Low Dose Cadmium Inhibits Proliferation of Human Renal Mesangial Cells via Activation of the JNK Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Cell Culture
2.2. Western Blotting
2.3. Cell Proliferation Assay
2.4. Cell Viability Assay
2.5. Annexin V-FITC/PI Analyses
2.6. Immunofluorescence
2.7. Phalloidin-Labelling
2.8. Statistical Analysis
3. Results
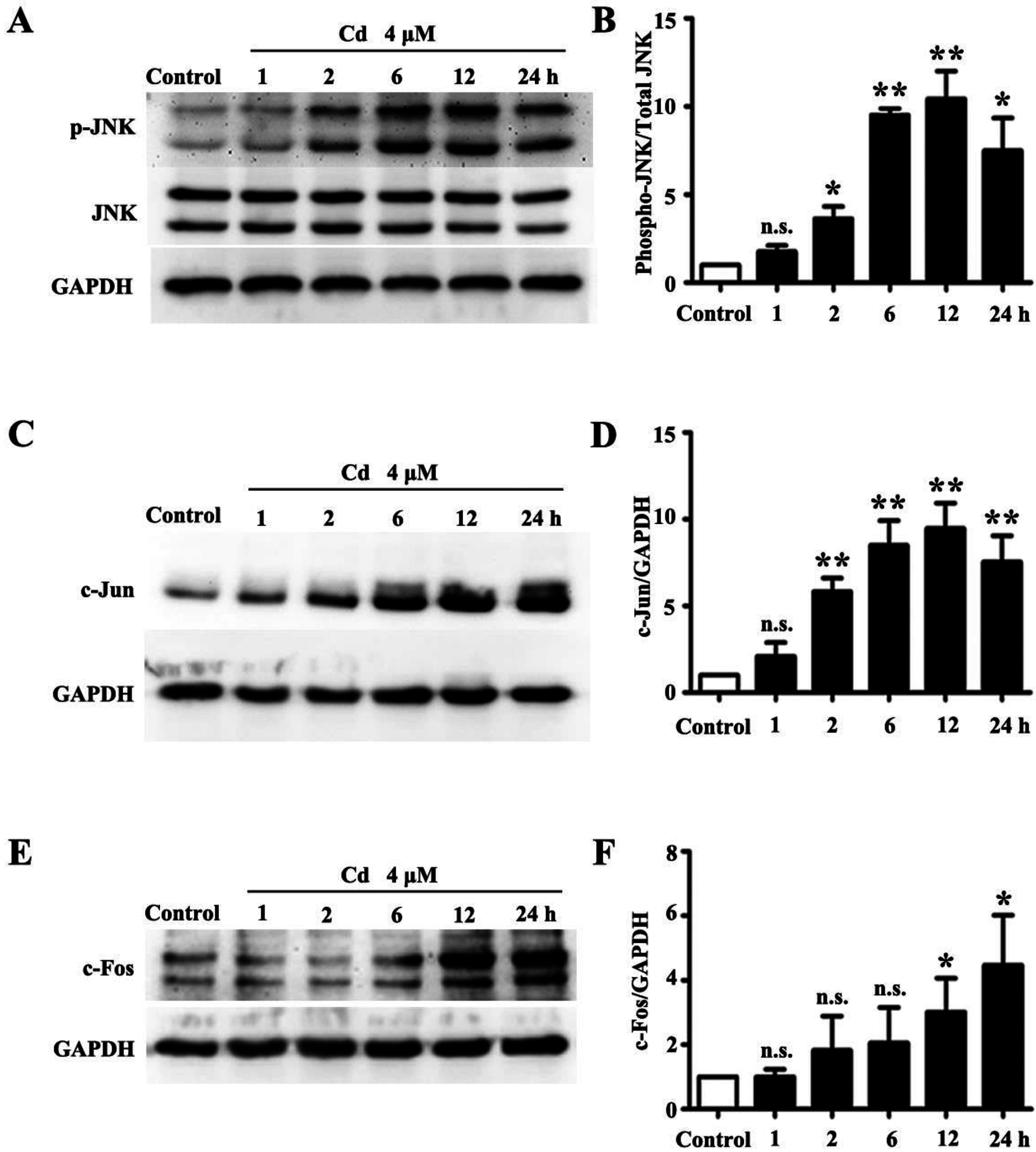
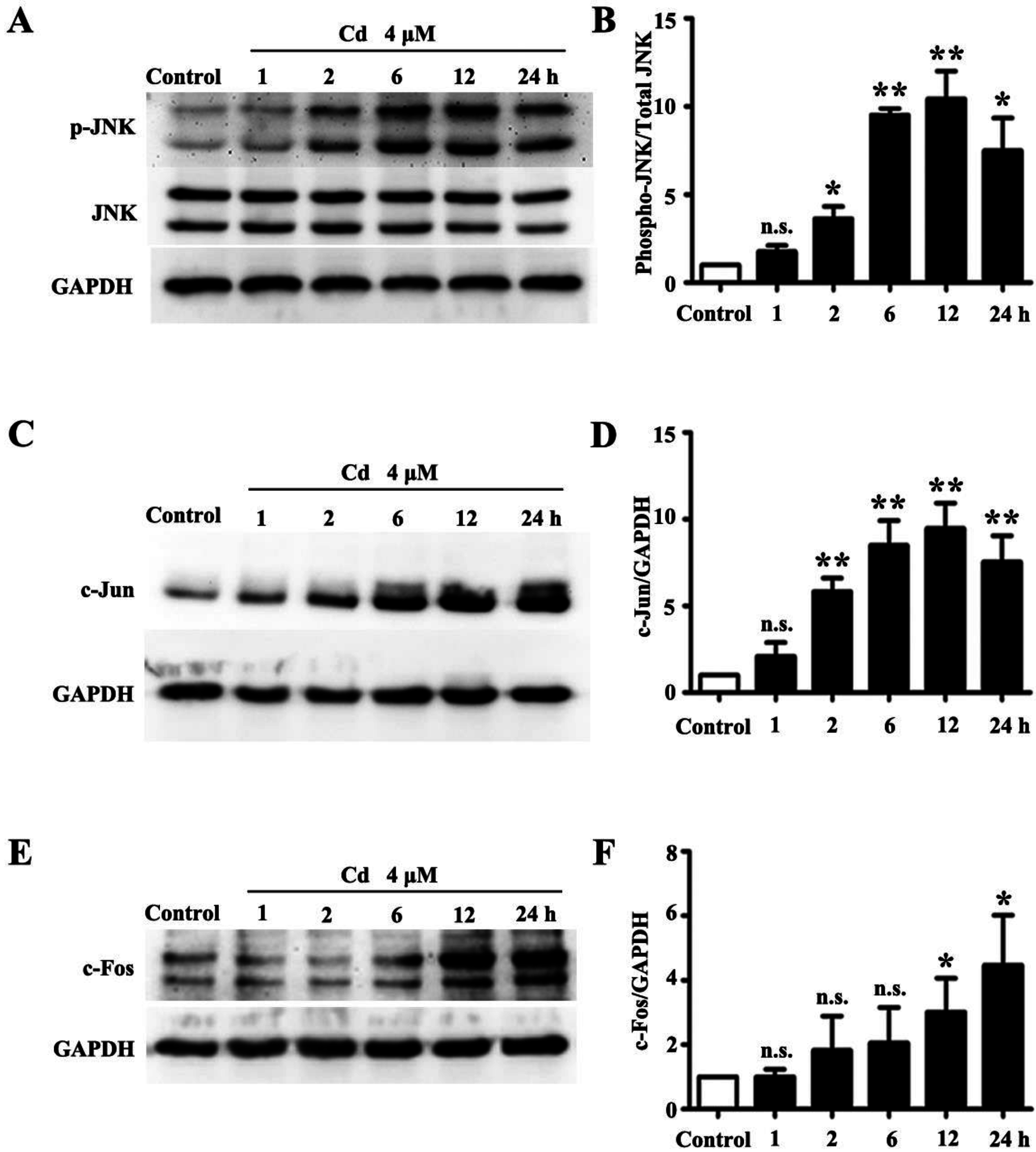
3.1. Low Dose Cd Activates JNK Pathway in HRMCs
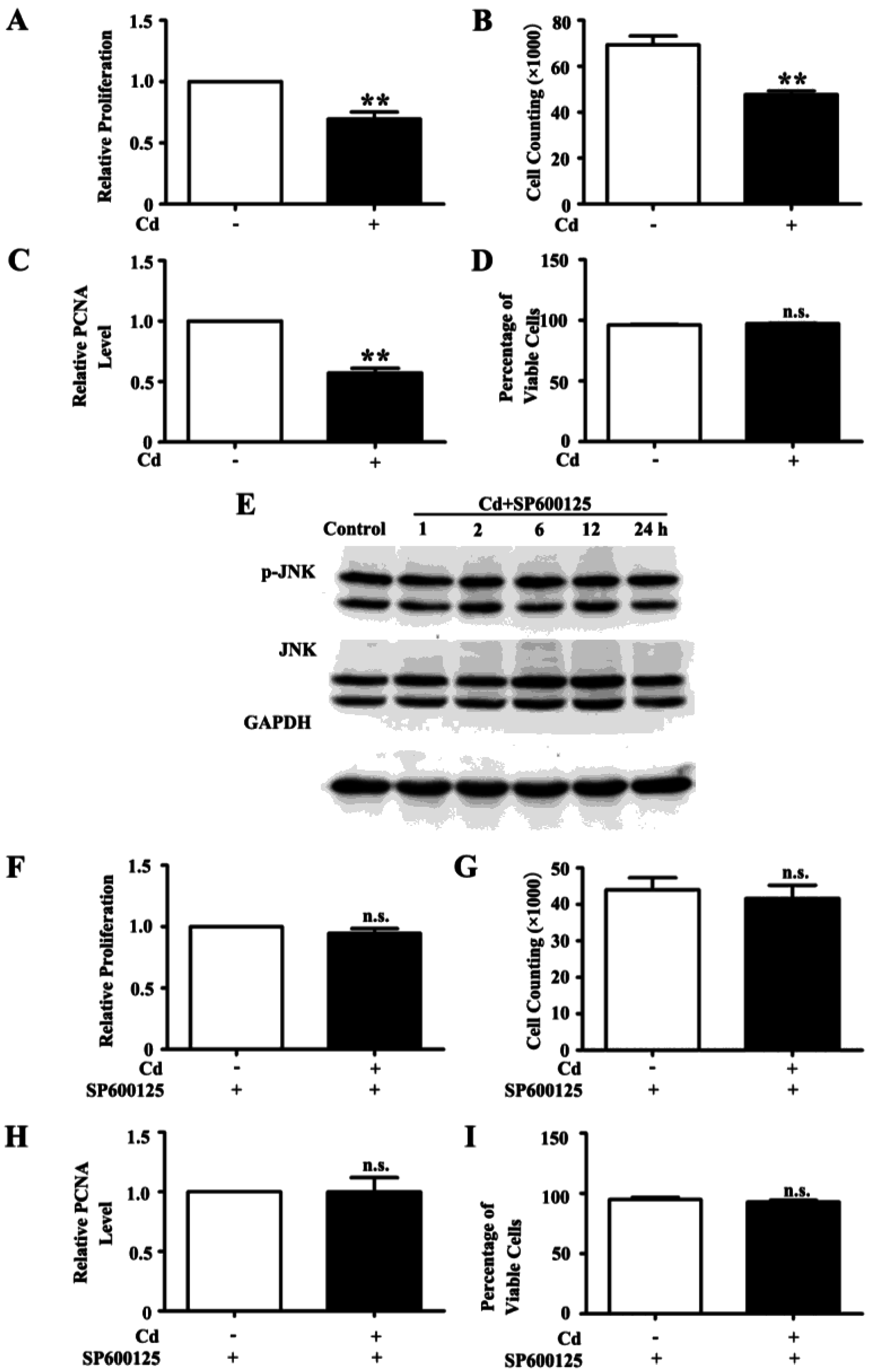
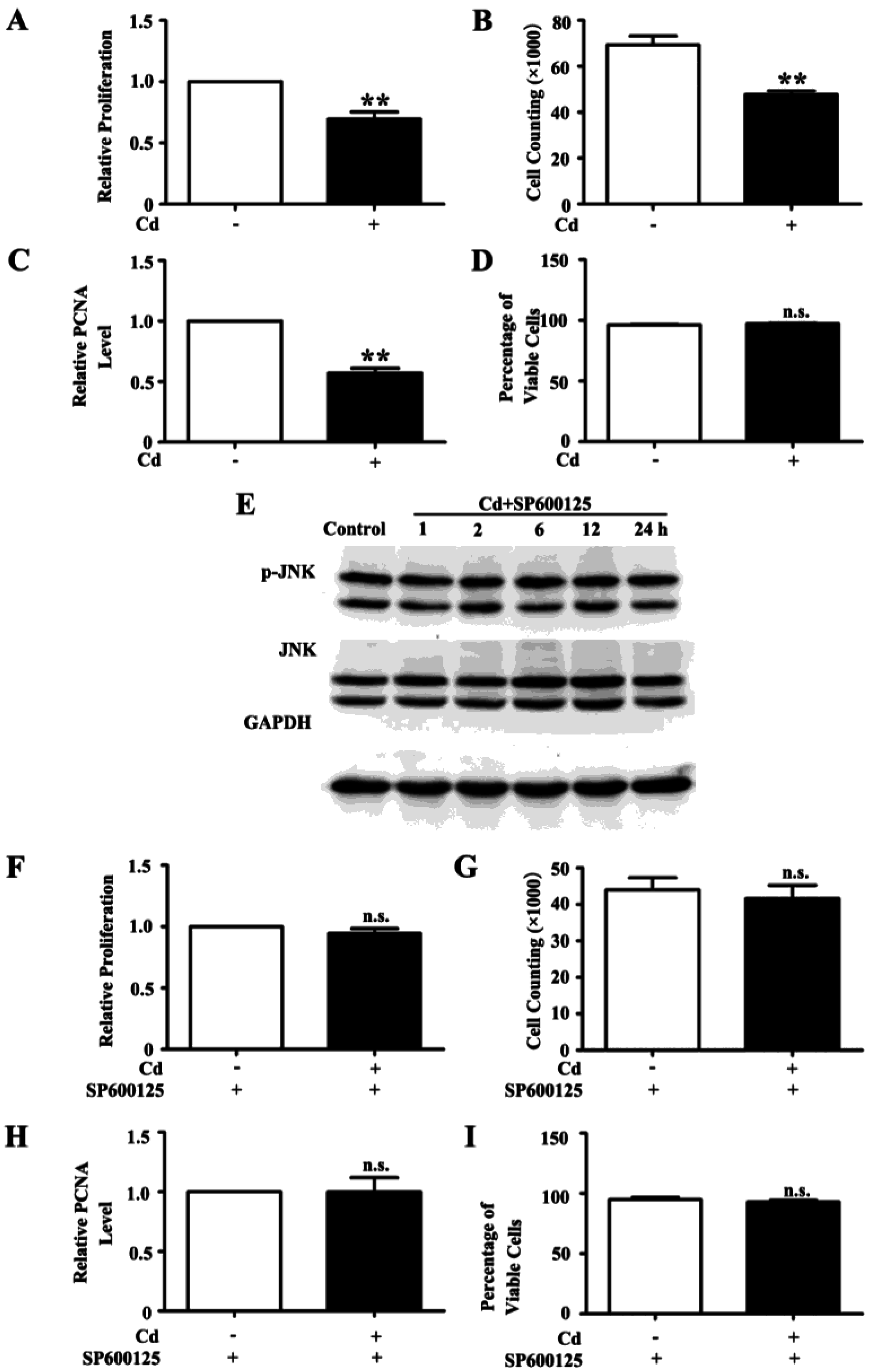
3.2. Low Dose Cd Inhibits Proliferation of HRMC via Activation of JNK Pathway
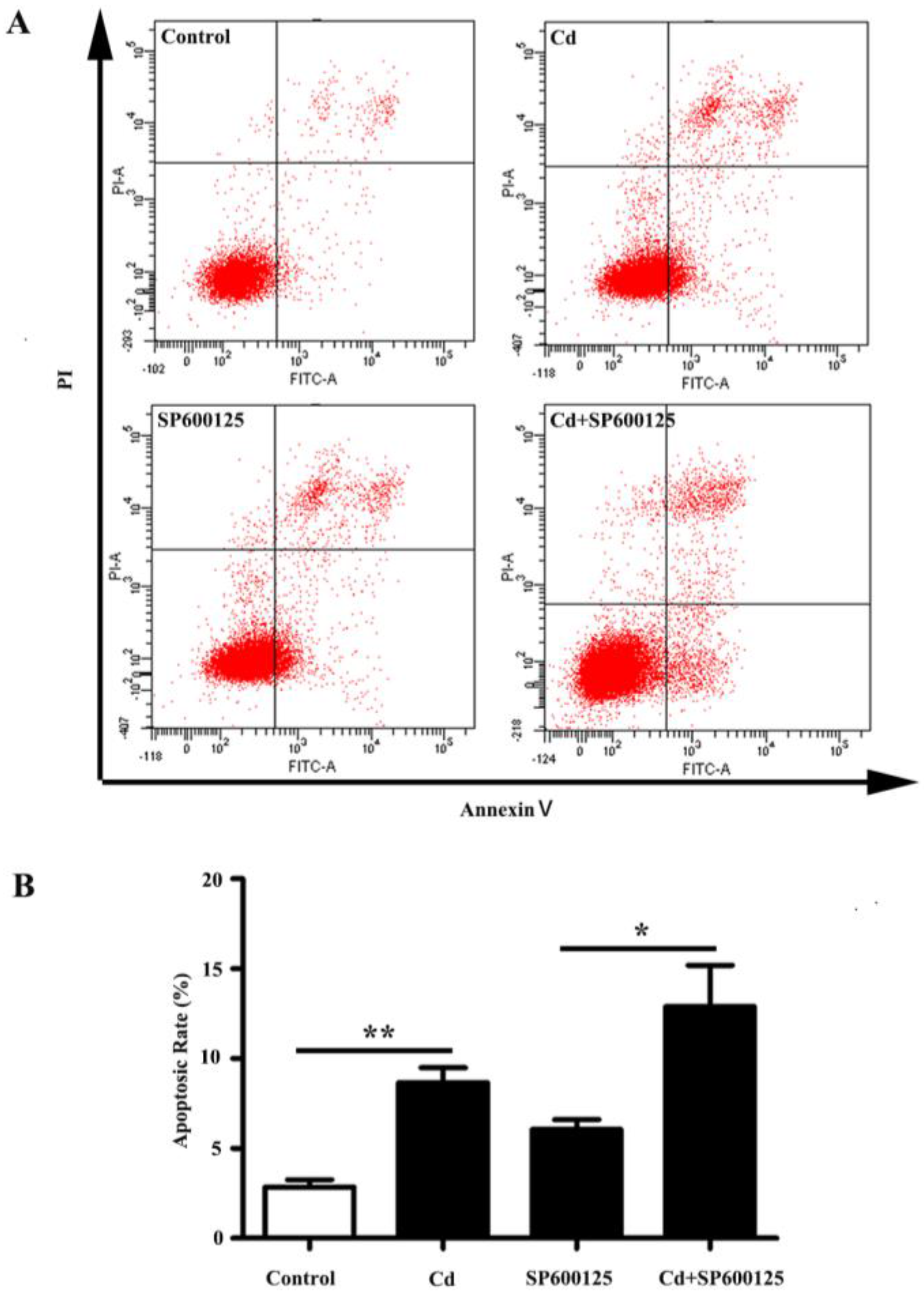
3.3. Effects of Low Dose Cd Exposure on Apoptosis of HRMCs.
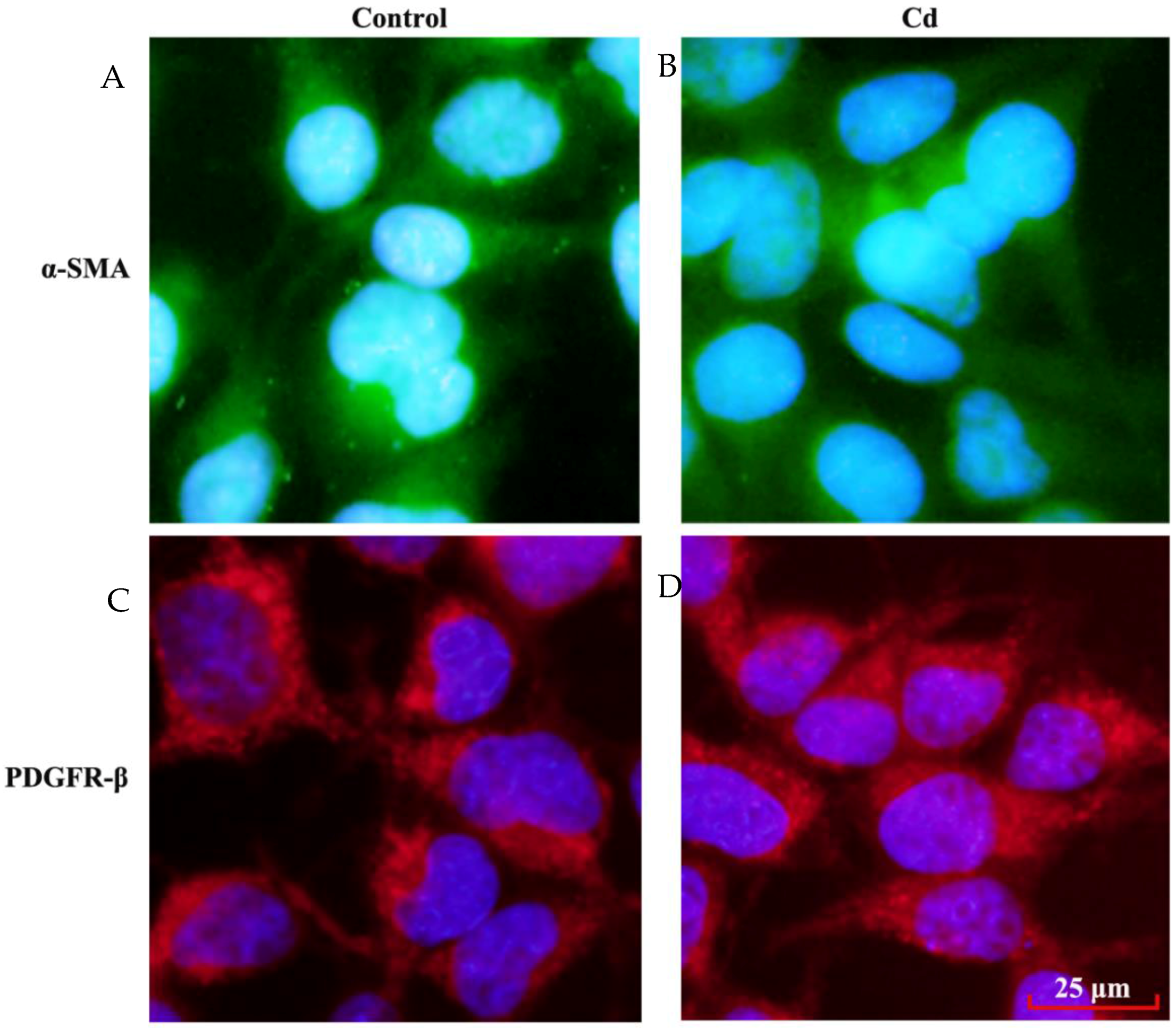
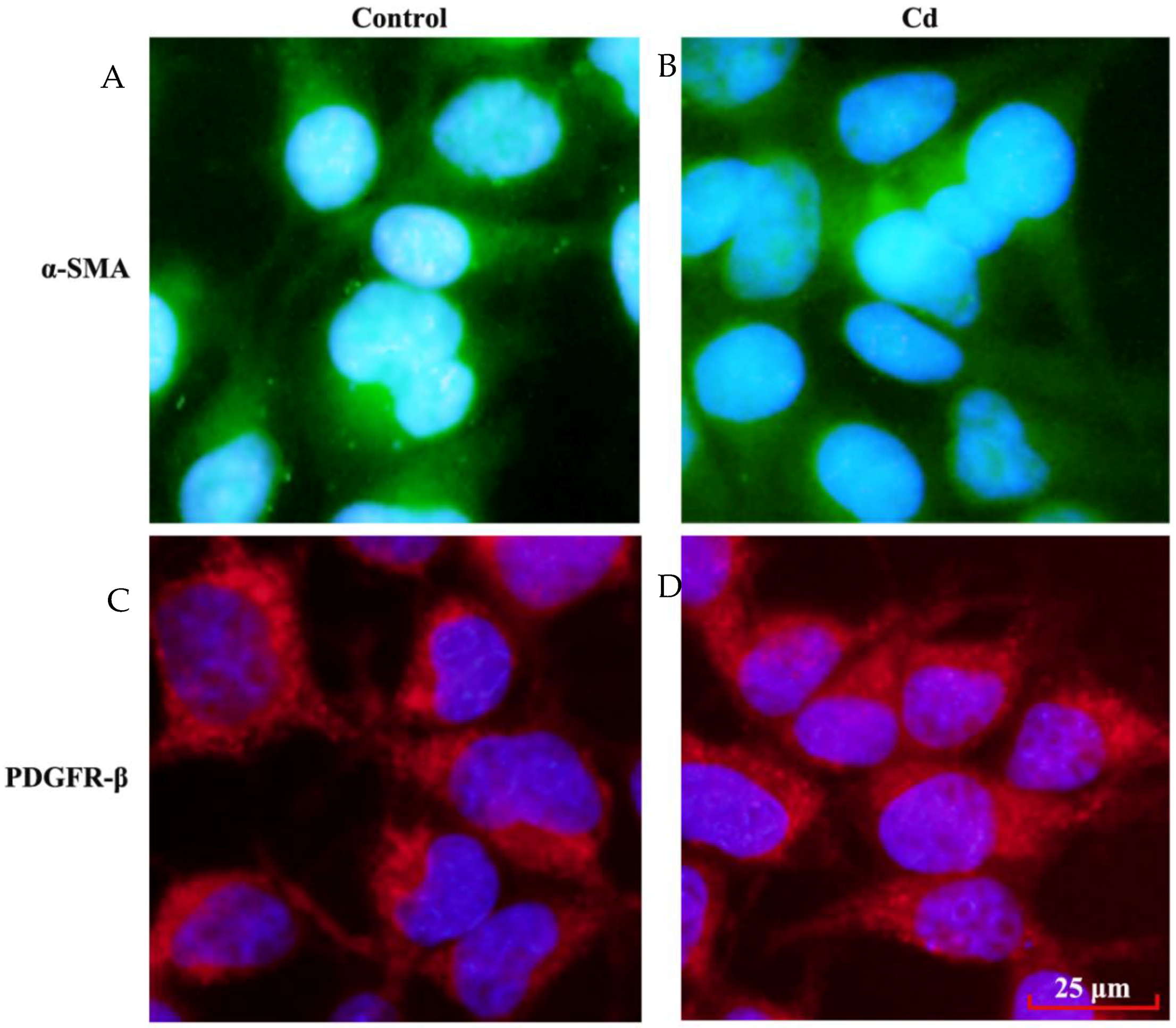
3.4. Effects of Low Dose Cd Exposure on theMesangial Cell Markers on HRMCs
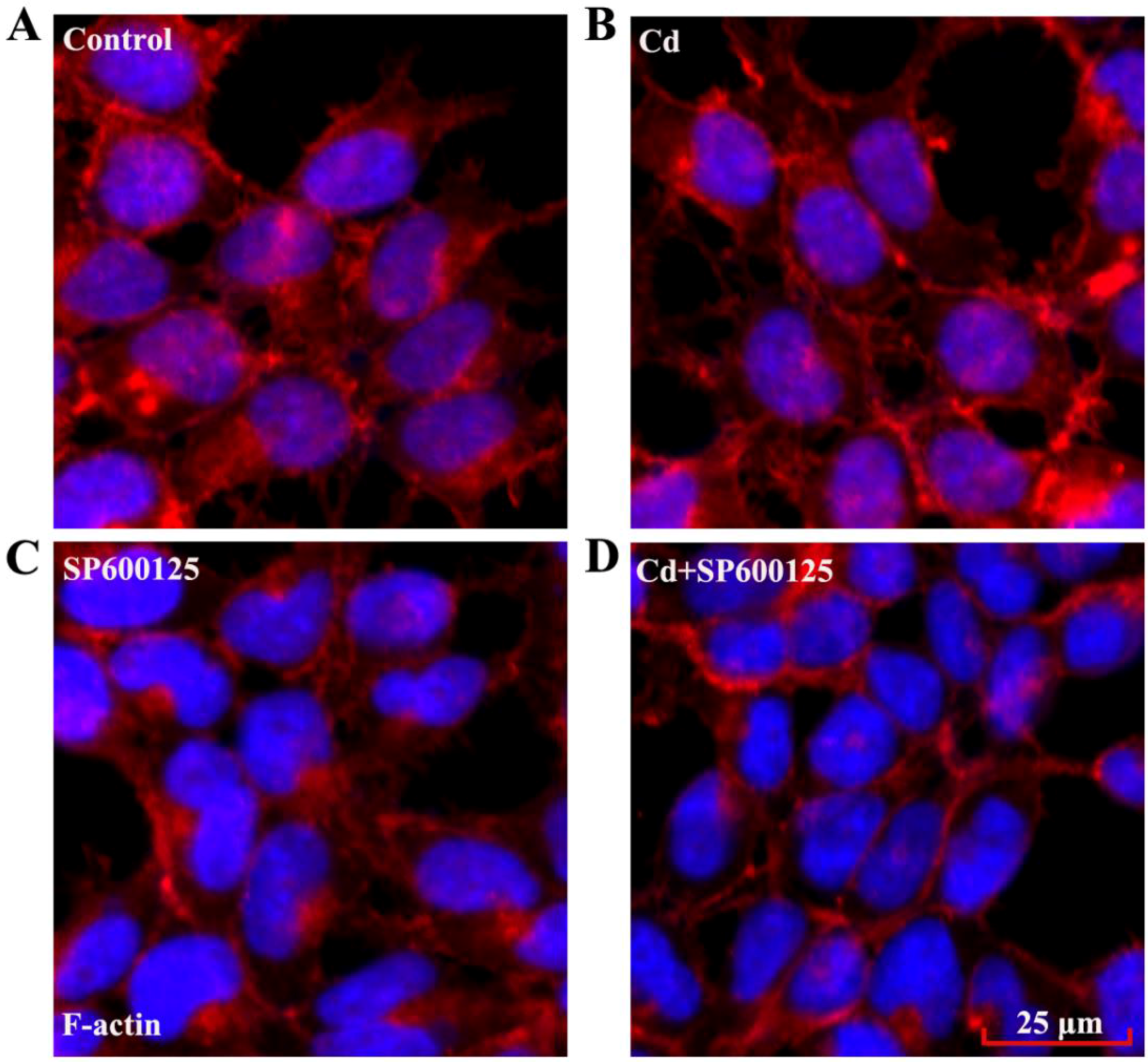
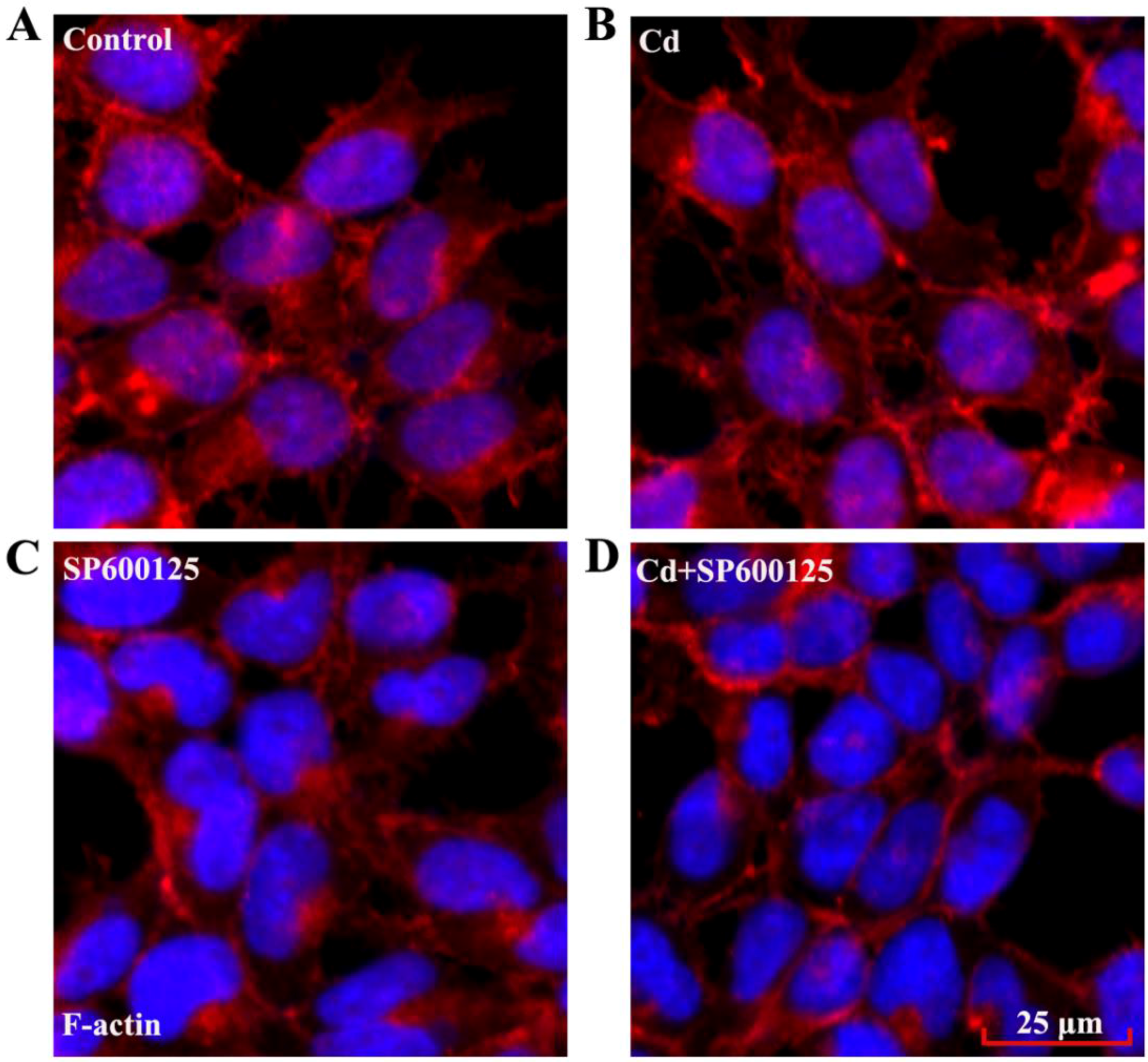
3.5. Effects of Low Dose Cd Exposure on the Actin Cytoskeleton of HRMCs
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nordberg, G.; Jin, T.; Wu, X.; Lu, J.; Chen, L.; Liang, Y.; Lei, L.; Hong, F.; Bergdahl, I.A.; Nordberg, M. Kidney dysfunction and cadmium exposure—Factors influencing dose-response relationships. J. Trace Elem. Med. Biol. 2012, 26, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Jarup, L.; Akesson, A. Current status of cadmium as an environmental health problem. Toxicol. Appl. Pharmacol. 2009, 238, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Garrett, S.H.; Sens, M.A.; Sens, D.A. Cadmium, environmental exposure, and health outcomes. Environ. Health Perspect. 2010, 118, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Prozialeck, W.C.; Edwards, J.R. Early biomarkers of cadmium exposure and nephrotoxicity. Biometals 2010, 23, 793–809. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y.; Klaassen, C.D. Nephrotoxicity of CdCl2 and Cd-metallothionein in cultured rat kidney proximal tubules and LLC-PK1 cells. Toxicol. Appl. Pharmacol. 1994, 128, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Prozialeck, W.C.; Edwards, J.R.; Woods, J.M. The vascular endothelium as a target of cadmium toxicity. Life Sci. 2006, 79, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.H. Glomerular basement membrane composition and the filtration barrier. Pediatr. Nephrol. 2011, 26, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Varagic, J. The ANG-(1–7)/ACE2/mas axis in the regulation of nephron function. Am. J. Physiol. Renal Physiol. 2010, 298, F1297–F1305. [Google Scholar] [CrossRef] [PubMed]
- Couser, W.G. New insights into mechanisms of immune glomerular injury. West J. Med. 1994, 160, 440–446. [Google Scholar] [PubMed]
- Mene, P.; Simonson, M.S.; Dunn, M.J. Physiology of the mesangial cell. Physiol. Rev. 1989, 69, 1347–1424. [Google Scholar] [PubMed]
- Ricono, J.M.; Xu, Y.C.; Arar, M.; Jin, D.C.; Barnes, J.L.; Abboud, H.E. Morphological insights into the origin of glomerular endothelial and mesangial cells and their precursors. J. Histochem. Cytochem. 2003, 51, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, P.; Hellstrom, M.; Kalen, M.; Karlsson, L.; Pekny, M.; Pekna, M.; Soriano, P.; Betsholtz, C. Paracrine PDGF-B/PDGF-Rbeta signaling controls mesangial cell development in kidney glomeruli. Development 1998, 125, 3313–3322. [Google Scholar] [PubMed]
- Templeton, D.M.; Liu, Y. Effects of cadmium on the actin cytoskeleton in renal mesangial cells. Can. J. Physiol. Pharmacol. 2013, 91, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Ding, G.; Huang, S.; Wu, Y.; Pan, X.; Guan, X.; Chen, R.; Yang, T. c-Jun NH2-terminal kinase mediation of angiotensin II-induced proliferation of human mesangial cells. Am. J. Physiol. Renal Physiol. 2005, 288, F1118–F1124. [Google Scholar] [CrossRef] [PubMed]
- Woodgett, J.R.; Avruch, J.; Kyriakis, J.M. Regulation of nuclear transcription factors by stress signals. Clin. Exp. Pharmacol. Physiol. 1995, 22, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal transduction by the JNK group of map kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Kang, Y.J.; Jeon, E.S.; Song, H.Y.; Woo, J.S.; Jung, J.S.; Kim, Y.K.; Kim, J.H. Role of c-Jun N-terminal kinase in the PDGF-induced proliferation and migration of human adipose tissue-derived mesenchymal stem cells. J. Cell. Biochem. 2005, 95, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Mengistu, M.; Brotzman, H.; Ghadiali, S.; Lowe-Krentz, L. Fluid shear stress-induced JNK activity leads to actin remodeling for cell alignment. J. Cell. Physiol. 2011, 226, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.R.; Degheselle, O.; Smeets, K.; Van Kerkhove, E.; Cuypers, A. Cadmium-induced pathologies: Where is the oxidative balance lost (or not)? Int. J. Mol. Sci. 2013, 14, 6116–6143. [Google Scholar] [CrossRef] [PubMed]
- Szymanska, E.; Skowronek, A.; Miaczynska, M. Impaired dynamin 2 function leads to increased AP-1 transcriptional activity through the JNK/c-Jun pathway. Cell. Signal. 2016, 28, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Jurikova, M.; Danihel, L.; Polak, S.; Varga, I. Ki67, PCNA, and MCM proteins: Markers of proliferation in the diagnosis of breast cancer. Acta Histochem. 2016, 118, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O′Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef] [PubMed]
- Diah, S.; Zhang, G.X.; Nagai, Y.; Zhang, W.; Gang, L.; Kimura, S.; Hamid, M.R.; Tamiya, T.; Nishiyama, A.; Hitomi, H. Aldosterone induces myofibroblastic transdifferentiation and collagen gene expression through the Rho-kinase dependent signaling pathway in rat mesangial cells. Exp. Cell Res. 2008, 314, 3654–3662. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Templeton, D.M. Cellular factors mediate cadmium-dependent actin depolymerization. Toxicol. Appl. Pharmacol. 1996, 139, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chin, T.A.; Templeton, D.M. Calcium-independent effects of cadmium on actin assembly in mesangial and vascular smooth muscle cells. Cell Motil. Cytoskel. 1996, 33, 208–222. [Google Scholar] [CrossRef]
- Danjo, Y.; Gipson, I.K. Actin “purse string” filaments are anchored by E-cadherin-mediated adherens junctions at the leading edge of the epithelial wound, providing coordinated cell movement. J. Cell Sci. 1998, 111 (Pt 22), 3323–3332. [Google Scholar] [PubMed]
- Gloushankova, N.A.; Krendel, M.F.; Alieva, N.O.; Bonder, E.M.; Feder, H.H.; Vasiliev, J.M.; Gelfand, I.M. Dynamics of contacts between lamellae of fibroblasts: Essential role of the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 1998, 95, 4362–4367. [Google Scholar] [CrossRef] [PubMed]
- Sens, P.; Plastino, J. Membrane tension and cytoskeleton organization in cell motility. J. Phys. Condens. Matter 2015. [Google Scholar] [CrossRef] [PubMed]
- Templeton, D.M.; Liu, Y. Multiple roles of cadmium in cell death and survival. Chem. Biol. Interact. 2010, 188, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Thevenod, F.; Lee, W.K. Toxicology of cadmium and its damage to mammalian organs. Metal Ions Life Sci. 2013, 11, 415–490. [Google Scholar] [PubMed]
- Liu, J.; Kapron, C.M. Differential induction of map kinase signalling pathways by cadmium in primary cultures of mouse embryo limb bud cells. Reprod. Toxicol. 2010, 29, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wang, B.; Li, L.; Dong, F.; Chen, X.; Li, Y.; Dong, X.; Wada, Y.; Kapron, C.M.; Liu, J. Low-dose cadmium upregulates VEGF expression in lung adenocarcinoma cells. Int. J. Environ. Res. Public Health 2015, 12, 10508–10521. [Google Scholar] [CrossRef] [PubMed]
- Prozialeck, W.C.; Edwards, J.R.; Nebert, D.W.; Woods, J.M.; Barchowsky, A.; Atchison, W.D. The vascular system as a target of metal toxicity. Toxicol. Sci. 2008, 102, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Guo, F.; Li, L.; Guo, L.; Hou, Y.; Hao, E.; Yan, S.; Allen, T.D.; Liu, J. Cadmium induces vascular permeability via activation of the p38 MAPK pathway. Biochem. Biophys. Res. Commun. 2014, 450, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Dong, F.; Xu, D.; Du, L.; Yan, S.; Hu, H.; Lobe, C.G.; Yi, F.; Kapron, C.M.; Liu, J. Short-term, low-dose cadmium exposure induces hyperpermeability in human renal glomerular endothelial cells. J. Appl. Toxicol. 2016, 36, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Eichler, T.E.; Ransom, R.F.; Smoyer, W.E. Differential induction of podocyte heat shock proteins by prolonged single and combination toxic metal exposure. Toxicol. Sci. 2005, 84, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.K.; Lee, W.K.; Molitor, M.; Wolff, N.A.; Thevenod, F. Cadmium induces Wnt signaling to upregulate proliferation and survival genes in sub-confluent kidney proximal tubule cells. Mol. Cancer 2010. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Shih, Y.L.; Lee, C.C.; Chen, W.L.; Lin, C.J.; Lin, Y.S.; Wu, K.H.; Shih, C.M. The role of endoplasmic reticulum in cadmium-induced mesangial cell apoptosis. Chem. Biol. Interact. 2009, 181, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Schlondorff, D. Roles of the mesangium in glomerular function. Kidney Int. 1996, 49, 1583–1585. [Google Scholar] [CrossRef] [PubMed]
- Floege, J.; Radeke, H.R.; Johnson, R.J. Glomerular cells in vitro versus the glomerulus in vivo. Kidney Int. 1994, 45, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, M.; Eminel, S.; Herdegen, T.; Waetzig, V. C-Jun N-terminal kinases (JNKs) and the cytoskeleton--functions beyond neurodegeneration. Int. J. Dev. Neurosci. 2004, 22, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Seger, R.; Krebs, E.G. The mapk signaling cascade. Faseb. J. 1995, 9, 726–735. [Google Scholar] [PubMed]
- Chuang, S.M.; Wang, I.C.; Yang, J.L. Roles of JNK, p38 and ERK mitogen-activated protein kinases in the growth inhibition and apoptosis induced by cadmium. Carcinogenesis 2000, 21, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, L.; Han, W.; Shen, T.; Ma, C.; Liu, Y.; Nie, X.; Liu, M.; Ran, Y.; Zhu, D. Activation of JNK/c-Jun is required for the proliferation, survival, and angiogenesis induced by EET in pulmonary artery endothelial cells. J. Lipid Res. 2012, 53, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Gururajan, M.; Chui, R.; Karuppannan, A.K.; Ke, J.; Jennings, C.D.; Bondada, S. c-Jun N-terminal kinase (JNK) is required for survival and proliferation of b-lymphoma cells. Blood 2005, 106, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian map kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, L.; Wang, Y.; Dong, F.; Chen, X.; Liu, F.; Xu, D.; Yi, F.; Kapron, C.M.; Liu, J. NF-κB signaling maintains the survival of cadmium-exposed human renal glomerular endothelial cells. Int. J. Mol. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, M.T.; Kanduri, K.; Lahdesmaki, H.J.; Lahesmaa, R.; Henttinen, T.A. Tubulin- and actin-associating GIMAP4 is required for IFN-γ secretion during Th cell differentiation. Immunol. Cell Biol. 2015, 93, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Jones, Y.; Ellisman, M.H.; Goldstein, L.S.; Karin, M. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev. Cell 2003, 4, 521–533. [Google Scholar] [CrossRef]
- Ispanovic, E.; Haas, T.L. JNK and PI3K differentially regulate MMP-2 and MT1-MMP mRNA and protein in response to actin cytoskeleton reorganization in endothelial cells. Am. J. Physiol. Cell Physiol. 2006, 291, C579–C588. [Google Scholar] [CrossRef] [PubMed]
- Buschhausen, L.; Seibold, S.; Gross, O.; Matthaeus, T.; Weber, M.; Schulze-Lohoff, E. Regulation of mesangial cell function by vasodilatory signaling molecules. Cardiovasc. Res. 2001, 51, 463–469. [Google Scholar] [CrossRef]
- Usatyuk, P.V.; Natarajan, V. Role of mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced actin remodeling and barrier function in endothelial cells. J. Biol. Chem. 2004, 279, 11789–11797. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Li, J.; Cheng, Z.; Xu, Y.; Wang, X.; Li, X.; Xu, D.; Kapron, C.M.; Liu, J. Low Dose Cadmium Inhibits Proliferation of Human Renal Mesangial Cells via Activation of the JNK Pathway. Int. J. Environ. Res. Public Health 2016, 13, 990. https://doi.org/10.3390/ijerph13100990
Chen X, Li J, Cheng Z, Xu Y, Wang X, Li X, Xu D, Kapron CM, Liu J. Low Dose Cadmium Inhibits Proliferation of Human Renal Mesangial Cells via Activation of the JNK Pathway. International Journal of Environmental Research and Public Health. 2016; 13(10):990. https://doi.org/10.3390/ijerph13100990
Chicago/Turabian StyleChen, Xiaocui, Jing Li, Zuowang Cheng, Yinghua Xu, Xia Wang, Xiaorui Li, Dongmei Xu, Carolyn M. Kapron, and Ju Liu. 2016. "Low Dose Cadmium Inhibits Proliferation of Human Renal Mesangial Cells via Activation of the JNK Pathway" International Journal of Environmental Research and Public Health 13, no. 10: 990. https://doi.org/10.3390/ijerph13100990
APA StyleChen, X., Li, J., Cheng, Z., Xu, Y., Wang, X., Li, X., Xu, D., Kapron, C. M., & Liu, J. (2016). Low Dose Cadmium Inhibits Proliferation of Human Renal Mesangial Cells via Activation of the JNK Pathway. International Journal of Environmental Research and Public Health, 13(10), 990. https://doi.org/10.3390/ijerph13100990