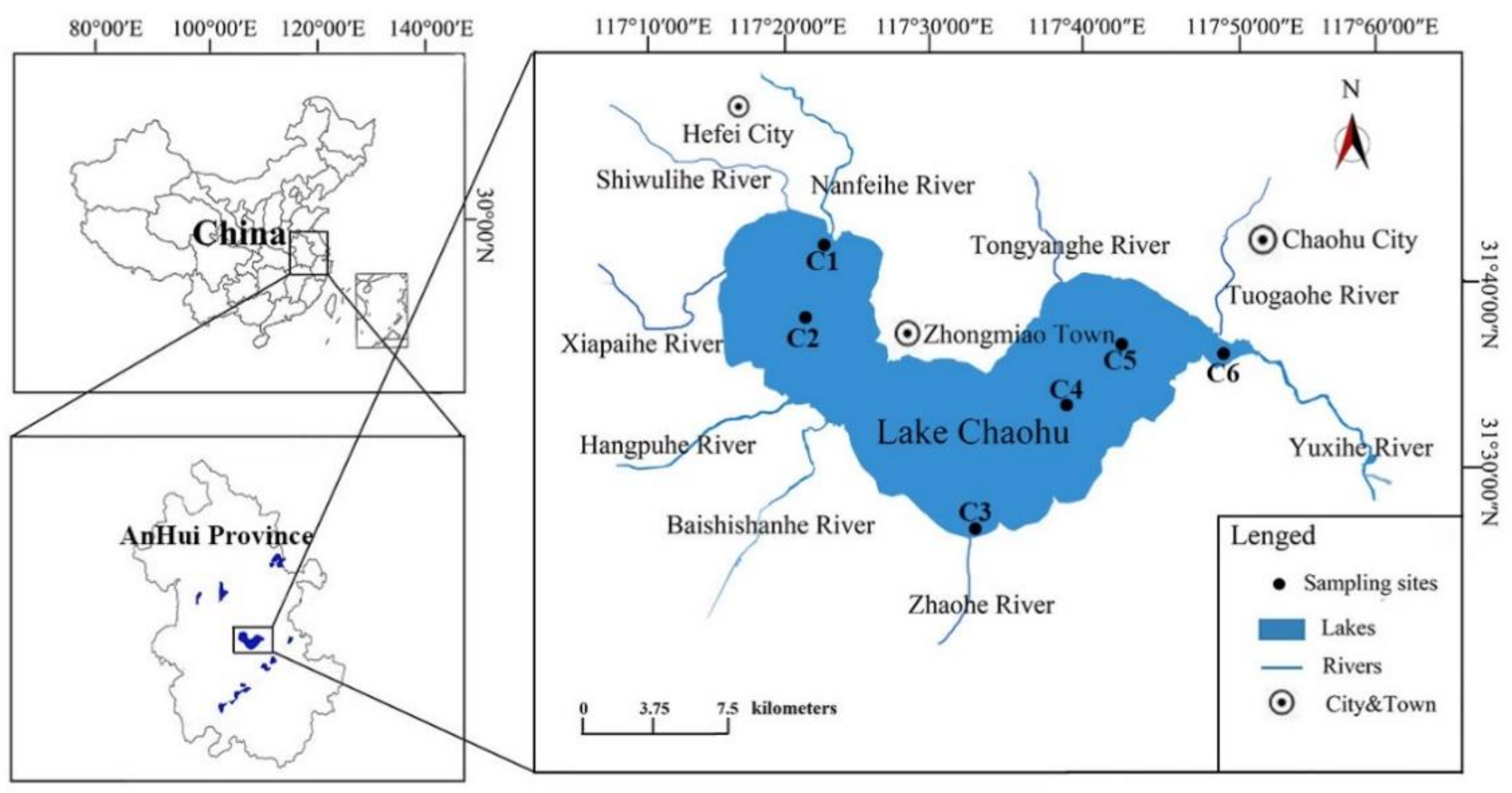
2.1. Study Site Description and Sampling Procedure
2.2. Chemical Analysis
2.3. DNA Extraction and Polymerase Chain Reaction (PCR) Amplification
2.4. Sequencing Data Optimization and Operational Taxonomic Units (OTU) Clustering Analysis
2.5. Statistical Analyses
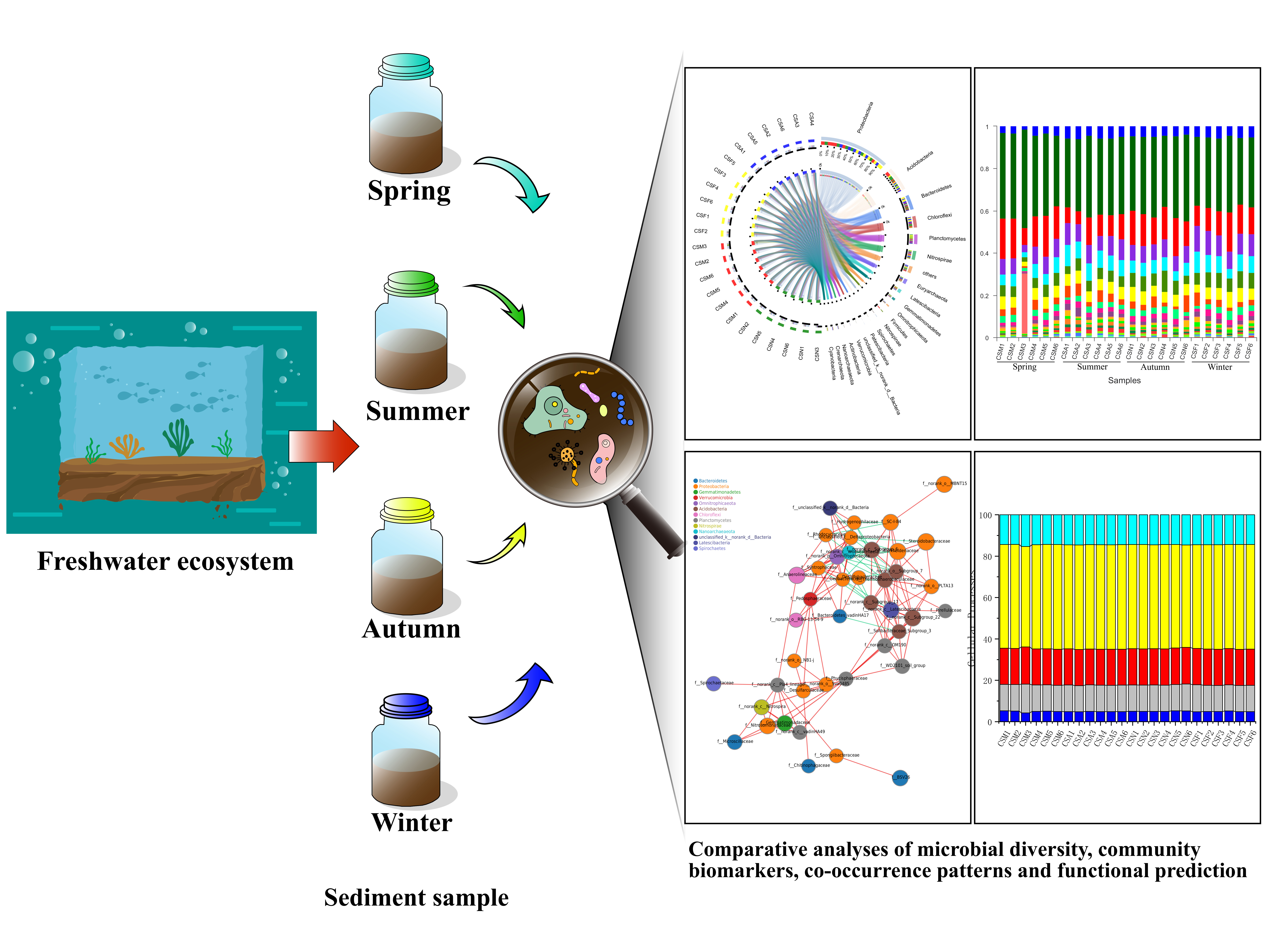
3. Results
3.1. Alpha Diversity
3.2. Beta Diversity
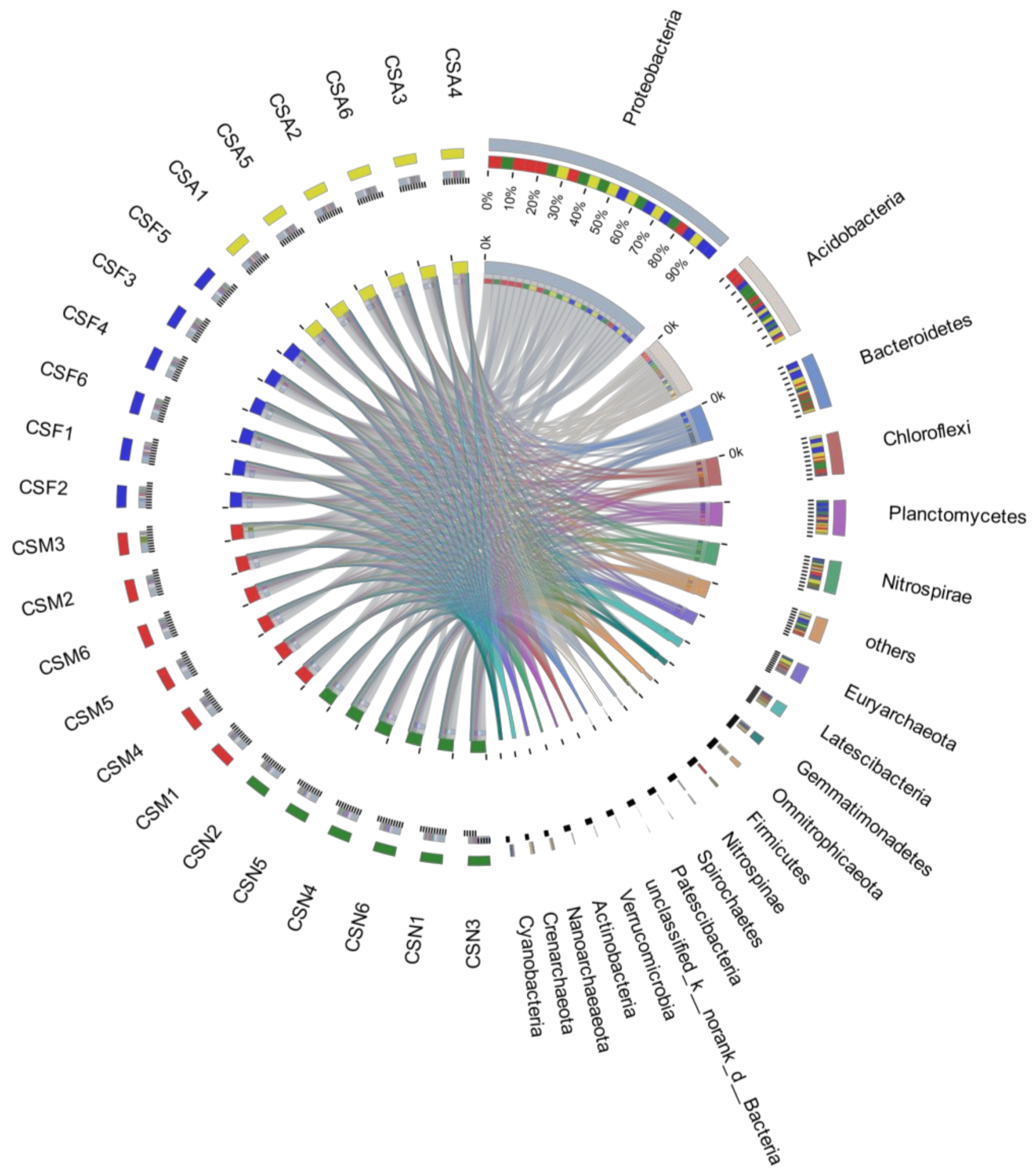
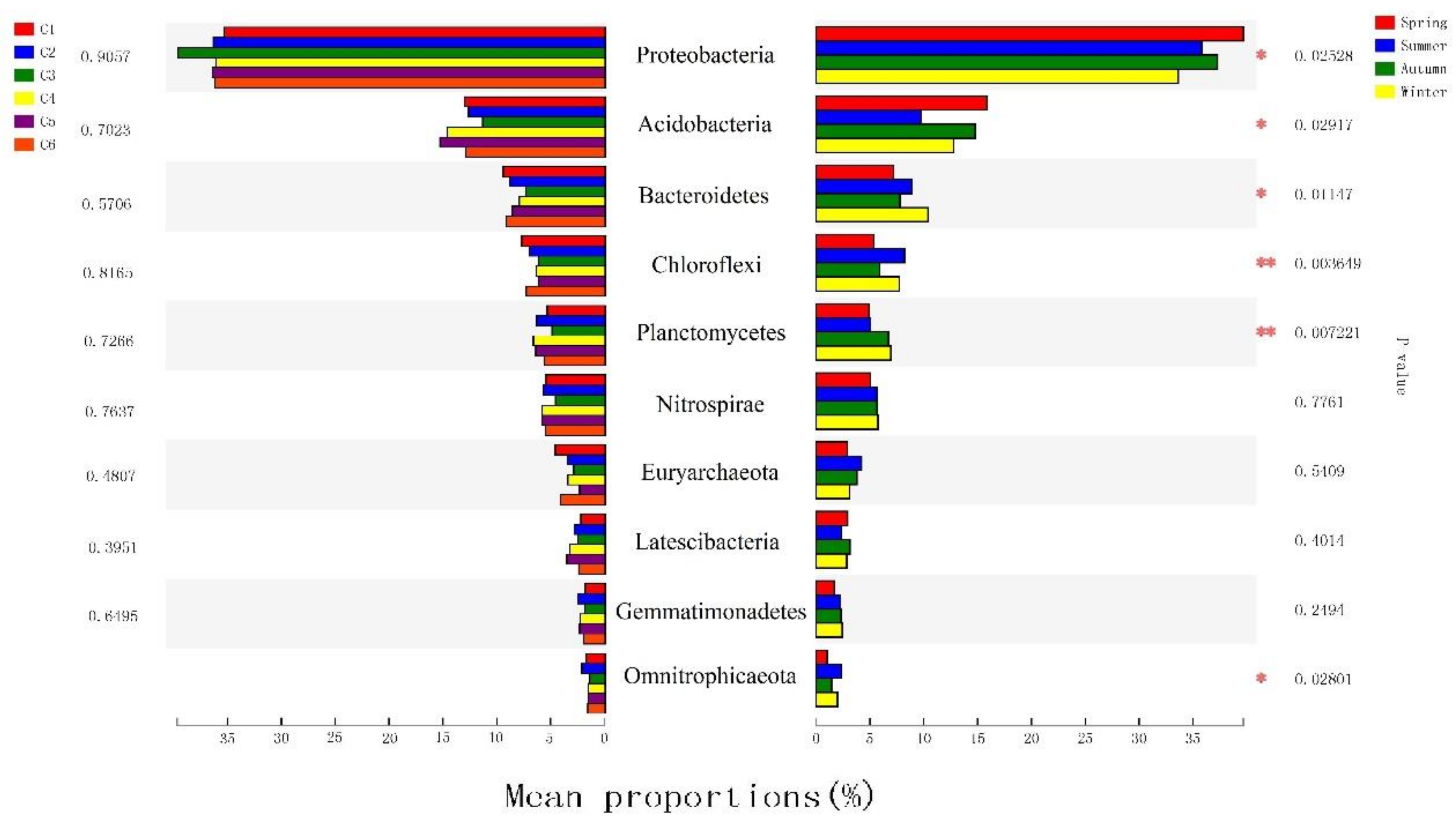
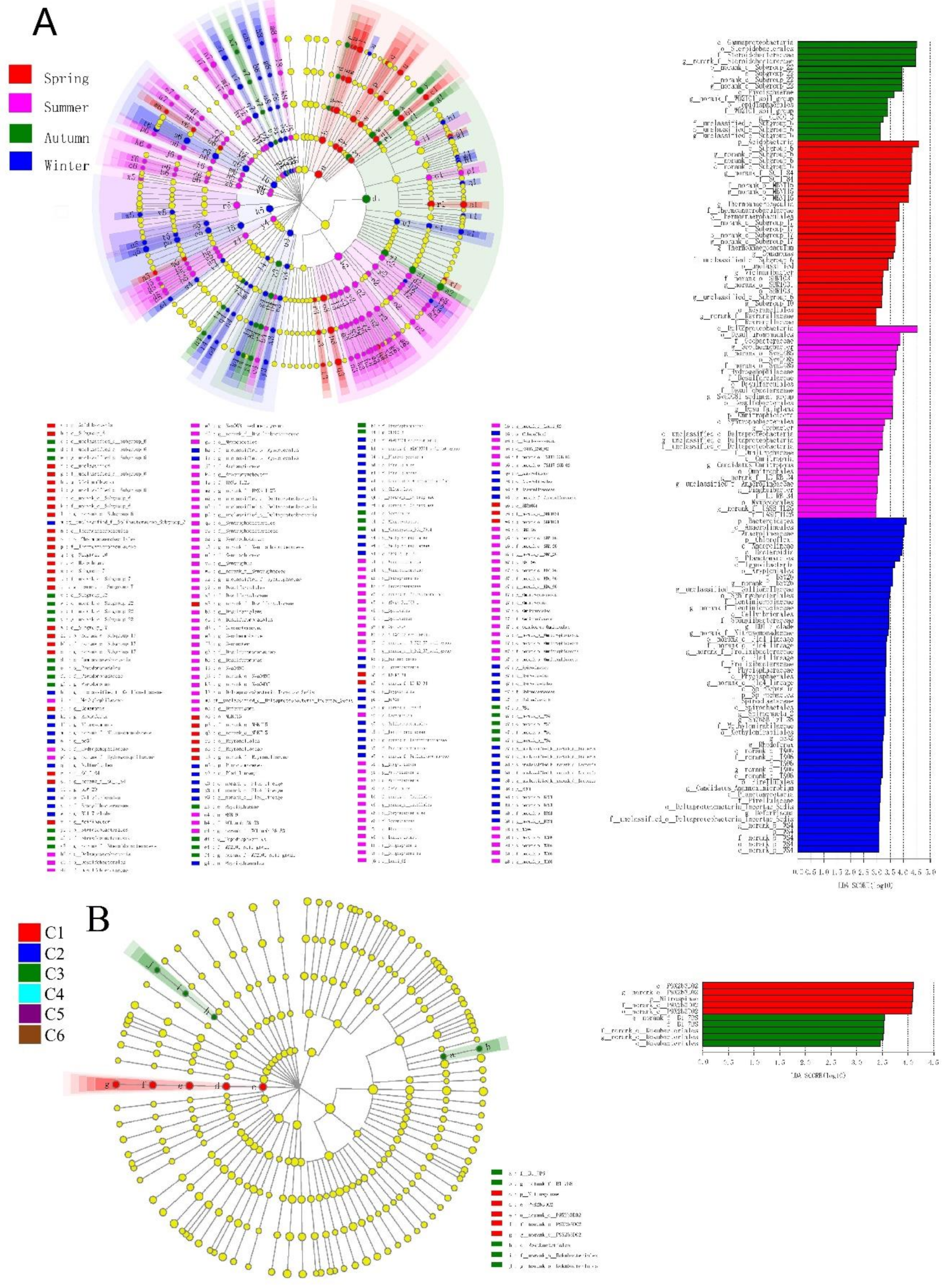
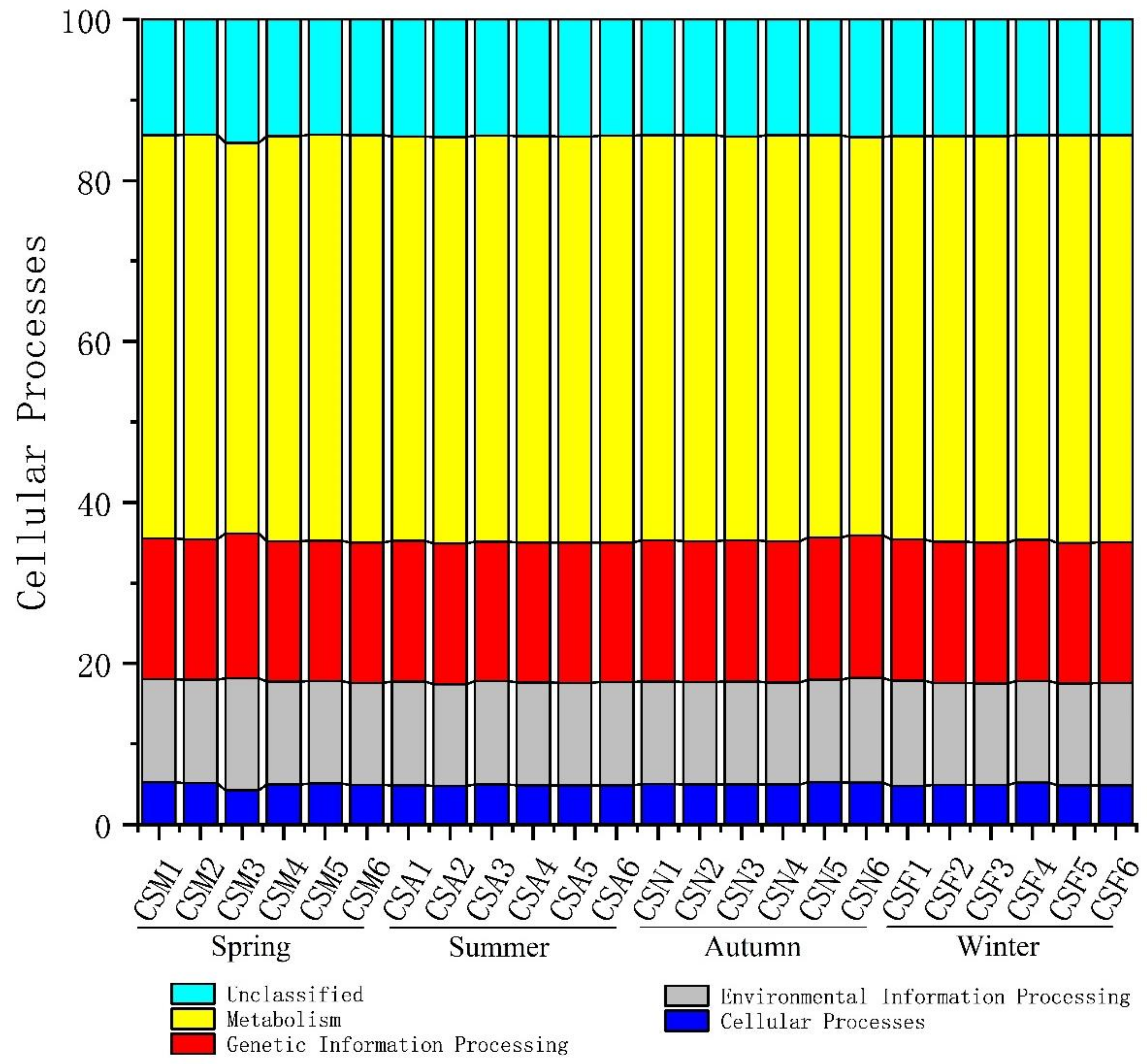
3.3. The Spatiotemporal Distribution of Bacterial Communities
3.4. Physical and Chemical Indicators Correlated with Bacterial Community Composition
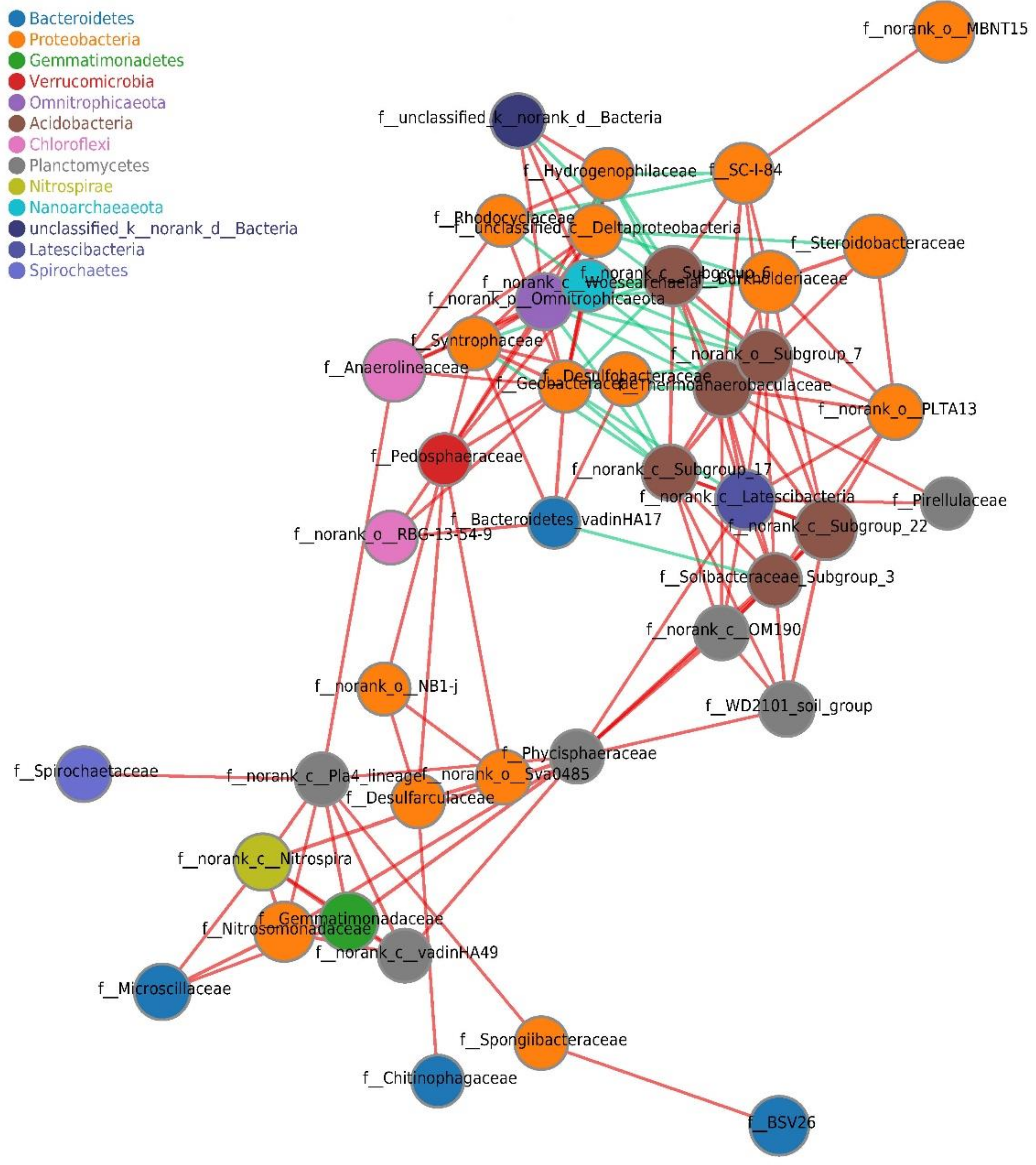
3.5. Co-Occurrence Network Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, B.; Liu, C.Q.; Maberly, S.C.; Wang, F.; Hartmann, J. Coupling of carbon and silicon geochemical cycles in rivers and lakes. Sci. Rep. 2016, 6, 35832. [Google Scholar] [CrossRef] [PubMed]
- Ávila, M.P.; Staehr, P.A.; Barbosa, F.A.R.; Chartone-Souza, E.; Nascimento, A.M.A. Seasonality of freshwater bacterioplankton diversity in two tropical shallow lakes from the brazilian atlantic forest. FEMS Microbiol. Ecol. 2016, 93, fiw218. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Ren, Z.; Shi, J.P.; Huang, X.Z.; Chen, J.R. Impact of agricultural non-point source pollution in eutrophic water body of taihu lake. Environ. Sci. Technol. 2010, 33, 106–111. [Google Scholar]
- Zan, F.; Huo, S.; Xi, B.; Zhu, C.; Liao, H.; Zhang, J.; Yeager, K.M. A 100-year sedimentary record of natural and anthropogenic impacts on a shallow eutrophic lake, lake chaohu, china. J. Environ. Monit. 2012, 14, 804–816. [Google Scholar] [CrossRef]
- Xiao, F.; Gulliver, J.S.; Simcik, M.F. Perfluorooctane sulfonate (pfos) contamination of fish in urban lakes: A prioritization methodology for lake management. Water Res. 2013, 47, 7264–7272. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Huang, G.; An, C.; Song, P.; Xin, X.; Yao, Y.; Zheng, R. Biophysiological and factorial analyses in the treatment of rural domestic wastewater using multi-soil-layering systems. J. Environ. Manag. 2018, 226, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Soldatova, E.A.; Guseva, N.V.; Shvartsev, S.L. Impact of human activity on the groundwater chemical composition of the south part of the poyang lake basin. IERI Procedia 2014, 8, 113–118. [Google Scholar] [CrossRef]
- Hudon, C.; Carignan, R. Cumulative impacts of hydrology and human activities on water quality in the st. Lawrence river (lake saint-pierre, quebec, canada). Can. J. Fish. Aquat. Sci. 2008, 65, 1165–1180. [Google Scholar] [CrossRef]
- Proulx, M.; Pick, F.R.; Mazumder, A.; Hamilton, P.B.; Lean, D.R.S. Experimental evidence for interactive impacts of human activities on lake algal species richness. Oikos 1996, 76, 191–195. [Google Scholar] [CrossRef]
- Hanna, S.; Kaarina, L.; Sihvonen, L.M.; Kaarina, S.; Mirja, L.; Matias, R.; Lars, P.; Christina, L. Bacteria contribute to sediment nutrient release and reflect progressed eutrophication-driven hypoxia in an organic-rich continental sea. PLoS ONE 2013, 8, e67061. [Google Scholar]
- Hou, J.; Song, C.; Cao, X.; Zhou, Y. Shifts between ammonia-oxidizing bacteria and archaea in? Relation to nitrification potential across trophic gradients in? Two large chinese lakes (lake taihu and lake chaohu). Water Res. 2013, 47, 2285–2296. [Google Scholar] [CrossRef] [PubMed]
- Méndez-García, C.; Mesa, V.; Sprenger, R.R.; Richter, M.; Diez, M.S.; Solano, J.; Gallego, J.R. Microbial stratification in low pH oxic and suboxic macroscopic growths along an acid mine drainage. ISME J. 2014, 8, 1259–1274. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Liu, L.; Zhang, Y.; Jiang, F. Occurrence and Succession of Bacterial Community in O3/BAC Process of Drinking Water Treatment. Int. J. Environ. Res. Public Health 2019, 16, 3112. [Google Scholar] [CrossRef] [PubMed]
- Chrost, R.J.; Adamczewski, T.; Kalinowska, K.R.; Skowronska, A. Inorganic phosphorus and nitrogen modify composition and diversity of microbial communities in water of mesotrophic lake. Pol. J. Microbiol. 2009, 58, 77–90. [Google Scholar]
- Song, H.; Coggins, L.; Reichwaldt, E.; Ghadouani, A. The importance of lake sediments as a pathway for microcystin dynamics in shallow eutrophic lakes. Toxins 2015, 7, 900–918. [Google Scholar] [CrossRef]
- Kim, H.; Bae, H.S.; Reddy, K.R.; Ogram, A. Distributions, abundances and activities of microbes associated with the nitrogen cycle in riparian and stream sediments of a river tributary. Water Res. 2016, 106, 51–61. [Google Scholar] [CrossRef]
- Moody, J. Race, school integration, and friendship segregation in america. Am. J. Sociol. 2001, 107, 679–716. [Google Scholar] [CrossRef]
- Ryan, L.; D’Angelo, A. Changing times: Migrants’ social network analysis and the challenges of longitudinal research. Soc. Netw. 2017, 53, 148–158. [Google Scholar] [CrossRef]
- Stephenson, K.; Lewin, D. Managing workforce diversity: Macro and micro level hr implications of network analysis. Int. J. Manpow. 1996, 17, 168–196. [Google Scholar] [CrossRef]
- Ramayo-Caldas, Y.; Mach, N.; Lepage, P.; Levenez, F.; Denis, C.; Lemonnier, G.; Leplat, J.J.; Billon, Y.; Berri, M.; Doré, J.E. Phylogenetic network analysis applied to pig gut microbiota identifies an ecosystem structure linked with growth traits. ISME J. 2016, 10, 2973–2977. [Google Scholar] [CrossRef]
- Wu, D.; Su, Y.; Xi, H.; Chen, X.; Xie, B. Urban and agriculturally influenced water contribute differently to the spread of antibiotic resistance genes in a mega-city river network. Water Res. 2019, 158, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Zhou, G.; Zhang, J.; Wang, W. Microbial community responses to biochar addition when a green waste and manure mix are composted: A molecular ecological network analysis. Bioresour. Technol. 2019, 273, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Yang, Z.H.; Zheng, Y.; Liu, J.B.; Xiong, W.P.; Zhang, Y.R.; Lu, Y.; Xue, W.J.; Fan, C.Z. Organic loading rate and hydraulic retention time shape distinct ecological networks of anaerobic digestion related microbiome. Bioresour. Technol. 2018, 262, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Frey, B.; Mayer, J.; Mäder, P.; Widmer, F. Distinct soil microbial diversity under long-term organic and conventional farming. ISME J. 2015, 9, 1177–1194. [Google Scholar] [CrossRef]
- Ho, C.W.; Ruehli, A.; Brennan, P. The modified nodal approach to network analysis. IEEE Trans. Circuits Syst. 1975, 22, 504–509. [Google Scholar]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef]
- Zhao, R.; Feng, J.; Yin, X.; Liu, J.; Fu, W.; Berendonk, T.U.; Zhang, T.; Li, X.; Li, B. Antibiotic resistome in landfill leachate from different cities of china deciphered by metagenomic analysis. Water Res. 2018, 134, 126–139. [Google Scholar] [CrossRef]
- Liang, X.; Huadong, R.; Sheng, L.; Xiuhui, L.; Xiaohua, Y. Soil bacterial community structure and co-occurrence pattern during vegetation restoration in karst rocky desertification area. Front. Microbiol. 2017, 8, 2377. [Google Scholar]
- Jiao, S.; Liu, Z.; Lin, Y.; Yang, J.; Chen, W.; Wei, G. Bacterial communities in oil contaminated soils: Biogeography and co-occurrence patterns. Soil Biol. Biochem. 2016, 98, 64–73. [Google Scholar] [CrossRef]
- Delafont, V.; Bouchon, D.; Héchard, Y.; Moulin, L. Environmental factors shaping cultured free-living amoebae and their associated bacterial community within drinking water network. Water Res. 2016, 100, 382–392. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Liu, Q.; Wang, L.; Li, J.; Li, S.; Zhang, X. The distribution and risk assessment of heavy metals in water, sediments, and fish of chaohu lake, china. Environ. Earth Sci. 2018, 77, 97. [Google Scholar] [CrossRef]
- Xu, Z.; Ji, J.; Shi, C. Water geochemistry of the chaohu lake basin rivers, china: Chemical weathering and anthropogenic inputs. Appl. Geochem. 2011, 26, S379–S383. [Google Scholar] [CrossRef]
- Jiang, Y.; Yang, Y.; Wu, Y.; Tao, J.; Cheng, B. Microcystin bioaccumulation in freshwater fish at different trophic levels from the eutrophic lake chaohu, china. Bull. Environ. Contam. Toxicol. 2017, 99, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Xu, F. Exergy and structural exergy as ecological indicators for the development state of the lake chaohu ecosystem. Ecol. Model. 1997, 99, 41–49. [Google Scholar] [CrossRef]
- Xu, F.L.; Jørgensen, S.E.; Tao, S.; Li, B.G. Modeling the effects of ecological engineering on ecosystem health of a shallow eutrophic chinese lake (lake chao). Ecol. Model. 1999, 117, 239–260. [Google Scholar] [CrossRef]
- Xu, F.-L.; Tao, S.; Xu, Z.R. The restoration of riparian wetlands and macrophytes in lake chao, an eutrophic chinese lake: Possibilities and effects. Hydrobiologia 1999, 405, 169–178. [Google Scholar] [CrossRef]
- Mingming, H.; Jianhua, M.; Xi, Z. Current situation and comprehensive control measures of water environment in the chaohu lake. Meteorol. Environ. Res. 2016, 7, 45. [Google Scholar]
- Shang, G.P.; Shang, J.C. Causes and control countermeasures of eutrophication in chaohu lake, china. Chin. Geogr. Sci. 2005, 15, 348–354. [Google Scholar] [CrossRef]
- Yang, B.; Jiang, Y.J.; He, W.; Liu, W.X.; Kong, X.Z.; Jørgensen, S.E.; Xu, F.L. The tempo-spatial variations of phytoplankton diversities and their correlation with trophic state levels in a large eutrophic chinese lake. Ecol. Indic. 2016, 66, 153–162. [Google Scholar] [CrossRef]
- Xu, F.L.; Tao, S.; Dawson, R.W.; Xu, Z.R. The distributions and effects of nutrients in the sediments of a shallow eutrophic chinese lake. Hydrobiologia 2003, 492, 85–93. [Google Scholar] [CrossRef]
- Yao, S.C.; Xue, B.; Li, S.J.; Liu, J.F.; Xia, W.F. Determination of Lake Sedimentation Rate in the Middle and Lower Reaches of the Yangtze River and Its Environmental Significance: A Case Study of Honghu, Chaohu and Taihu Lakes (in China). Resour. Environ. Yangtze River Basin 2006, 05, 569–573. [Google Scholar]
- Jin, X.; Tu, Q. The Standard Methods for Observation and Analysis in Lake Eutrophication; Chinese Environmental Science Press: Beijing, China, 1990; p. 240. [Google Scholar]
- Edgar, R.C. Search and clustering orders of magnitude faster than blast. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Samarajeewa, A.D.; Hammad, A.; Masson, L.; Khan, I.U.H.; Scroggins, R.; Beaudette, L.A. Comparative assessment of next-generation sequencing, denaturing gradient gel electrophoresis, clonal restriction fragment length polymorphism and cloning-sequencing as methods for characterizing commercial microbial consortia. J. Microbiol. Methods 2015, 108, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Vegan: Community Ecology Package. Available online: http://cran.r-project.org/package=vegan (accessed on 15 October 2019).
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Nagendra, H. Opposite trends in response for the shannon and simpson indices of landscape diversity. Appl. Geogr. 2002, 22, 175–186. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.I. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Ganzert, L.; Bajerski, F.; Wagner, D. Bacterial community composition and diversity of five different permafrost-affected soils of Northeast Greenland. FEMS Microbiol. Ecol. 2014, 89, 426–441. [Google Scholar] [CrossRef]
- Huang, W.; Jiang, X. Profiling of Sediment Microbial Community in Dongting Lake before and after Impoundment of the Three Gorges Dam. Int. J. Environ. Res. Public Health 2016, 13, 617. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Qin, H.; Guo, S.; Chen, W.; Liang, J. Bacterial communities of four adjacent fresh lakes at different trophic status. Ecotoxicol. Environ. Saf. 2018, 157, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Kurilkina, M.I.; Zakharova, Y.R.; Galachyants, Y.P.; Petrova, D.P.; Bukin, Y.S.; Domysheva, V.M.; Blinov, V.V.; Likhoshway, Y.V. Bacterial community composition in the water column of the deepest freshwater lake baikal as determined by next-generation sequencing. FEMS Microbiol. Ecol. 2016, 92, fiw094. [Google Scholar] [CrossRef]
- Hua, G.; Cheng, Y.; Kong, J.; Li, M.; Zhao, Z. High-throughput sequencing analysis of bacterial community spatiotemporal distribution in response to clogging in vertical flow constructed wetlands. Bioresour. Technol. 2017, 248, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, H.; Chen, X.; Wan, D.; Mai, W.; Sun, C. Cometabolic degradation of low-strength coking wastewater and the bacterial community revealed by high-throughput sequencing. Bioresour. Technol. 2017, 245, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Mei, J.; Jia, W.; Chen, J.; Li, X.; Ji, B.; Wu, H. Treatment of heavily polluted river water by tidal-operated biofilters with organic/inorganic media: Evaluation of performance and bacterial community. Bioresour. Technol. 2019, 279, 34–42. [Google Scholar] [CrossRef]
- Yan, Q.; Stegen, J.C.; Yu, Y.; Deng, Y.; Li, X.; Wu, S.; Dai, L.; Zhang, X.; Li, J.; Wang, C. Nearly a decade-long repeatable seasonal diversity patterns of bacterioplankton communities in the eutrophic lake donghu (wuhan, china). Mol. Ecol. 2017, 26, 3839–3850. [Google Scholar] [CrossRef]
- Haller, L.; Tonolla, M.; Zopfi, J.; Peduzzi, R.; Wildi, W.; Poté, J. Composition of bacterial and archaeal communities in freshwater sediments with different contamination levels (lake geneva, switzerland). Water Res. 2011, 45, 1213–1228. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, A.N.; Wang, J.; Liu, S.; Jiang, X.; Dang, C.; Ma, T.; Liu, S.; Chen, Q.; Xie, S. Integrated biogeography of planktonic and sedimentary bacterial communities in the yangtze river. Microbiome 2018, 6, 16. [Google Scholar] [CrossRef]
- Staley, C.; Gould, T.J.; Wang, P.; Phillips, J.; Cotner, J.B.; Sadowsky, M.J. Bacterial community structure is indicative of chemical inputs in the upper mississippi river. Front. Microbiol. 2014, 5, 524. [Google Scholar] [CrossRef]
- Wittebolle, L.; Marzorati, M.; Clement, L.; Balloi, A.; Daffonchio, D.; Heylen, K.; De Vos, P.; Verstraete, W.; Boon, N. Initial community evenness favours functionality under selective stress. Nature 2009, 458, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Lennon, J.T.; Jones, S.E. Microbial seed banks: The ecological and evolutionary implications of dormancy. Nat. Rev. Microbiol. 2011, 9, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Yong, L.; Zhang, J.; Lei, Z.; Zhang, X.; Xie, S. Spatial distribution of bacterial communities in high-altitude freshwater wetland sediment. Limnology 2014, 15, 249–256. [Google Scholar]
- Rodionov, D.A.; Dubchak, I.; Arkin, A.; Alm, E.; Gelfand, M.S. Reconstruction of regulatory and metabolic pathways in metal-reducing δ-proteobacteria. Genome Biol. 2004, 5, R90. [Google Scholar] [CrossRef]
- Bai, Y.; Shi, Q.; Wen, D.; Li, Z.; Jefferson, W.A.; Feng, C.; Tang, X. Bacterial communities in the sediments of dianchi lake, a partitioned eutrophic waterbody in china. PLoS ONE 2012, 7, e37796. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Yong, L.; Xie, S.; Liu, Y. Bacterioplankton communities in a high-altitude freshwater wetland. Ann. Microbiol. 2014, 64, 1405–1411. [Google Scholar] [CrossRef]
- Jingxu, Z.; Yuyin, Y.; Lei, Z.; Yuzhao, L.; Shuguang, X.; Yong, L. Distribution of sediment bacterial and archaeal communities in plateau freshwater lakes. Appl. Microbiol. Biotechnol. 2015, 99, 3291–3302. [Google Scholar]
- Liu, W.; Meng, T.; Wang, M.; Cheng, W. Spatial distribution characteristics of nutrients and microbial communities in sediments of water source reservoirs. Bull. Soil Water Conserv. 2016, 36, 24–29. [Google Scholar]
- Zhang, M.Q.; Chen, J. Investigation on pollution situation of phosphorus in the main rivers entering chaohu lake. Trace Elem. Sci. 2003, 10, 41–44. [Google Scholar]
- On Countermeasures for Agricultural Non-Point Source Pollution Control in Chaohu Lake Basin. Available online: http://en.cnki.com.cn/Article_en/CJFDTOTAL-HFGS200601024.htm (accessed on 15 October 2019).
- Sht, A.; Khaĭdarov, S.A. Hygienic study of the stability of methylnitrophos and pentachloronitrobenzene used in agriculture. Gig. I Sanit. 1983, 2, 74–75. [Google Scholar]
- Bolam, D.N.; Koropatkin, N.M. Glycan recognition by the bacteroidetes sus-like systems. Curr. Opin. Struct. Biol. 2012, 22, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Gómez, B.; Richter, M.; Schüler, M.; Pinhassi, J.; Acinas, S.G.; González, J.M.; Pedrós-Alió, C. Ecology of marine bacteroidetes: A comparative genomics approach. ISME J. 2013, 7, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.; Hehemann, J.H.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and gut bacteroidetes: The food connection. Front. Microbiol. 2011, 2, 93. [Google Scholar] [CrossRef] [PubMed]
- Margarete, B.; Michael, K.; Hanno, T.; Michael, R.; Thierry, L.; Elke, A.; Würdemann, C.A.; Christian, Q.; Heiner, K.; Florian, K. Whole genome analysis of the marine bacteroidetes’gramella forsetii’ reveals adaptations to degradation of polymeric organic matter. Environ. Microbiol. 2010, 8, 2201–2213. [Google Scholar]
- Campbell, B.J. The Family Acidobacteriaceae; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Rmm, A.; Safi, N.J.; De, B.D.; El-Nahhal, Y.; Rullkotter, J. Microbial diversity of a heavily polluted microbial mat and its community changes following degradation of petroleum compounds. Appl. Environ. Microbiol. 2002, 68, 1674–1683. [Google Scholar]
- Choudhari, S.; Lohia, R.; Grigoriev, A. Comparative metagenome analysis of an alaskan glacier. J. Bioinform. Comput. Biol. 2014, 12, 1441003. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Xing, C.; Kun, W.; Chen, J.; Zheng, B.; Xia, J. Comparison among the microbial communities in the lake, lake wetland, and estuary sediments of a plain river network. Microbiologyopen 2018, 8, e00644. [Google Scholar]
- Branco, R.; Chung, A.P.; Ver’Issimo, A.N.; Morais, P.V. Impact of chromium-contaminated wastewaters on the microbial community of a river. FEMS Microbiol. Ecol. 2010, 54, 35–46. [Google Scholar] [CrossRef]
- Xie, X.Y.; Qian, X.; Zhang, Y.C.; Qian, Y. Effect on chaohu lake water environment of water transfer from yangtze river to chaohu lake. Res. Environ. Sci. 2009, 22, 897–901. [Google Scholar]
- Chen, Y.F. In Study on the response of lake chaohu eutrophication to yangtze river—Lake chaohu water transfer project. In Proceedings of the World Congress on Computer Science & Information Engineering, Los Angeles, CA, USA, 31 March–2 April 2009; Volume 5, pp. 614–619. [Google Scholar]
- Fan, L.; Song, C.; Meng, S.; Qiu, L.; Zheng, Y.; Wu, W.; Qu, J.; Li, D.; Zhang, C.; Hu, G. Spatial distribution of planktonic bacterial and archaeal communities in the upper section of the tidal reach in yangtze river. Sci. Rep. 2016, 6, 39147. [Google Scholar] [CrossRef]
- Liu, M.; Tian, X.; Wu, Y.; Zhou, F.; Huang, H.; Bao, S.; Zhang, W. Temporal distribution of bacterial community structure in the changjiang estuary hypoxia area and the adjacent east china sea. Environ. Res. Lett. 2012, 7, 33–40. [Google Scholar] [CrossRef]
- Ludwig, W.; Schleifer, K.H.; Whitman, W.B. Taxonomic Outline of the Phylum Firmicutes; Springer: New York, NY, USA, 2015. [Google Scholar]
- Franz, C.M.A.P.; Huch, M.; Cho, G.S.; Toit, M.D. The Family Streptococcaceae; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Pascal, H.; Frédéric, H.; Laetitia, F.; BenoîT, G.; Deborah, P.; Nathalie, L.B.; Bernard, D.; Alexander, B.; Christine, D.; Ehrlich, S.D.; et al. New insights in the molecular biology and physiology of streptococcus thermophilus revealed by comparative genomics. FEMS Microbiol. Rev. 2005, 29, 435–463. [Google Scholar]
- Octavia, S.; Lan, R. The Family Enterobacteriaceae; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Dann, L.M.; Smith, R.J.; Jeffries, T.C.; Mckerral, J.C.; Fairweather, P.G.; Oliver, R.L.; Mitchell, J.G. Persistence, loss and appearance of bacteria upstream and downstream of a river system. Mar. Freshw. Res. 2017, 68, 851–862. [Google Scholar] [CrossRef]
- Duarte, S.; Cássio, F.; Ferreira, V.; Canhoto, C.; Pascoal, C. Seasonal variability may affect microbial decomposers and leaf decomposition more than warming in streams. Microb. Ecol. 2016, 72, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Cardona, C.; Weisenhorn, P.; Henry, C.; Gilbert, J.A. Network-based metabolic analysis and microbial community modeling. Curr. Opin. Microbiol. 2016, 31, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A. Microbial community structure and its functional implications. Nature 2009, 459, 193–199. [Google Scholar] [CrossRef]
- Röling, W.F.M. The Family Geobacteraceae; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Evans, T.G.; Hofmann, G.E. Defining the limits of physiological plasticity: How gene expression can assess and predict the consequences of ocean change. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 1733–1745. [Google Scholar] [CrossRef]
- Lenski, E.R. Experimental evolution and the dynamics of adaptation and genome evolution in microbial populations. ISME J. 2017, 11, 2181–2194. [Google Scholar] [CrossRef]
- Meyer, A.F.; Lipson, D.A.; Martin, A.P.; Schadt, C.W.; Schmidt, S.K. Molecular and metabolic characterization of cold-tolerant alpine soil pseudomonas sensu stricto. Appl. Environ. Microbiol. 2004, 70, 483–489. [Google Scholar] [CrossRef]
- Aiba, M.; Takafumi, H.; Hiura, T. Interspecific differences in determinants of plant species distribution and the relationships with functional traits. J. Ecol. 2012, 100, 950–957. [Google Scholar] [CrossRef]
- Alimov, A.F. Structural and functional-characteristics of aquatic animal communities. Int. Rev. Hydrobiol. 2010, 76, 169–182. [Google Scholar] [CrossRef]
- Pedrósalió, C. Marine microbial diversity: Can it be determined? Trends Microbiol. 2006, 14, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Ateequr, R.; Patricia, L.; Andreas, N.; Stephan, H.; Stefan, S.; Ott, S.J. Transcriptional activity of the dominant gut mucosal microbiota in chronic inflammatory bowel disease patients. J. Med. Microbiol. 2010, 59, 1114–1122. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Groups | Statistic | P value | Permutation Number |
|---|---|---|---|---|
| ANOSIM | Seasonal | 0.319 | 0.001 | 999 |
| Spatial | −0.083 | 0.929 | 999 |
| Taxon | No. of Bacterial Taxa with Significant Seasonal Variation | Total no. of Bacterial Taxa in LEfSe Analysis | No. of Bacterial Taxa Enriched in the Sediment During the 4 Seasons | |||
|---|---|---|---|---|---|---|
| Spring | Summer | Autumn | Winter | |||
| Phylum | 8 | 15 | 1 | 1 | 0 | 6 |
| Class | 16 | 32 | 3 | 2 | 3 | 8 |
| Order | 31 | 65 | 7 | 8 | 4 | 12 |
| Family | 34 | 78 | 8 | 9 | 4 | 13 |
| Genus | 42 | 64 | 101 | 11 | 5 | 15 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Cheng, Y.; Gao, G.; Jiang, J. Spatial-Temporal Variation of Bacterial Communities in Sediments in Lake Chaohu, a Large, Shallow Eutrophic Lake in China. Int. J. Environ. Res. Public Health 2019, 16, 3966. https://doi.org/10.3390/ijerph16203966
Zhang L, Cheng Y, Gao G, Jiang J. Spatial-Temporal Variation of Bacterial Communities in Sediments in Lake Chaohu, a Large, Shallow Eutrophic Lake in China. International Journal of Environmental Research and Public Health. 2019; 16(20):3966. https://doi.org/10.3390/ijerph16203966
Chicago/Turabian StyleZhang, Lei, Yu Cheng, Guang Gao, and Jiahu Jiang. 2019. "Spatial-Temporal Variation of Bacterial Communities in Sediments in Lake Chaohu, a Large, Shallow Eutrophic Lake in China" International Journal of Environmental Research and Public Health 16, no. 20: 3966. https://doi.org/10.3390/ijerph16203966
APA StyleZhang, L., Cheng, Y., Gao, G., & Jiang, J. (2019). Spatial-Temporal Variation of Bacterial Communities in Sediments in Lake Chaohu, a Large, Shallow Eutrophic Lake in China. International Journal of Environmental Research and Public Health, 16(20), 3966. https://doi.org/10.3390/ijerph16203966