Chitosan Nanoparticles Act as an Adjuvant to Promote both Th1 and Th2 Immune Responses Induced by Ovalbumin in Mice


Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Chemicals and Cell Line
2.3. CNP Preparation and Characterization
2.4. Toxicity Assays
2.5. Immunization
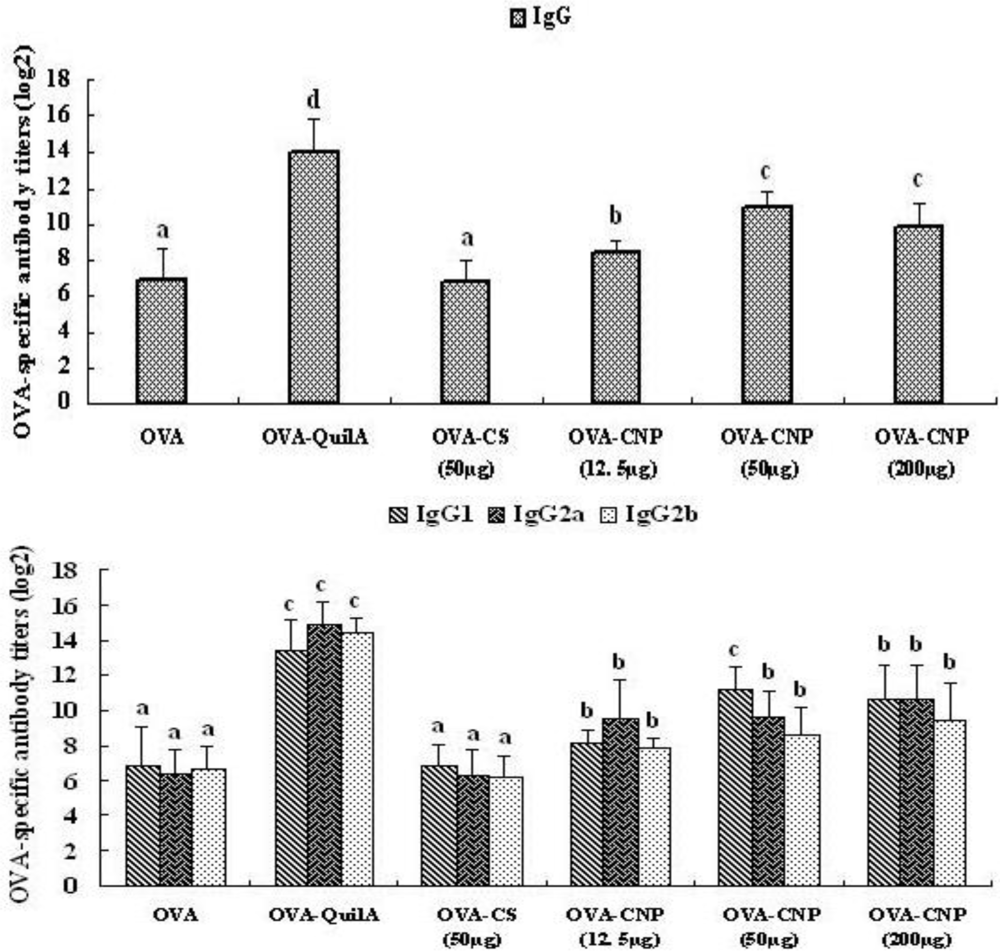
2.6. Measurement of OVA-Specific IgG and Subclasses
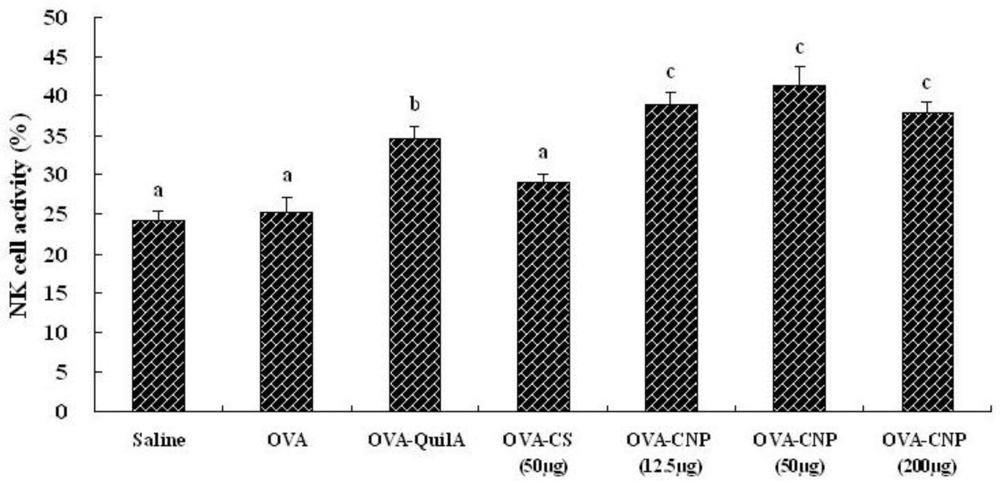
2.7. Assay of Natural Killer (NK) Cell Activity
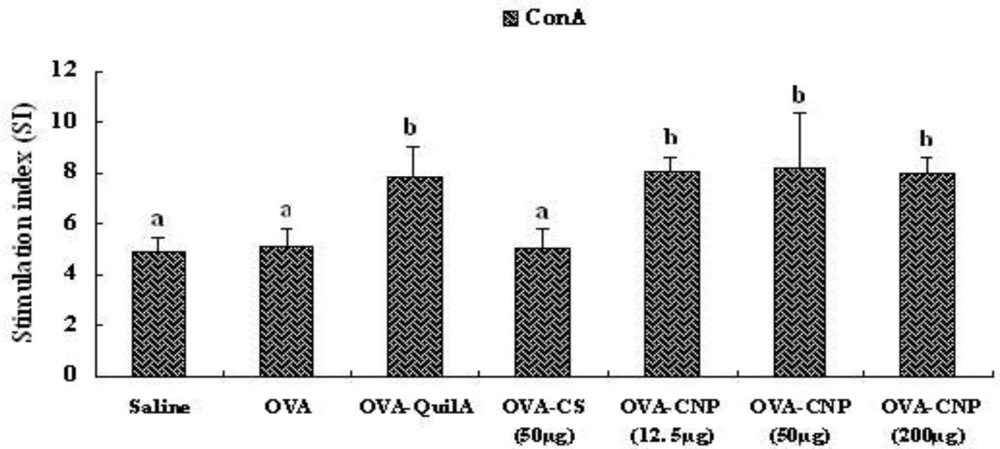
2.8. Splenocyte Proliferation Assay
2.9. Cytokine Determination in the Cultured Supernatants of Splenocytes by ELISA
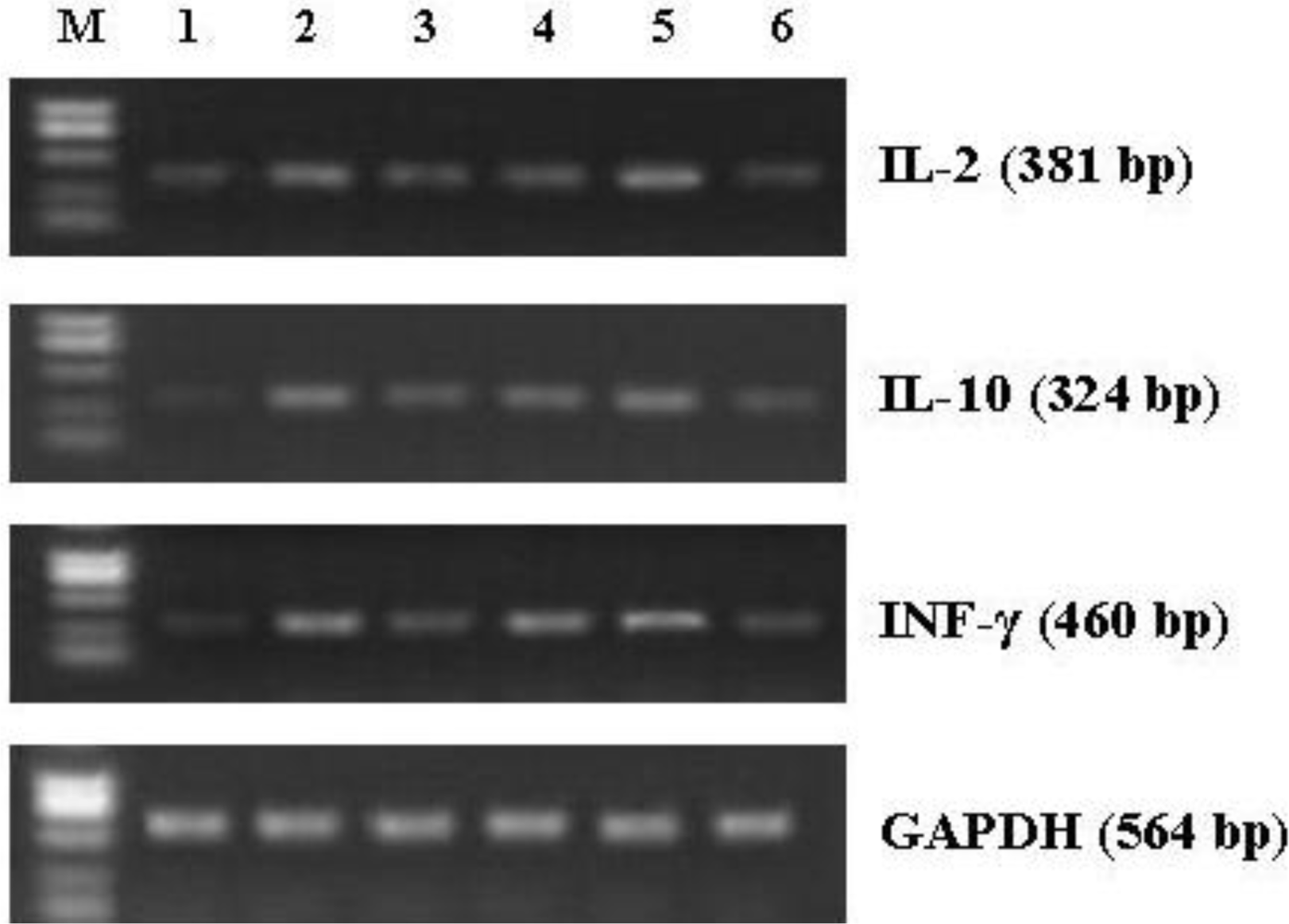
2.10. Reversed Transcript-Polymerase Chain Reaction (RT-PCR) for Cytokines Gene Expression
2.11. Statistical Analysis
3. Results
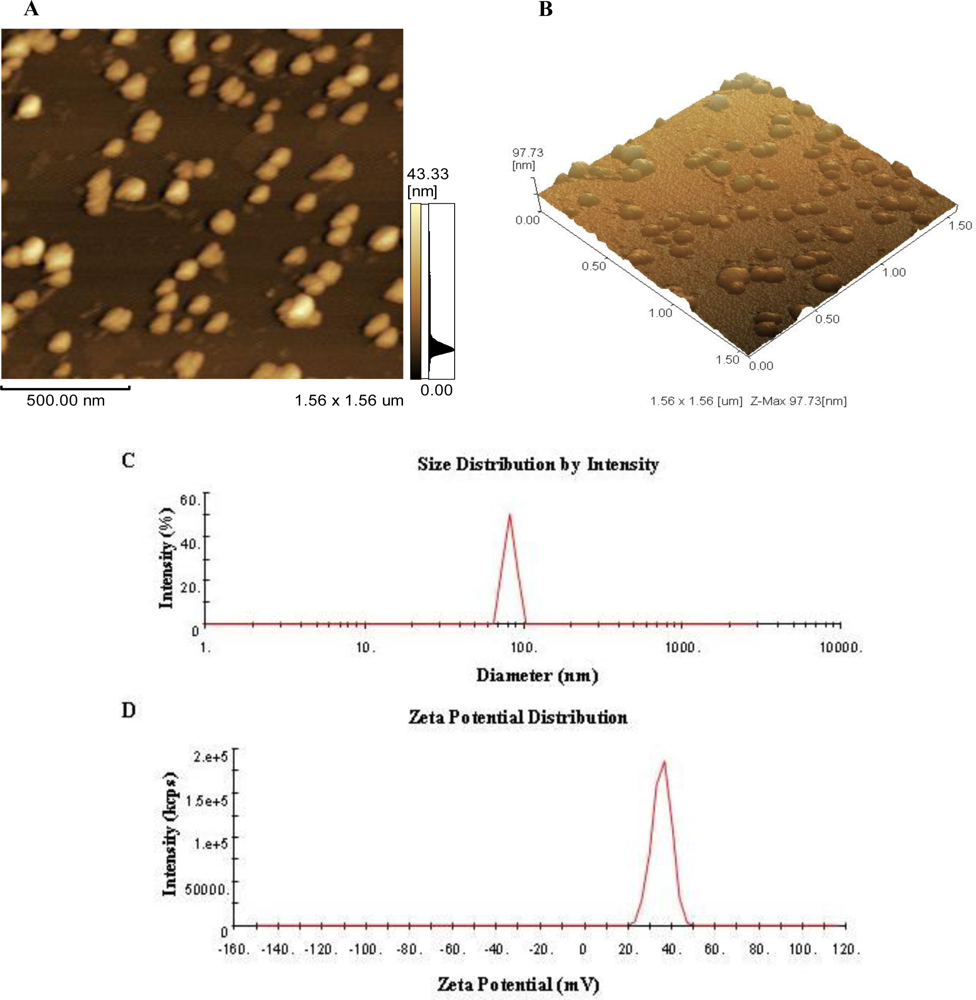
3.1. Morphology, Size and Zeta Potential of CNP
3.2. Toxicity of CNP
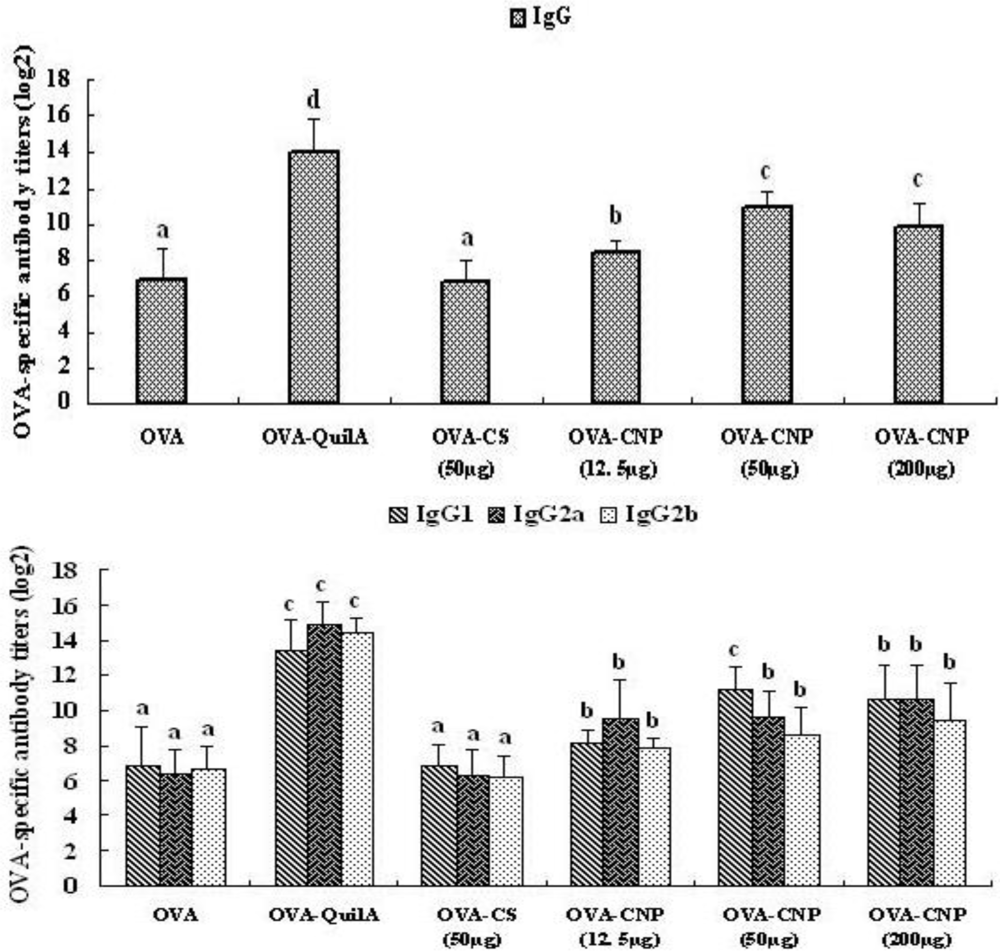
3.3. Effect of CNP on the OVA-Specific Serum Antibody Response
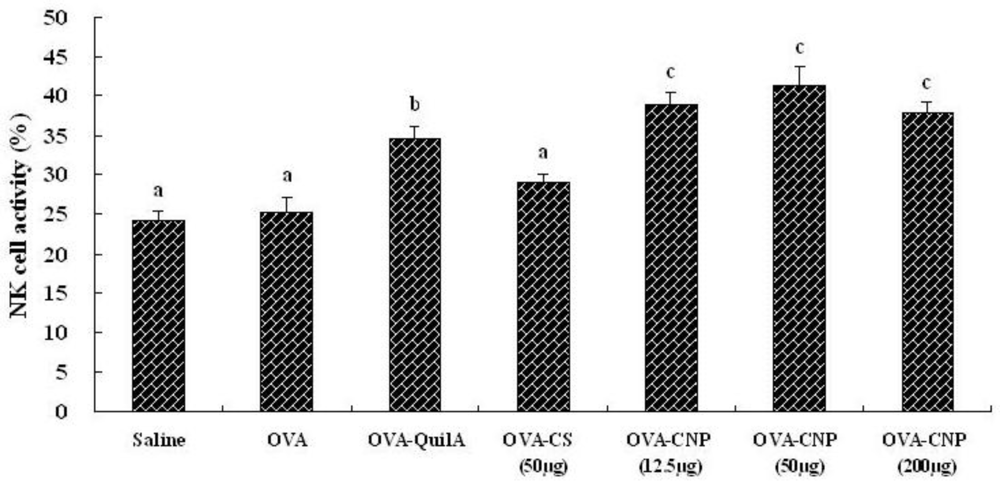
3.4. Effect of CNP on Natural Killer (NK) Cell Activity in OVA-Immunized Mice
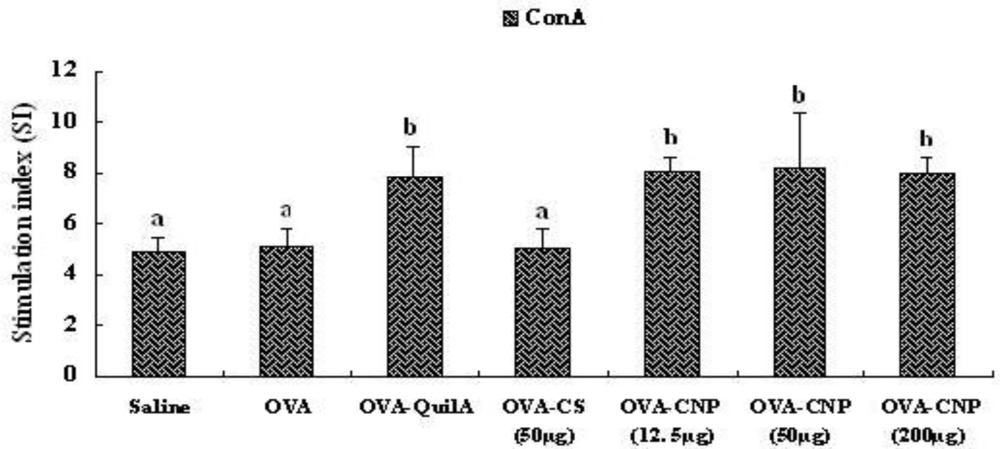
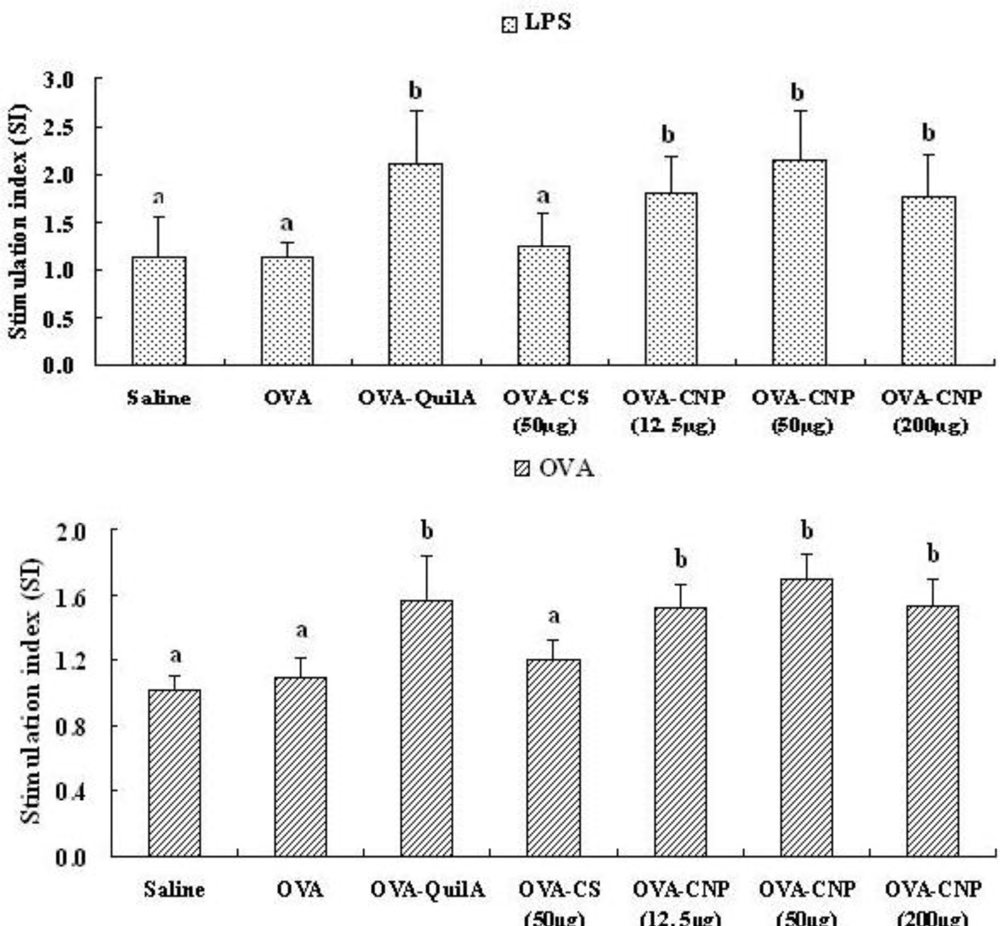
3.5. Effect of CNP on Splenocyte Proliferation in OVA-Immunized Mice
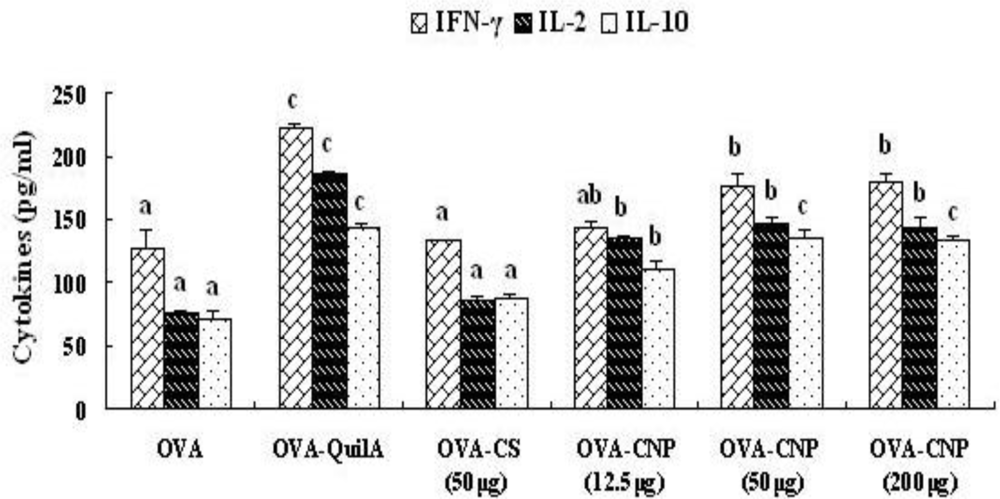
3.6. Effect of CNP on Cytokines Level in Splenocytes from the OVA-Immunized Mice
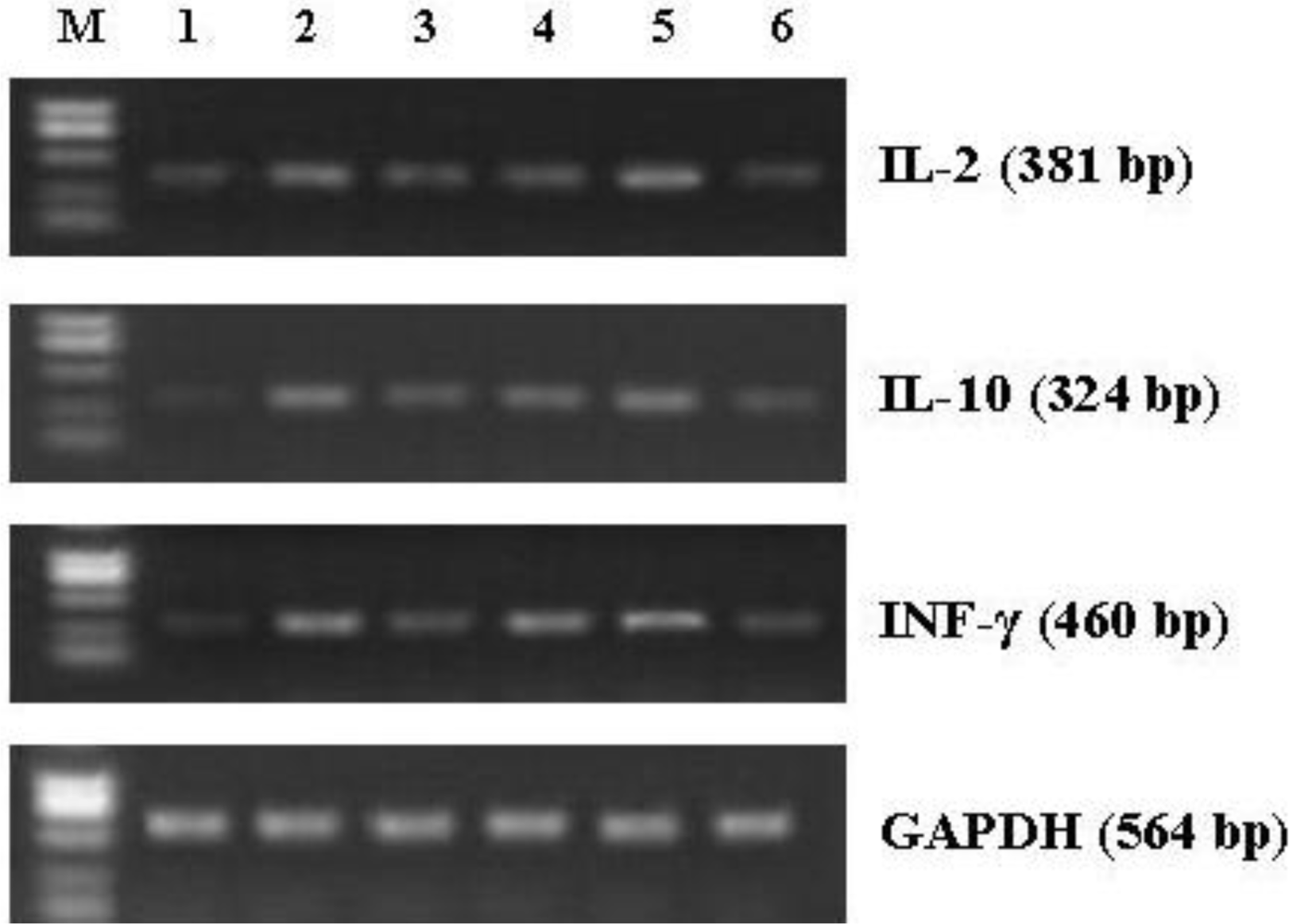
3.7. Effect of CNP on mRNA Expression of Cytokines in Splenocytes from the Immunized Mice
4. Discussion
5. Conclusions
Acknowledgments
- Samples Availability: Available from the authors.
References
- Kammer, AR; Amacker, M; Rasi, S; Westerfeld, N; Gremion, C; Neuhaus, D; Zurbriggen, R. A new and versatile virosomal antigen delivery system to induce cellular and humoral immune responses. Vaccine 2007, 25, 7065–7074. [Google Scholar]
- Kensil, CR; Kammer, R. QS-21: Awater-soluble triterpene glycoside adjuvant. Expert Opin Investig Drugs 1998, 7, 1475–1482. [Google Scholar]
- Skene, CD; Sutton, P. Saponin-adjuvanted particulate vaccines for clinical use. Methods 2006, 40, 53–59. [Google Scholar]
- Gupta, RK; Relyveld, EH; Lindblad, EB; Bizzini, B; Ben-Efraim, S; Gupta, CK. Adjuvants: A balance between toxicity and adjuvanticity. Vaccine 1993, 11, 293–306. [Google Scholar]
- Kensil, CR; Patel, U; Lennick, M; Marciani, D. Separation and characterization of saponins with adjuvant activity from Quillaja saponaria Molina cortex. J Immunol 1991, 146, 431–437. [Google Scholar]
- Kensil, CR; Wu, JY; Soltysik, S. Structural and immunological characterization of the vaccine adjuvant QS-21. Pharm Biotechnol 1995, 6, 525–541. [Google Scholar]
- Waite, DC; Jacobson, EW; Ennis, FA; Edelman, R; White, B; Kammer, R; Anderson, C; Kensil, CR. Three double-blind, randomized trials evaluating the safety and tolerance of different formulations of the saponin adjuvant QS-21. Vaccine 2001, 19, 3957–3967. [Google Scholar]
- Janeway, CA, Jr. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today 1992, 13, 11–16. [Google Scholar]
- Freund, J; Casals, J; Hosmer, E. Sensitization and antibody formation after injection of tubercle bacili and paraffin oil. Proc Soc Exp Biol Med 1937, 37, 509–513. [Google Scholar]
- Pascual, DM; Morales, RD; Gil, ED; Muñoz, LM; López, JE; Casanueva, OLJ. Adjuvants: Present regulatory challenges. Vaccine 2006, 24, S88–S89. [Google Scholar]
- HogenEsch, H. Mechanisms of stimulation of the immune response by aluminum adjuvants. Vaccine 2002, 20, S34–S39. [Google Scholar]
- Schirmbeck, R; Melber, K; Kuhrber, A; Janowicz, ZA; Reimann, J. Immunization with soluble hepatitis B virus surface protein elicits murine H-2 class I-restricted CD8+ cytotoxic T lymphocyte responses in vivo. J Immunol 1994, 152, 1110–1119. [Google Scholar]
- Petrovsky, N. Novel human polysaccharide adjuvants with dual Th1 and Th2 potentiating activity. Vaccine 2006, 24, S26–S29. [Google Scholar]
- Glenny, A; Pope, C; Waddington, H; Falacce, U. The antigenic value of toxoid precipitated by potassium alum. J Pathol Bacteriol 1926, 29, 31–40. [Google Scholar]
- Morein, B; Sundquist, B; Hoglund, S; Dalsgaad, K; Osterhaus, A. Iscom, a novel structure for antigenic presentation of membrane proteins from enveloped viruses. Nature 1984, 308, 457–460. [Google Scholar]
- Pearse, MJ; Drane, D. ISCOMATRIX adjuvant for antigen delivery. Adv Drug Deliv Rev 2005, 57, 465–474. [Google Scholar]
- Sharp, FA; Ruane, D; Claass, B; Creagh, E; Harris, J; Malyala, P; Singh, M; O’Hagan, DT; Pétrilli, V; Tschopp, J; et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci USA 2009, 106, 870–875. [Google Scholar]
- Sailaja, AK; Amareshwar, P; Chakravarty, P. Chitosan nanoparticles as a drug delivery system. Res J Pharm Biol Chem Sci 2010, 1, 474–484. [Google Scholar]
- Dinarello, CA. Blocking IL-1 in systemic inflammation. J Exp Med 2005, 201, 1355–1359. [Google Scholar]
- van der Lubben, IM; Verhoef, JC; Borchard, G; Junginger, HE. Chitosan and its derivatives in mucosal drug and vaccine delivery. Eur J Pharm Sci 2001, 14, 201–207. [Google Scholar]
- Knorr, D. Use of chitinous polymers in foodda challenge for food research and development. Food Technol 1984, 38, 85–97. [Google Scholar]
- Kurita, K. Chemistry and application of chitin and chitosan. Polym Degrad Stab 1998, 59, 117–120. [Google Scholar]
- Razdan, A; Pettersson, D. Effect of chitin and chitosan on nutrient digestibility and plasma lipid concentrations in broiler chickens. Br J Nutr 1994, 72, 277–288. [Google Scholar]
- van der Lubben, IM; Verhoef, JC; Borchard, G; Junginger, HE. Chitosan microparticles for oral vaccination: Preparation, characterization and preliminary in vivo uptake studies in murine Peyer’s patches. Biomaterials 2001, 22, 687–694. [Google Scholar]
- De Campos, AM; Sanchez, A; Alonso, MJ. Chitosan nanoparticles: A new vehicle for the improvement of the delivery of drugs to the ocular surface. Application to cyclosporin A. Int J Pharm 2001, 224, 159–168. [Google Scholar]
- Illum, L; Jabbal-Gill, I; Hinchcliffe, M; Fisher, AN; Davis, SS. Chitosan as a novel nasal delivery system for vaccines. Adv Drug Deliv Rev 2001, 51, 81–96. [Google Scholar]
- Zhu, B; Qie, Y; Wang, J; Zhang, Y; Wang, Q; Xu, Y; Wang, H. Chitosan microspheres enhance the immunogenicity of an Ag85B-based fusion protein containing multiple T-cell epitopes of Mycobacterium tuberculosis. Eur J Pharm Biopharm 2007, 66, 318–326. [Google Scholar]
- Nishimura, K; Nishimura, S; Nishi, N; Saiki, I; Tokura, S; Azuma, I. Immunological activity of chitin and its derivatives. Vaccine 1984, 2, 93–99. [Google Scholar]
- Suzuki, K; Mikami, T; Okawa, Y; Tokoro, A; Suzuki, S; Suzuki, M. Antitumor effect of hexa-N-acetylchitohexaose and chitohexaose. Carbohydr Res 1986, 151, 403–408. [Google Scholar]
- Tokoro, A; Tatewaki, N; Suzuki, K; Mikami, T; Suzuki, S; Suzuki, M. Growth-inhibitory effect of hexa-N-acetylchitohexaose and chitohexaose against meth-A solid tumor. Chem Pharm Bull 1988, 36, 784–790. [Google Scholar]
- Calvo, P; Remunan-Lopez, C; Vila-Jato, JL; Alonso, MJ. Chitosan and chitosan: Ethylene oxide-propylene oxide block copolymer nanoparticles as novel carriers and vaccines. Pharm Res 1997, 14, 1431–1436. [Google Scholar]
- Shimbata, Y; Foster, LA; Metzger, WJ; Myrvik, QN. Alveolar macrophage priming by intravenous administration of chitin particles, polymers of N-acetyl-d-glucosamine, in mice. Infect Immun 1997, 65, 1734–1741. [Google Scholar]
- Seferian, PG; Martinez, ML. Immune stimulating activity of two new chitosan containing adjuvant formulations. Vaccine 2000, 19, 661–668. [Google Scholar]
- Qurashi, T; Blair, HS; Alen, SJJ. Studies on modified chitosan membranes. I. Preparation and characterization. J Appl Polym Sci 1992, 46, 255–261. [Google Scholar]
- Qi, L; Xu, Z; Jiang, X; Hu, C; Zou, X. Preparation and antibacterial activity of chitosan nanoparticles. Carbohydr Res 2004, 339, 2693–2700. [Google Scholar]
- Tu, J; Sun, HX; Ye, YP. Immunomodulatory and antitumor activity of triterpenoid fractions from the rhizomes of Astilbe chinensis. J Ethnopharmacol 2008, 119, 266–271. [Google Scholar]
- Sun, HX; Ye, YP; Pan, HJ; Pan, YJ. Adjuvant effect of Panax notoginseng saponins on the immune responses to ovalbumin in mice. Vaccine 2004, 22, 3882–3889. [Google Scholar]
- O’Hagan, DT; MacKichan, ML; Singh, M. Recent developments in adjuvants for vaccines against infectious diseases. Biomol Eng 2001, 18, 69–85. [Google Scholar]
- Aucouturier, J; Dupuis, L; Ganne, V. Adjuvants designed for veterinary and human vaccines. Vaccine 2001, 19, 2666–2672. [Google Scholar]
- Hunter, RL. Overview of vaccine adjuvants: Present and future. Vaccine 2002, 20(Suppl. 3), S7–S12. [Google Scholar]
- Suzuki, K; Okawa, Y; Hashimoto, K; Suzuki, S; Suzuki, M. Protecting effect of chitin and chitosan on experimentally induced murine candidiasis. Microbiol Immunol 1984, 28, 903–912. [Google Scholar]
- Peluso, G; Petillo, O; Ranieri, M; Santin, M; Ambrosio, L; Calabro, D; Avallone, B; Balsamo, G. Chitosan-mediated stimulation of macrophage function. Biomaterials 1994, 15, 1215–1220. [Google Scholar]
- Villiers, C; Chevallet, M; Diemer, H; Couderc, R; Freitas, H; Van Dorsselaer, A; Marche, PN; Rabilloud, T. From secretome analysis to immunology: Chitosan induces major alterations in the activation of dendritic cells via a TLR4-dependent mechanism. Mol Cell Proteomics 2009, 8, 1252–1264. [Google Scholar]
- van der Lubben, IM; Verhoef, JC; Borchard, G; Junginger, HE. Chitosan for mucosal vaccination. Adv Drug Deliv Rev 2001, 52, 139–144. [Google Scholar]
- Sun, HX; Wang, H; Xu, HS; Ni, Y. Novel polysaccharide adjuvant from the roots of Actinidia eriantha with dual Th1 and Th2 potentiating activity. Vaccine 2009, 27, 3984–3991. [Google Scholar]
- Gupta, RK; Siber, GR. Adjuvants for humans vaccines—current status, problems and future prospects. Vaccine 1995, 13, 1263–1276. [Google Scholar]
- Audibert, FM; Lise, LD. Adjuvants: Current status, clinical perspectives and future prospects. Immunol Today 1993, 14, 281–284. [Google Scholar]
- Sedlik, C. Th1 and Th2 subsets of T lymphocytes: Characteristics, physiological role and regulation. Bull Inst Pasteur 1996, 94, 173–200. [Google Scholar]
- Livingston, PO; Adluri, S; Helling, F; Yao, TJ; Kensil, CR; Newman, MJ; Marciani, D. Phase 1 trial of immunological adjuvant QS-21 with a GM2 ganglioside-keyhole limpet haemocyanin conjugate vaccine in patients with malignant melanoma. Vaccine 1994, 12, 1275–1280. [Google Scholar]
- Cher, DJ; Mosmann, TR. Two types of murine helper T-cell clones. II. Delayed-type hypersensitivity is mediated by Th1 clones. J Immunol 1987, 138, 3688–3694. [Google Scholar]
- Mosmann, TR; Sad, S. The expanding universe of T-cells subsets: Th1, Th2 and more. Immunol Today 1996, 17, 138–146. [Google Scholar]
- Coffman, RL; Seymour, BWP; Lebman, DA; Hiraki, DD; Christiansen, JA; Shrader, B; Cherwinski, HM; Savelkoul, HFJ; Finkelman, FD; Bond, MW; et al. The role of helper T cell products in mouse B cell differentiation and isotype regulation. Immunol Rev 1988, 102, 5–28. [Google Scholar]
- McKee, AS; Munks, MW; Marrack, P. How do adjuvants work? Important considerations for new generation adjuvants. Immunity 2007, 27, 687–690. [Google Scholar]
- Roitt, I; Brostoff, J; Male, D. Immunology; Mosby: London, UK, 2000; pp. 121–135. [Google Scholar]
- Boon, T; Cerottini, JC; Van den Eynde, B; van der Bruggen, P; van Pel, A. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol 1994, 12, 337–365. [Google Scholar]
- Moretta, L; Bottino, C; Cantoni, C; Mingari, MC; Moretta, A. Human natural killer cell function and receptors. Curr Opin Pharmacol 2001, 1, 387–391. [Google Scholar]
- Rappuoli, R. Bridging the knowledge gaps in vaccine design. Nat Biotechnol 2007, 25, 1361–1366. [Google Scholar]
- Kos, FJ; Engleman, EG. Immune regulation: A critical link between NK cells and CTLs. Immunol Today 1996, 17, 174–176. [Google Scholar]
- Medzhitov, R; Janeway, CA. Innate immunity: Impact on the adaptive immune response. Curr Opin Immunol 1997, 9, 4–9. [Google Scholar]
- Orange, JS. Formation and function of the lytic NK-cell immunological synapse. Nat Rev Immunol 2008, 8, 713–725. [Google Scholar]
- Zhang, J; Sun, R; Wei, H; Tian, Z. Antitumor effects of recombinant human prolactin in human adenocarcinoma-bearing SCID mice with human NK cell xenograft. Int Immunopharmacol 2005, 5, 417–425. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer sequence | Product size (bp) |
|---|---|---|
| IL-2 | 5′-CTCTACAGCGGAAGCACAGC-3′ 5′-CATCTCCTCAGAAAGTCCACCA-3′ | 381 |
| IFN-γ | 5′-TGAACGCTACACACTGCATCTTGG-3′ 5′-CGACTCCTTTTCCGCTTCCTGAG-3′ | 460 |
| IL-10 | 5′-CCAGTTTTACCTGGTAGAAGTGATG-3′ 5′-TGTCTAGGTCCTGGAGTCCAGCAGACTCAA-3′ | 324 |
| GAPDH | 5′-CCCACAGTAAATTCAACGGCAC-3′ 5′-CATTGGGGTTAGGAACACGGA-3′ | 564 |
| Gene | OVA | OVA-QuilA | OVA-CNP | OVA-CS | ||
|---|---|---|---|---|---|---|
| (12.5 μg) | (50 μg) | (200 μg) | (50 μg) | |||
| IL-2 | 15.0 ± 1.4 a | 45.6 ± 2.1 c | 26.6 ± 1.4 b | 38.8 ± 2.1 bc | 50.6 ± 2.2 c | 16.0 ± 1.2 a |
| IFN-γ | 15.8 ± 1.6 a | 40.4 ± 2.3 c | 29.0 ± 1.4 b | 38.8 ± 2.3 bc | 44.0 ± 2.5 c | 16.0 ± 1.6 a |
| IL-10 | 15.0 ± 1.4 a | 45.0 ± 2.7 c | 22.4 ± 1.8 ab | 30.4 ± 2.2 b | 44.8 ± 2.7 c | 18.2 ± 1.7 a |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wen, Z.-S.; Xu, Y.-L.; Zou, X.-T.; Xu, Z.-R. Chitosan Nanoparticles Act as an Adjuvant to Promote both Th1 and Th2 Immune Responses Induced by Ovalbumin in Mice. Mar. Drugs 2011, 9, 1038-1055. https://doi.org/10.3390/md9061038
Wen Z-S, Xu Y-L, Zou X-T, Xu Z-R. Chitosan Nanoparticles Act as an Adjuvant to Promote both Th1 and Th2 Immune Responses Induced by Ovalbumin in Mice. Marine Drugs. 2011; 9(6):1038-1055. https://doi.org/10.3390/md9061038
Chicago/Turabian StyleWen, Zheng-Shun, Ying-Lei Xu, Xiao-Ting Zou, and Zi-Rong Xu. 2011. "Chitosan Nanoparticles Act as an Adjuvant to Promote both Th1 and Th2 Immune Responses Induced by Ovalbumin in Mice" Marine Drugs 9, no. 6: 1038-1055. https://doi.org/10.3390/md9061038
APA StyleWen, Z.-S., Xu, Y.-L., Zou, X.-T., & Xu, Z.-R. (2011). Chitosan Nanoparticles Act as an Adjuvant to Promote both Th1 and Th2 Immune Responses Induced by Ovalbumin in Mice. Marine Drugs, 9(6), 1038-1055. https://doi.org/10.3390/md9061038