3. Experimental Section
3.1. General
Optical rotations were measured using a Jasco P-1030 polarimeter. Melting points (mp) were measured using a Yazawa melting point apparatus BY-2 and are uncorrected. IR spectra were recorded using a Jasco FT-IR/620 spectrometer. UV spectra were recorded using a Jasco V-550 spectrophotometer. Circular dichroism (CD) spectra were measured with a Jasco J-720 spectropolarimeter. 1H- and 13C-NMR spectra were recorded on a Bruker DRX-400 or a Bruker Biospin AV-600 spectrometer. Chemical shifts are given on the δ (ppm) scale using tetramethylsilane (TMS) as the internal standard (s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; m, multiplet; br, broad). High resolution ESIMS (HRESIMS) spectra were obtained using a Micromass LCT spectrometer. Elemental analysis data were obtained using an Elemental Vavio EL. Flash column chromatography was performed using Kanto Chemical Silica Gel 60N (spherical, neutral) 40–50 μm.
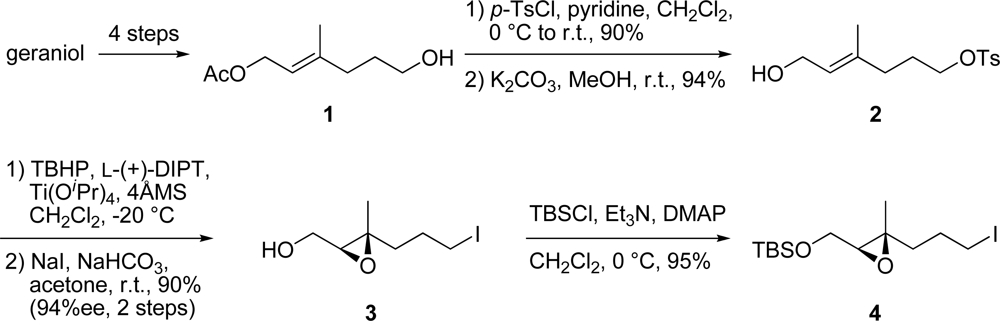
3.2. (E)-6-Hydroxy-4-methylhex-4-enyl 4-methylbenzenesulfonate (2)
To a solution of (
E)-6-hydroxy-3-methylhex-2-enyl acetate [
6] (
1, 530 mg, 3.08 mmol) in CH
2Cl
2 (10.3 mL) were added pyridine (374 μL, 4.62 mmol) and
p-toluenesulfonyl chloride (705 mg, 3.70 mmol) at 0 °C. After stirring for 5 hr at r.t., the mixture was diluted with Et
2O, washed with H
2O and brine, and then dried. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 2:1) to generate tosylate (905 mg, 90% yield) as a colorless oil. IR (neat) 2924, 1733, 1359 cm
−1;
1H-NMR (400 MHz, CDCl
3) δ ppm: 7.77 (2H, d,
J = 8.2 Hz), 7.33 (2H, d,
J = 8.2 Hz), 5.25 (1H, m), 4.52 (2H, d,
J = 7.2 Hz), 4.01 (2H, t,
J = 6.4 Hz), 2.44 (3H, s), 2.05 (2H, t,
J = 7.2 Hz), 2.03 (3H, s), 1.77 (2H, m), 1.64 (3H, s);
13C-NMR (100 MHz, CDCl
3) δ ppm: 170.9, 144.7, 133.3, 129.8, 127.8, 119.6, 69.8, 61.0, 35.0, 26.8, 21.5, 20.9, 16.2; HRESIMS (
m/
z) calcd. for C
16H
23O
5S (M+H)
+ 349.1086, found 349.1086; Anal. Calcd. for C
16H
22O
5S: C, 58.87; H, 6.79. Found: C, 58.94; H, 6.75.
To a solution of the above tosylate (9.47 g, 29.0 mmol) in MeOH (290 mL) was added K2CO3 (4.81 g, 34.8 mmol) at r.t. After stirring for 30 min at the same temperature, the mixture was diluted with Et2O and then filtered through a silica gel pad. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 2:1) to generate allylic alcohol 2 (7.74 g, 94% yield) as a colorless oil. IR (neat) 3387, 2923, 1354 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 7.75 (2H, d, J = 8.2 Hz), 7.32 (2H, d, J = 8.2 Hz), 5.31 (1H, m), 4.07 (2H, d, J = 6.5 Hz), 4.00 (2H, t, J = 6.5 Hz), 2.42 (3H, s), 2.02 (2H, t, J = 7.8 Hz), 1.76 (2H, m), 1.59 (3H, s), 1.52 (1H, br s); 13C-NMR (100 MHz, CDCl3) δ ppm: 144.7, 137.2, 133.2, 129.8, 127.8, 124.7, 69.8, 59.0, 35.0, 26.6, 21.5, 15.9; HRESIMS (m/z) calcd. for C14H21O4S (M+H)+ 307.0980, found 307.0994; Anal. Calcd. for C14H20O4S: C, 59.13; H, 7.09. Found: C, 59.07; H, 7.06.
3.3. ((2S,3S)-3-(3-Iodopropyl)-3-methyloxiran-2-yl)methanol (3)
To a cold (−20 °C) suspension of 4Å molecular sieves (114 mg) in CH2Cl2 (1.6 mL) were added L-(+)-DIPT (5.2 μL, 24.8 μmol), Ti(OiPr)4 (6.2 μL, 21.0 μmol) and TBHP (164 μL, 101 mmol, 6.17 M in CH2Cl2 solution). After stirring for 30 min at the same temperature, a solution of allylic alcohol 2 (54.4 mg, 191 μmol) in CH2Cl2 (500 μL) was added over 5 min. After stirring at −20 °C for 15 min, NaOH (13.0 μL, 30% in saturated aqueous NaCl) was added. The mixture was diluted with Et2O, warmed to r.t. and stirred for 10 min. MgSO4 (11.6 mg) and Celite (1.4 mg) were then added and after stirring for 15 min, the mixture was filtered through a Celite pad and the filtrate was concentrated under reduced pressure to afford the crude epoxide. To a solution of the crude epoxide in acetone (1.9 mL) were added NaHCO3 (17.7 mg, 210 μmol) and NaI (286 mg, 1.91 mmol) at r.t. After stirring for 8 hr at the same temperature, the mixture was diluted with Et2O and then filtered through a silica gel pad. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 1:2) to generate epoxy iodide 3 (44.0 mg, 90% yield) as a yellow oil. [α]D28 −10.0 (c 1.03, CHCl3); IR (neat) 3418, 2929 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 3.79 (1H, m), 3.68 (1H, m), 3.18 (2H, m), 2.97 (2H, t, J = 5.4 Hz), 1.99 (1H, br s), 1.93 (2H, m), 1.64 (2H, m), 1.29 (3H, s); 13C-NMR (100 MHz, CDCl3) δ ppm: 62.6, 61.2, 60.3, 39.0, 29.0, 16.8, 5.8; HRESIMS (m/z) calcd. for C7H12IO (M-OH)+ 238.9933, found 238.9930; Anal. Calcd. for C7H13IO2: C, 32.83; H, 5.12. Found: C, 33.06; H, 5.26.
3.4. tert-Butyl(((2S,3S)-3-(3-iodopropyl)-3-methyloxiran-2-yl)methoxy)dimethylsilane (4)
To a solution of epoxy iodide 3 (387 mg, 1.51 mmol) in CH2Cl2 (1.5 mL) were added Et3N (253 mg, 1.82 mmol), DMAP (185 mg, 1.51 mmol) and TBSCl (251 mg, 1.82 mmol) and the mixture was stirred at r.t. for 30 min. The mixture was diluted with Et2O, washed with saturated aqueous NaHCO3 solution, H2O and brine, and then dried. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 7:1) to generate epoxy iodide 4 (530 mg, 95% yield) as a colorless oil. [α]D25 +6.9 (c 1.06, CHCl3); IR (neat) 2929 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 3.76 (1H, dd, J = 11.5, 5.5 Hz), 3.69 (1H, dd, J = 11.5, 5.5 Hz), 3.19 (2H, t, J = 7.0 Hz), 2.91 (1H, t, J = 5.5 Hz), 1.95 (2H, m), 1.64 (2H, m), 1.26 (3H, s), 0.91 (9H, s), 0.07 (6H, s); 13C-NMR (100 MHz, CDCl3) δ ppm: 62.8, 62.0, 59.5, 39.0, 29.2, 25.9, 18.3, 16.7, 5.8, −5.2, −5.4; HRESIMS (m/z) calcd. for C13H28IO2Si (M+H)+ 371.0903, found 371.0921; Anal. Calcd. for C13H27IO2Si: C, 42.16; H, 7.35. Found: C, 42.37; H, 7.23.
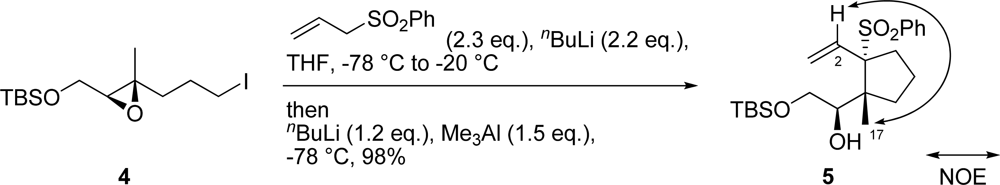
3.5. (R)-1-[(1R,2S)-2-Benzenesulfonyl-1-methyl-2-vinylcyclopentyl]-2-(tert-butyldimethylsiloxy) ethanol (5)
To a solution of allyl phenyl sulfone (95.2 mg, 0.552 mmol) in THF (3.0 mL) was added nBuLi (317 μL, 0.500 mmol, 1.58 M in hexane solution) at −78 °C and the mixture was warmed to 0 °C. The mixture was stirred for 30 min at the same temperature. After cooling to −78 °C, a solution of epoxy iodide 4 (84.1 mg, 0.227 mmol) in THF (1.6 mL) was added and the mixture was warmed to −20 °C. The mixture was stirred for 30 min at the same temperature. After cooling to −78 °C, nBuLi (173 μL, 0.273 mmol, 1.58 M in hexane solution) was added and the mixture was warmed to −20 °C. The mixture was stirred for 30 min at the same temperature. After cooling to −78 °C, Me3Al (331 μL, 0.341 mmol, 1.03 M in hexane) was added. After stirring for 1 hr at the same temperature, the mixture was diluted with Et2O, washed with saturated aqueous NH4Cl solution, H2O and brine, and then dried. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 10:1) to generate cyclopentane 5 (94.4 mg, 98% yield) as a white solid. mp 125–126 °C; [α]D25 −114 (c 0.81, CHCl3); IR (KBr) 3560, 2952 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 7.78 (2H, m), 7.57 (1H, m), 7.45 (2H, m), 6.19 (1H, dd, J = 17.4, 10.9 Hz), 5.25 (1H, d, J = 10.9 Hz), 4.73 (1H, d, J = 17.4 Hz), 4.56 (1H, dt, J = 6.2, 2.9 Hz), 4.29 (1H, dd, J = 9.5, 3.5 Hz), 3.67 (1H, t, J = 8.8 Hz), 3.23 (1H, d, J = 2.9 Hz), 2.57 (1H, m), 2.06 (3H, m), 1.73 (2H, m), 0.99 (3H, s), 0.93 (9H, s), 0.14 (6H, s); 13C-NMR (100 MHz, CDCl3) δ ppm: 137.4, 135.3, 133.3, 130.6, 127.9, 120.3, 81.0, 74.3, 64.2, 54.6, 37.2, 30.8, 25.9, 20.0, 19.6, 18.2, −5.1, −5.3; HRESIMS (m/z) calcd. for C22H37O4SSi (M+H)+ 425.2182, found 425.2179; Anal. Calcd. for C22H36O4SSi: C, 62.22; H, 8.54. Found: C, 62.11; H, 8.41.
3.6. {(1S,2R)-2-[(R)-1,2-Bis(tert-butyldimethylsiloxy)ethyl]-2-methyl-1-vinylcyclopentanesulfonyl} benzene (6)
To a solution of cyclopentane 5 (7.01 g, 16.5 mmol) in CH2Cl2 (16.5 mL) were added 2,6-lutidine (17.7 g, 165 mmol) and TBSOTf (7.02 g, 26.6 mmol) and the mixture was stirred at 0 °C for 30 min. The mixture was diluted with Et2O, washed with saturated aqueous NaHCO3 solution, H2O and brine, and then dried. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 5:1) to generate bis-silyl ether 6 (8.89 g, quantitative yield) as a colorless oil. [α]D25 −77.9 (c 1.62, CHCl3); IR (neat) 2954, 1133 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 7.78 (2H, m), 7.57 (1H, m), 7.45 (2H, m), 6.37 (1H, dd, J = 17.4, 10.9 Hz), 5.27 (1H, d, J = 10.9), 4.72 (1H, d, J = 17.4 Hz), 4.63 (1H, dd, J = 5.9, 1.8 Hz), 4.15 (1H, dd, J = 10.5, 1.8 Hz), 3.87 (1H, dd, J = 10.5, 5.9), 2.51 (1H, m), 2.12 (1H, m), 2.01 (1H, m), 1.90 (3H, m), 0.92 (21H, m), 0.16 (12H, m); 13C-NMR (100 MHz, CDCl3) δ ppm: 137.3, 135.3, 133.2, 130.7, 127.9, 120.7, 81.1, 77.2, 66.5, 56.3, 40.0, 31.6, 26.3, 26.2, 19.8, 19.0, 18.5, −3.3, −4.6, −5.1, −5.4; HRESIMS (m/z) calcd. for C28H51O4SSi2 (M+H)+ 539.3047, found 539.3086; Anal. Calcd. for C28H50O4SSi2: C, 62.40; H, 9.35. Found: C, 62.37; H, 9.11.
3.7. (S)-1-[(R)-1,2-Bis(tert-butyldimethylsiloxy)ethyl]-2-ethylidene-1-methylcyclopentane (7)
To a solution of bis-silyl ether 6 (9.29 g, 17.2 mmol) in MeOH (344 mL) were added Na2HPO4 (17.1 g, 120.5 mmol) and 5% Na(Hg) (31.6 g). After stirring for 1 hr at r.t., the mixture was diluted with Et2O and filtered through silica gel. The filtrate was then concentrated under reduced pressure. The resultant residue was then purified by silica gel column chromatography (hexane only) to generate E-olefin 7 (6.86 g, quantitative yield) as a colorless oil. [α]D25 +19.0 (c 1.35, CHCl3); IR (neat) 2955 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 5.17 (1H, m), 3.78 (1H, dd, J = 10.3, 2.8 Hz), 3.52 (1H, dd, J = 9.2, 1.8 Hz), 3.46 (1H, dd, J = 10.3, 5.8 Hz), 2.35 (1H, m), 2.08 (2H, m), 1.67 (1H, m), 1.58 (3H, d, J = 6.7 Hz), 1.53 (2H, m), 1.22 (1H, m), 0.97 (3H, s), 0.88 (9H, s), 0.86 (9H, s), 0.07 (3H, s), 0.03 (3H, s), 0.03 (3H, s), 0.00 (3H, s); 13C-NMR (100 MHz, CDCl3) δ ppm: 150.2, 114.1, 79.1, 66.3, 49.5, 35.7, 30.0, 26.1, 26.0, 23.9, 22.5, 18.4, 18.3, 14.7, −3.9, −5.0, −5.3; HRESIMS (m/z) calcd. for C22H46O2Si2Na (M+Na)+ 421.2934, found 421.2914; Anal. Calcd. for C22H46O2Si2: C, 66.26; H, 11.63. Found: C, 66.36; H, 11.50.
3.8. (S)-1-{(1R,2S)-2-[(R)-2,2-Dimethyl-[1,3]dioxolan-4-yl]-2-methylcyclopentyl}ethanol (9)
To a solution of E-olefin 7 (3.86 g, 9.69 mmol) in THF (19.4 mL) was added catecholborane (6.40 mL, 60.1 mmol) dropwise at 0 °C. After stirring for 12 hr at the same temperature, 1M NaOH solution (12.9 mL) and 35% aqueous H2O2 solution (36.9 mL) were added to the mixture at r.t. After stirring for 2 hr, the resultant mixture was diluted with CHCl3, washed with H2O and brine, dried and then concentrated to afford a mixture of diol 8 and triol 8a. To a solution of the crude alcohols in acetone (96.9 mL) was added p-TsOH·H2O (735 mg, 3.88 mmol) at r.t. After stirring for 2 hr at the same temperature, the mixture was diluted with Et2O, washed with saturated aqueous NaHCO3 solution, H2O and brine, and then dried. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/acetone = 4:1) to generate acetonide 9 (2.11 g, 95% yield) as a colorless oil. [α]D25 +4.0 (c 0.58, CHCl3); IR (neat) 3443, 2956 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 4.42 (1H, dd, J = 8.5, 6.5 Hz), 4.01 (1H, m), 3.96 (1H, dd, J = 7.8, 6.5 Hz), 3.66 (1H, t, J = 8.2 Hz), 2.07 (1H, s), 1.89 (1H, m), 1.69–1.50 (4H, m), 1.41 (3H, s), 1.35 (3H, s), 1.37–1.33 (2H, m), 1.20 (3H, d, J = 6.2 Hz), 1.07 (3H, s); 13C-NMR (100 MHz, CDCl3) δ ppm: 108.5, 79.9, 69.3, 66.9, 59.1, 45.1, 35.0, 31.2, 27.8, 26.5, 25.3, 24.2, 23.3; HRESIMS (m/z) calcd. for C13H25O3 (M+H)+ 229.1804, found 229.1810; Anal. Calcd. for C13H24O3: C, 68.38; H, 10.59. Found: C, 68.43; H, 10.59.
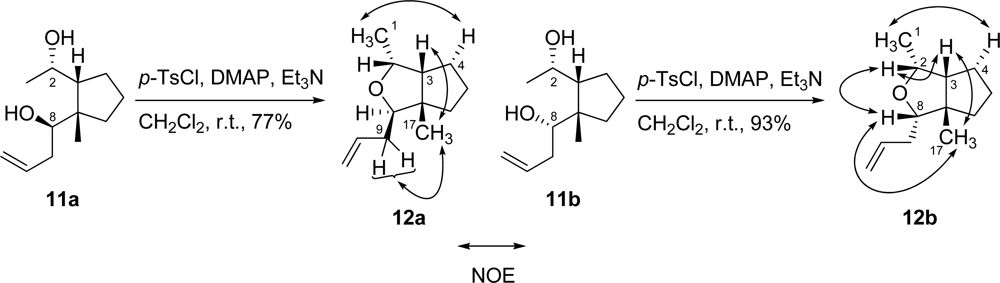
3.9. (R)-1-[(1S,2R)-2-((S)-1-Hydroxyethyl)-1-methylcyclopentyl]but-3-en-1-ol (11a) and (S)-1-[(1S,2R)-2-((S)-1-hydroxyethyl)-1-methylcyclopentyl]but-3-en-1-ol (11b)
To a solution of acetonide 9 (2.11 g, 9.24 mmol) in THF was added a solution of HIO42H2O (12.6 g, 55.4 mmol) in H2O (93.0 mL) at r.t. After stirring for 3 hr at 45 °C, the mixture was diluted with Et2O, washed with H2O and brine, dried and then concentrated to afford crude hemiacetal 10. To a solution of crude hemiacetal 10 in Et2O (93.0 mL) was added allyl magnesium bromide (33.0 mL, 32.3 mmol, 1.0 M in Et2O solution) at −78 °C. After stirring for 1 hr at 0 °C, the mixture was diluted with Et2O, washed with saturated aqueous NH4Cl solution, H2O and brine, and then dried. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 8:1) to generate diol 11a (914 mg, 50% yield) as a colorless oil and diol 11b (650 mg, 36 % yield) as a white solid. Compound 11a: [α]D25 −5.0 (c 0.39, CHCl3); IR (neat) 3306, 2955 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 5.87 (1H, m), 5.19 (1H, d, J = 10.4 Hz), 5.18 (1H, d, J = 16.8 Hz), 3.86 (1H, m), 3.68 (1H, dd, J =10.5, 2.3 Hz), 3.18 (2H, br s), 2.34 (1H, m), 2.13 (1H, m), 1.80 (1H, m), 1.62–1.34 (5H, m), 1.22 (1H, m), 1.17 (3H, d, J = 6.2 Hz), 1.09 (3H, s); 13C-NMR (100 MHz, CDCl3) δ ppm: 136.1, 118.7, 72.5, 69.2, 58.8, 47.8, 39.5, 37.1, 30.2, 22.7, 22.5, 22.4; HRESIMS (m/z) calcd. for C12H23O2 (M+H)+ 199.1698, found 199.1713; Anal. Calcd. for C12H22O2: C, 72.68; H, 11.18. Found: C, 72.40; H, 10.99. Compound 11b: mp 93–95 °C; [α]D25 −35.0 (c 0.20, CHCl3); IR (KBr) 3351, 2953 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 5.30 (1H, m), 5.18 (1H, d, J = 10.2 Hz), 5.17 (1H, d, J = 16.9 Hz), 4.11 (1H, m), 3.76 (1H, dd, J = 10.8, 2.1 Hz), 2.48 (1H, m), 2.27 (2H, br s), 2.13 (1H, m), 1.85 (1H, m), 1.67 (2H, m), 1.60 (2H, m), 1.45 (2H, m,), 1.22 (3H, d, J = 6.2 Hz), 1.19 (3H, s); 13C-NMR (100 MHz, CDCl3) δ ppm: 136.2, 118.6, 75.7, 69.5, 59.5, 47.5, 37.8, 36.5, 32.0, 29.5, 23.9, 23.4; HRESIMS (m/z) calcd. for C12H23O2 (M+H)+ 199.1698, found 199.1681; Anal. Calcd. for C12H22O2: C, 72.68; H, 11.18. Found: C, 72.65; H, 10.91.
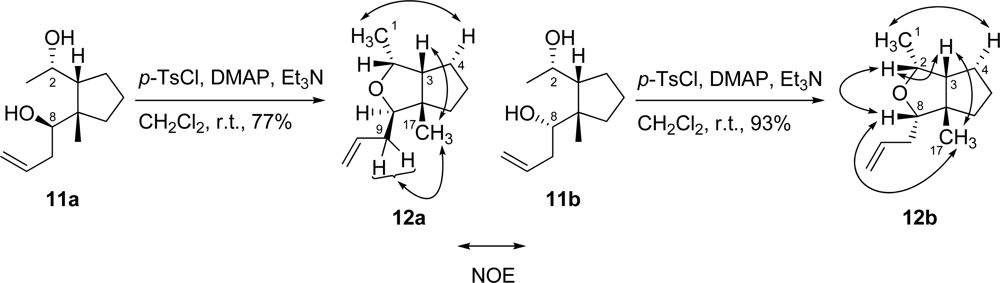
3.10. (1R,2R,3aS,3R)-3-Allyl-1,3a-dimethylhexahydrocyclopenta[c]furan (12a)
To a solution of diol 11a (34.4 mg, 0.174 mmol) in CH2Cl2 (1.7 mL) were added Et3N (105 mg, 1.04 mmol), DMAP (106 mg, 0.868 mmol) and p-toluenesulfonyl chloride (132 mg, 0.694 mmol) at r.t. The mixture was stirred for two days at the same temperature. The mixture was diluted with Et2O, washed with saturated aqueous NH4Cl solution, H2O and brine, and then dried. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 10:1) to generate tetrahydrofuran 12a (24.1 mg, 77% yield) as a colorless oil. [α]D26 +21.5 (c 1.58, CHCl3); IR (neat) 2953, 2870 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 5.84 (1H, m), 5.09 (1H, m), 5.03 (1H, m), 4.25 (1H, quint., J = 6.5 Hz), 3.64 (1H, dd, J = 9.0, 4.8 Hz), 2.30–2.15 (2H, m), 2.07 (1H, m), 1.68–1.59 (5H, m), 1.46 (1H, m), 1.16 (3H, d, J = 6.5 Hz), 1.07 (3H, s); NOESY correlations (H/H): H-1/H-4; H-3/H-17; H-9/H-17; 13C-NMR (100 MHz, CDCl3) δ ppm: 136.7, 116.4, 85.0, 74.5, 57.0, 39.1, 35.4, 27.3, 26.5, 22.1, 17.3; HRESIMS (m/z) calcd. for C12H21O (M+H)+ 181.1592, found 181.1590; Anal. Calcd. for C12H20O: C, 79.94; H, 11.18. Found: C, 80.11; H, 11.10.
3.11. (1R,2R,3aS,3S)-3-Allyl-1,3a-dimethylhexahydrocyclopenta[c]furan (12b)
To a solution of diol 11b (32.4 mg, 0.163 mmol) in CH2Cl2 (1.6 mL) were added Et3N (82.8 mg, 0.817 mmol), DMAP (79.9 mg, 0.654 mmol) and p-toluenesulfonyl chloride (93.5 mg, 0.490 mmol) at r.t. The mixture was stirred for 2 days at the same temperature. The mixture was diluted with Et2O, washed with saturated aqueous NH4Cl solution, H2O and brine, and then dried. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 5:1) to generate tetrahydrofuran 12b (27.1 mg, 93% yield) as a colorless oil. [α]D26 −35.1 (c 1.14, CHCl3); IR (neat) 2951, 2866 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 5.87 (1H, m), 5.12 (1H, m), 5.04 (1H, m), 3.84 (1H, quint., J = 6.4 Hz), 3.32 (1H, dd, J = 8.4, 4.9 Hz), 2.36–2.23 (2H, m), 1.97 (1H, m), 1.63–1.59 (6H, m), 1.19 (3H, d, J = 6.5 Hz), 1.07 (3H, s); NOESY correlations (H/H): H-1/H-4; H-2/H-3; H-2/H-8; H-3/H-17; H-8/H-17; 13C-NMR (100 MHz, CDCl3) δ ppm: 136.1, 116.3, 86.9, 75.4, 56.1, 35.1, 34.7, 30.0, 27.7, 26.8, 25.9, 15.5; HRESIMS (m/z) calcd. for C12H21O (M+H)+ 181.1592, found 181.1577; Anal. Calcd. for C12H20O: C, 79.94; H, 11.18. Found: C, 79.91; H, 11.14.
3.12. Conversion from diol 11b to diol 11a
To a solution of diol 11b (50.0 mg, 0.252 mmol) in DMF (252 μL) were added imidazole (21.0 mg, 0.308 mmol) and TBDPSCl (83.5 mg, 0.304 mmol) and the mixture was stirred at r.t. for 1 hr. The mixture was diluted with Et2O, washed with saturated aqueous NaHCO3 solution, H2O and brine and then dried. The crude mixture was diluted with Et2O and filtered through a silica gel pad. The filtrate was concentrated to afford the crude alcohol. To a solution of the crude alcohol in CH3CN (2.5 mL) was added IBX (212 mg, 0.757 mmol) at r.t. After stirring for 30 min at 80 °C, the mixture was diluted with Et2O and then filtered through a Celite pad. Removal of the solvent gave a residue which was filtered through a silica gel pad to afford the crude ketone. To a solution of the crude ketone in MeOH (2.5 mL) was added NaBH4 (28.6 mg, 0.756 mmol) at r.t. After stirring for 2 hr under reflux, the mixture was diluted with Et2O, washed with H2O and brine and then dried. Removal of the solvent gave a residue which was then filtered through a silica gel pad to afford the crude alcohols. To a solution of the crude alcohols in THF (2.5 mL) was added TBAF (760 μL, 0.760 mmol, 1.0 M in THF solution) at r.t. After stirring for 12 hr at 40 °C, the mixture was diluted with Et2O, washed with H2O and brine and then dried. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 4:1) to generate diol 11a (34.6 mg, 69%) and diol 11b (6.9 mg, 14 %).
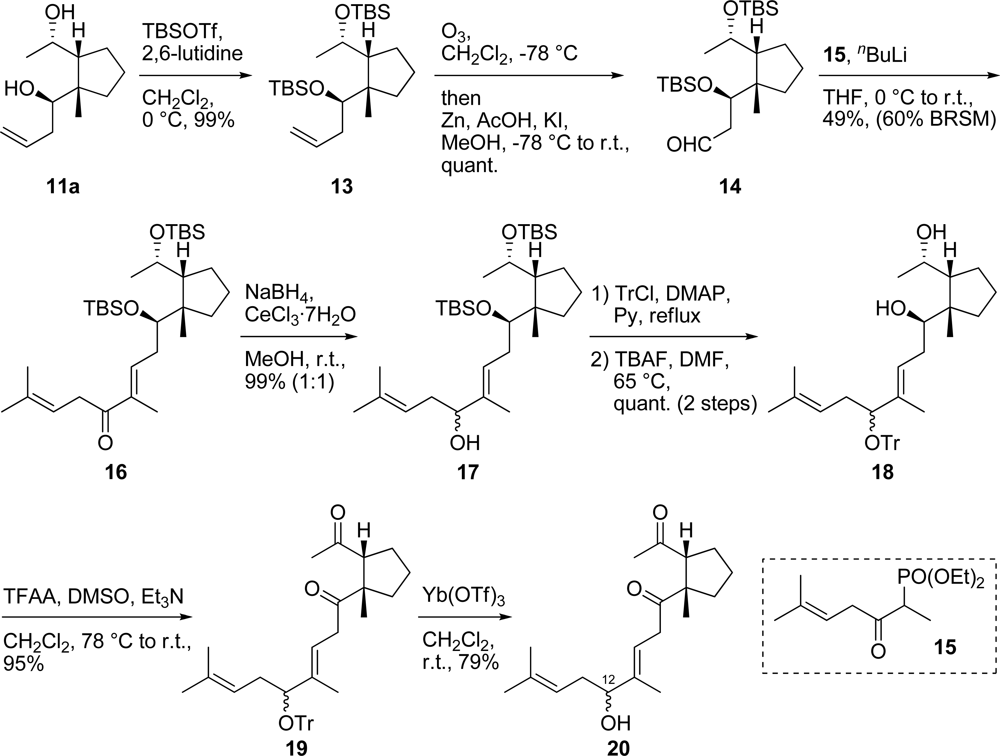
3.13. (1S,2R)-1-[(R)-1-(tert-Butyldimethylsiloxy)but-3-enyl]-2-[(S)-1-(tert-butyldimethylsiloxy)ethyl]-1-methylcyclopentane (13)
To a solution of diol 11a (988 mg, 4.98 mmol) in CH2Cl2 (2.7 mL) were added 2,6-lutidine (2.67 g, 24.9 mmol) and TBSOTf (3.95 g, 14.9 mmol) and the mixture was stirred at 0 °C for 30 min. The mixture was diluted with Et2O, washed with saturated aqueous NaHCO3 solution, H2O and brine, and then dried. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane only) to generate bis-silyl ether 13 (2.11 g, 99% yield) as a colorless oil. [α]D25 +0.97 (c 1.07, CHCl3); IR (neat) 2956, 2885, 1471 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 5.90 (1H, m), 5.00 (2H, m), 4.13 (1H, quint, J = 6.1 Hz), 3.80 (1H, dd, J = 6.5, 3.7 Hz), 2.38 (1H, m), 2.22 (1H, m), 1.85–1.57 (6H, m), 1.19 (1H, m), 1.09 (3H, d, J = 6.1 Hz), 1.04 (3H, s), 0.89 (9H, s), 0.88 (9H, s), 0.06 (12H, m); 13C-NMR (100 MHz, CDCl3) δ ppm: 137.7, 115.8, 77.2, 76.8, 69.1, 56.6, 50.1, 40.3, 35.1, 27.4, 27.1, 26.2, 26.0, 22.5, 18.4, 18.1, −3.0, −3.4, −3.6, −4.0; HRESIMS (m/z) calcd. for C24H51O2Si2 (M+H)+ 427.3428, found 427.3437; Anal. Calcd. for C24H50O2Si2: C, 67.54; H, 11.81. Found: C, 67.44; H, 11.66.
3.14. (R)-3-(tert-Butyldimethylsiloxy)-3-{(1S,2R)-2-[(S)-1-(tert-butyldimethylsiloxy)ethyl]-1-methyl-cyclopentyl}propionaldehyde (14)
A cold (−78 °C) solution of bis-silyl ether 13 (482 mg, 1.13 mmol) in CH2Cl2 (56.5 mL) was treated with ozone until the blue color generated persisted for more than 15 min. Excess ozone was removed using an argon flow. To the mixture were then added MeOH (56.5 mL), Zn powder (739 mg, 11.3 mmol), KI (1.88 g, 11.3 mmol) and AcOH (682 mg, 11.4 mmol). The mixture was allowed to warm to r.t., stirred for 1 hr at the same temperature and then concentrated under reduced pressure. The resultant residue was diluted with Et2O, washed with saturated aqueous NaHCO3 solution, H2O and brine and then dried. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 20:1) to generate aldehyde 14 (484 mg, quantitative yield) as a colorless oil. [α]D25 −0.56 (c 1.08, CHCl3); IR (neat) 2955, 2857, 1727 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 9.85 (1H, dd, J = 2.7, 1.3 Hz), 4.42 (1H, dd, J = 6.0, 4.1 Hz), 4.10 (1H, quint, J = 6.1 Hz), 2.67 (2H, m), 1.84 (1H, m), 1.74 (2H, m), 1.61 (3H, m), 1.23 (1H, m), 1.12 (3H, d, J = 6.1 Hz), 1.05 (3H, s), 0.88 (9H, s), 0.88 (9H, s), 0.06 (12H, m); 13C-NMR (100 MHz, CDCl3) δ ppm: 201.9, 70.9, 69.0, 56.3, 50.5, 49.6, 35.4, 27.3, 26.7, 26.0, 22.4, 22.3, −3.4, −3.7, −4.0, −4.1; HRESIMS (m/z) calcd. for C23H48O3Si2Na (M+Na)+ 451.3040, found 451.3052; Anal. Calcd. for C23H48O3Si2: C, 64.42; H, 11.28. Found: C, 64.40; H, 11.10.
3.15. (6E,9R)-9-(tert-Butyldimethylsiloxy)-9-{(1R,2S)-2-[(S)-1-(tert-butyldimethylsiloxy)ethyl]-1-methylcyclopentyl}-2,6-dimethylnona-2,6-dien-5-one (16)
To a solution of phosphonate
15 [
5] (306 mg, 1.17 mmol) in THF (700 μL) was added
nBuLi (591 μL, 0.935 mmol, 1.58 M in hexane solution) at 0 °C. The mixture was stirred for 1 hr at the same temperature and a solution of aldehyde
14 (200 mg, 0.467 mmol) in THF (4.0 mL) was added dropwise at r.t. After stirring for 5 hr, the mixture was diluted with Et
2O, washed with saturated aqueous NH
4Cl solution, H
2O and brine, and then dried. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/benzene = 4:1) to generate α,β-unsaturated ketone
16 (123 mg, 49% yield) as a white solid and recovered aldehyde
14 (36.5 mg, 18% yield). Compound
16: mp 55–58 °C; [α]
D25 +11.2 (
c 0.57, CHCl
3); UV (MeOH) λ
max (ɛ) nm: 234 (18800); IR (KBr) 2956, 2931, 2856, 1674 cm
−1;
1H-NMR (400 MHz, CDCl
3) δ ppm: 6.83 (1H, t,
J = 6.1 Hz), 5.34 (1H, m), 4.10 (1H, quint,
J = 6.2 Hz), 3.97 (1H, t,
J = 5.6 Hz), 3.38 (2H, d,
J = 7.0 Hz), 2.49 (2H, m), 1.85 (1H, m), 1.78 (3H, s), 1.74 (3H, s), 1.80–1.74 (2H, m), 1.64 (3H, s), 1.64–1.57 (4H, m), 1.11 (3H, d,
J = 6.1 Hz), 1.06 (3H, s), 0.90 (9H, s), 0.88 (9H, s), 0.08 (6H, d,
J = 12.6 Hz), 0.06 (6H, d,
J = 6.5 Hz);
13C-NMR (100 MHz, CDCl
3) δ ppm: 199.9, 141.5, 136.9, 134.6, 117.1, 77.2, 75.9, 69.1, 56.5, 50.1, 37.1, 35.6, 35.1, 27.2, 27.1, 26.1, 26.0, 25.7, 22.5, 22.4, 18.3, 18.1, 11.9, −3.2, −3.4, −3.8, −3.9; HRESIMS (
m/
z) calcd. for C
31H
61O
3Si
2 (M+H)
+ 537.4159, found 537.4168; Anal. Calcd. for C
31H
60O
3Si
2: C, 69.34; H, 11.26. Found: C, 69.40; H, 11.05.
3.16. (5R,6E,9R)-9-(tert-Butyldimethylsiloxy)-9-{(1R,2S)-2-[(S)-1-(tert-butyldimethylsiloxy)ethyl]-1-methylcyclopentyl}-2,6-dimethylnona-2,6-dien-5-ol and (5S,6E,9R)-9-(tert-butyldimethylsiloxy)-9-{(1R,2S)-2-[(S)-1-(tert-butyldimethylsiloxy)ethyl]-1-methylcyclopentyl}-2,6-dimethylnona-2,6-dien-5-ol (17)
To a solution of CeCl3·7H2O (144 mg, 0.386 mmol) in MeOH (9.3 mL) was added NaBH4 (11.0 mg, 0.290 mmol) at 0 °C. The mixture was then added to a solution of α,β-unsaturated ketone 16 (104 mg, 0.193 mmol) in MeOH (10.0 mL) at 0 °C and stirred for 30 min at the same temperature. The mixture was diluted with Et2O, washed with H2O and brine, and then dried. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (CHCl3 only) to generate a diastereomeric mixture of allylic alcohol 17 (103 mg, 99% yield) as a colorless oil. IR (neat) 3353, 2957 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 5.54 (1H, m), 5.11 (1H, m), 4.14 (1H, m), 4.00 (1H, m), 3.80 (1H, m), 2.42–2.14 (4H, m), 1.86 (1H, m), 1.80–1.57 (5H, m), 1.72 (3H, s), 1.64 (3H, s), 1.62 (3H, s), 1.15 (1H, m), 1.09 (3H, d, J = 6.1 Hz), 1.02 (1.5 H, s), 1.02 (1.5H, s), 0.88 (9H, d, J = 6.9 Hz), 0.88 (9H, d, J = 5.4 Hz), 0.06 (12H, m); 13C-NMR (100 MHz, CDCl3) δ ppm: 136.6, 136.5, 134.8, 134.7, 125.6, 125.5, 120.2, 120.2, 77.2, 77.2, 68.9, 68.9, 56.2, 56.2, 50.2, 50.2, 35.1, 35.0, 34.1, 34.0, 26.7, 26.6, 26.5, 26.3, 26.2, 26.0, 25.9, 22.5, 22.1, 22.1, 22.1, 18.4, 18.1, 18.0, 12.1, 12.1, −3.1, −3.2, −3.4, −3.5, −3.7, −3.7, −4.0, −4.0; HRESIMS (m/z) calcd. for C31H62O3Si2Na (M+Na)+ 561.4135, found 561.4156; Anal. Calcd. for C31H62O3Si2: C, 69.08; H, 11.59. Found: C, 68.92; H, 11.30.
3.17. (1R,3E,5R)-1-[(1S,2R)-2-((S)-1-Hydroxyethyl)-1-methylcyclopentyl]-4,8-dimethyl-5-trityloxy-nona-3,7-dien-1-ol and (1R,3E,5S)-1-[(1S,2R)-2-((S)-1-hydroxyethyl)-1-methylcyclopentyl]-4,8-dimethyl-5-trityloxynona-3,7-dien-1-ol (18)
To a solution of the diastereomeric mixture of allylic alcohol 17 (40.0 mg, 0.074 mmol) in pyridine (740 μL) were added DMAP (5.0 mg, 0.041 mmol) and TrCl (103 mg, 0.395 mmol) at r.t. After stirring for 4 days at 80 °C, the mixture was diluted with Et2O, washed with H2O and brine, and then dried. Removal of the solvent gave a residue which was filtered through a short-path silica gel pad (hexane/AcOEt = 20:1). The filtrate was then concentrated to afford the crude trityl ether. To a solution of the crude trityl ether in DMF (1.5 mL) was added TBAF (1.5 mL, 0.150 mmol, 1.0 M in THF solution) at r.t. After stirring for 2 days at 50 °C, the mixture was diluted with Et2O, washed with H2O and brine and then dried. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 10:1) to generate a diastereomeric mixture of diol 18 (40.9 mg, quantitative yield) as a colorless oil. IR (neat) 3344, 2961 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 7.51 (6H, m), 7.30–7.20 (9H, m), 4.93 (1H, t, J = 6.6 Hz), 4.77 (0.5H, dd, J = 6.7, 6.7 Hz), 4.68 (0.5H, dd, J = 9.0, 5.5 Hz), 4.00 (1H, dd, J = 5.7, 5.7 Hz), 3.79 (0.5H, m), 3.73 (0.5H, m), 3.47–3.36 (1H, m), 2.31 (0.5H, m), 2.12–1.88 (3.5 H, m), 1.82–1.73 (2.5H, m), 1.63 (3H, m), 1.54 (3H, m), 1.48 (3H, m), 1.57–1.43 (2.5H, m), 1.41–1.31 (2H, m), 1.17 (1.5H, m), 1.14 (1.5H, m), 1.07 (3H, m); 13C-NMR (100 MHz, CDCl3) δ ppm: 145.1, 140.4, 140.3, 133.7, 133.2, 129.0, 127.5, 127.5, 126.9, 126.9, 122.1, 121.4, 120.6, 120.4, 87.2, 87.2, 79.5, 78.5, 77.2, 72.6, 72.3, 69.0, 68.9, 59.1, 47.3, 47.2, 39.6, 33.1, 33.0, 30.3, 30.1, 30.1, 29.6, 29.2, 26.1, 25.8, 25.7, 22.4, 22.4, 22.3, 22.3, 22.2, 17.7, 12.7, 11.6; HRESIMS (m/z) calcd. for C38H48O3Na (M+Na)+ 575.3501, found 575.3522; Anal. Calcd. for C38H48O3: C, 82.56; H, 8.75. Found: C, 82.52; H, 8.60.
3.18. (3E,5R)-1-((1S,2R)-2-Acetyl-1-methylcyclopentyl)-4,8-dimethyl-5-trityloxynona-3,7-dien-1-one and (3E,5S)-1-((1S,2R)-2-acetyl-1-methylcyclopentyl)-4,8-dimethyl-5-trityloxynona-3,7-dien-1-one (19)
To a cold (−78 °C) solution of TFAA (67.6 mg, 0.322 mmol) in CH2Cl2 (100 μL) was added DMSO (33.6 mg, 0.430 mmol) in CH2Cl2 (100 μL). The mixture was stirred at 78 °C for 30 min, treated with a solution of the diastereomeric mixture of diol 18 (29.6 mg, 0.054 mmol) in CH2Cl2 (340 μL), stirred for 2 hr and then Et3N (54.3 mg, 0.537 mmol) was added. The mixture was warmed to r.t. and stirred for 30 min. The mixture was diluted with Et2O, washed with saturated aqueous NaHCO3 solution, H2O and brine and then dried. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 6:1) to generate a diastereomeric mixture of diketone 19 (27.9 mg, 95% yield) as a colorless oil. IR (neat) 2965, 1705 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 7.49 (6H, m), 7.26–7.16 (9H, m), 4.95 (1H, t, J = 6.1 Hz), 4.81 (1H, t, J = 7.3 Hz), 3.92 (1H, dd, J = 8.8, 4.7 Hz), 2.92 (2H, m), 2.78 (1H, dd, J = 8.6, 5.0 Hz), 2.23–1.72 (6H, m), 2.15 (3H, s), 1.63–1.55 (2H, m), 1.58 (3H, s), 1.48 (3H, s), 1.42 (3H, s), 1.21 (3H, s); 13C-NMR (100 MHz, CDCl3) δ ppm: 212.2, 210.7, 145.2, 137.6, 132.4, 129.2, 127.5, 126.8, 120.5, 118.7, 87.2, 79.3, 77.2, 60.9, 59.8, 37.9, 35.4, 33.5, 29.9, 27.6, 25.8, 25.2, 22.4, 17.8, 12.1; HRESIMS (m/z) calcd. for C38H44O3Na (M+Na)+ 571.3188, found 571.3196. Anal. Calcd. for C38H44O3: C, 83.17; H, 8.08. Found: C, 83.12; H, 8.21.
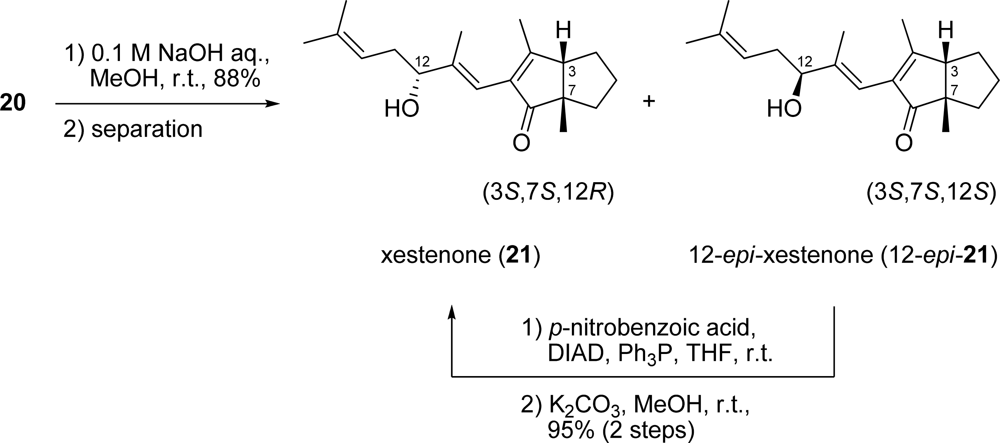
3.19. Diastereomeric mixture of secoxestenone (20)
To a solution of the diastereomeric mixture of diketone 19 (76.2 mg, 0.139 mmol) in CH2Cl2 (14 mL) was added Yb(OTf)3 (172 mg, 0.277 mmol) at r.t. After stirring for 30 min, the mixture was diluted with Et2O. To this was added NaHCO3 and the mixture was then filtered through a silica gel pad. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 1:1) to generate a diastereomeric mixture of secoxestenone 20 (33.5 mg, 79% yield) as a colorless oil. IR (neat) 3448, 2965, 1706 cm−1; 1H-NMR (400 MHz, CDCl3) δ ppm: 5.60 (1H, m), 5.10 (1H, m), 4.05 (1H, m), 3.27 (2H, m), 2.85 (1H, m), 2.27 (3H, m), 2.16 (3H, m), 2.09 (1H, m), 1.91–1.75 (3H, m), 1.71 (3H, s), 1.66 (1H, m), 1.63 (3H, s), 1.63 (3H, s), 1.28 (3H m); 13C-NMR (100 MHz, CDCl3) δ ppm: 212.8, 210.8, 148.5, 141.9, 134.7, 120.8, 120.1, 118.2, 77.2, 76.9, 61.3, 60.4, 59.7, 37.8, 35.9, 35.5, 34.0, 30.2, 29.8, 27.8, 27.7, 25.9, 25.7, 25.6, 25.3, 22.4, 18.0, 12.3, 12.1; HRESIMS (m/z) calcd. for C19H30O3Na (M+Na)+ 329.2093, found 329.2085. Anal. Calcd. for C19H30O3: C, 74.47; H, 9.87. Found: C, 74.29; H, 9.85.
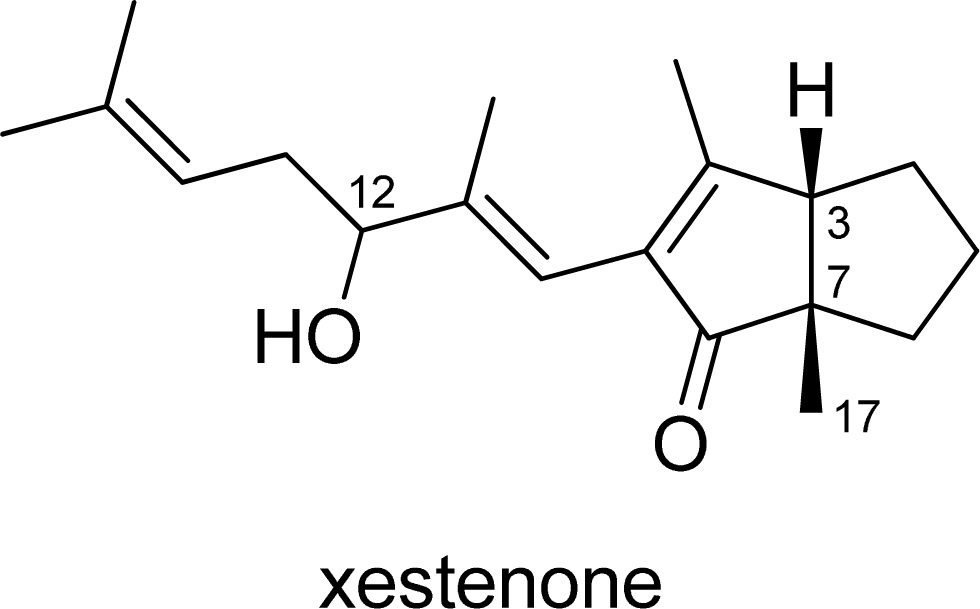
3.20. Xestenone (21) and 12-epi-xestenone (12-epi-21)
To a solution of the diastereomeric mixture of secoxestenone 20 (26.5 mg, 0.087 mmol) in MeOH (6.7 mL) was added 0.1 M NaOH aqueous solution (21.6 mL) at r.t. The mixture was stirred for 30 min, neutralized with 1.0 M HCl aqueous solution, diluted with Et2O, washed with H2O and brine, dried and then concentrated under reduced pressure. The resultant residue was purified by silica gel column chromatography (hexane/AcOEt = 4:1) to give a mixture of 21 and 12-epi-21 (22.0 mg, 88% yield) as a colorless oil. The above mixture was subjected to HPLC (CHIRALPAK IA, 1.0 cm × 25 cm, hexane/EtOH = 95:5, flow rate: 1.0 mL/min) to give xestenone 21 (tR = 12.0 min) and 12-epi-21 (tR = 15.0 min); 21: [α]D25 +2.2 (c 0.075, MeOH); UV (sh, MeOH) λmax nm (ɛ): 257 (6100); CD (MeOH) λext nm [θ]: 323 (+87,000), 258 (−129,000); IR (neat) 3419, 1685 cm−1; 1H-NMR (600 MHz, CDCl3) δ ppm: 5.93 (1H, s), 5.18 (1H, br t, J = 6.9 Hz), 4.18 (1H, t, J = 6.4 Hz), 2.71 (1H, d, J = 9.1 Hz), 2.35 (2H, m), 1.96 (3H, s), 1.93 (1H, m), 1.90 (1H, br s), 1.82 (1H, m), 1.75 (3H, s), 1.69 (1H, m), 1.67 (3H, s), 1.64 (1H, m), 1.55 (3H, s), 1.35 (1H, m), 1.25 (1H, m), 1.21 (3H, s); 13C-NMR (150 MHz, CDCl3) δ ppm: 212.7, 172.2, 144.3, 137.4, 134.9, 119.9, 115.6, 76.4, 56.7, 54.8, 37.5, 34.2, 28.9, 25.9, 24.8, 22.5, 18.0, 16.7, 14.4; HRESIMS (m/z) calcd. for C19H28O2Na (M+Na)+ 311.1987, found 311.1981. Anal. Calcd. for C19H28O2: C, 79.12; H, 9.78. Found: C, 78.97; H, 9.74. 12-epi-21: [α]D25 −113.7 (c 0.085, MeOH); UV (sh, MeOH) λmax nm (ɛ): 254 (2,100); CD (MeOH) λext nm [θ]: 320 (+116,000), 256 (−104,000); IR (neat) 3418, 1686 cm−1; 1H-NMR (600 MHz, CDCl3) δ ppm: 5.92 (1H, s), 5.16 (1H, t, J = 7.2 Hz), 4.19 (1H, t, J = 6.3 Hz), 2.71 (1H, d, J = 9.1 Hz), 2.35 (2H, m), 1.96 (3H, s), 1.93 (1H, m), 1.81 (1H, m), 1.74 (3H, s), 1.69 (1H, m), 1.67 (3H, s), 1.62 (1H, m), 1.55 (3H, s), 1.35 (1H, m), 1.25 (1H, m), 1.22 (3H, s); 13C-NMR (150 MHz, CDCl3) δ ppm: 212.8, 172.2, 144.3, 137.4, 134.8, 119.9, 115.9, 76.5, 56.7, 54.7, 37.5, 34.1, 28.9, 25.9, 24.8, 22.5, 18.0, 16.7, 14.1; HRESIMS (m/z) calcd. for C19H28O2Na (M+Na)+ 311.1987, found 311.1975. Anal. Calcd. for C19H28O2: C, 79.12; H, 9.78. Found: C, 79.03; H, 9.88.
3.21. General procedure for the synthesis of MPA ester
To a solution of xestenone (21) in CH2Cl2 were added DCC, DMAP and (S)-(+)- or (R)-(−)-α-methoxyphenylacetic acid at r.t. After stirring for 30 min at 40 °C the mixture was concentrated. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 6:1) to generate (S)- or (R)-MPA ester. (S)-MPA ester: 1H-NMR (400 MHz, CDCl3) δ ppm: 7.44–7.27 (5H, m), 5.66 (1H, s), 5.26 (1H, dd, J = 7.9, 5.7 Hz), 5.06 (1H, m), 4.75 (1H, s), 3.43 (3H, s), 2.62 (1H, d, J = 9.2 Hz), 2.39 (2H, m), 1.88 (1H, m), 1.76 (1H, m), 1.70 (3H, s), 1.68 (3H, s), 1.61 (3H, s), 1.59 (2H, m), 1.30 (1H, m), 1.26 (3H, br s), 1.19 (1H, m), 1.15 (3H, s); 13C-NMR (150 MHz, CDCl3) δ ppm: 211.9, 169.8, 139.2, 137.0, 134.6, 128.8, 128.5, 128.2, 127.2, 119.0, 118.1, 117.8, 82.6, 79.0, 56.6, 54.7, 37.4, 31.8, 29.7, 28.8, 25.8, 24.7, 22.5, 18.0, 16.5, 14.4; HRESIMS (m/z) calcd. for C28H36O4Na (M+Na)+ 459.2511, found 459.2521; (R)-MPA ester: 1H-NMR (400 MHz, CDCl3) δ ppm: 7.45–7.29 (5H, m), 5.88 (1H, s), 5.24 (1H, dd, J = 7.7, 5.7 Hz), 4.80 (1H, m), 4.77 (1H, s), 3.43 (3H, s), 2.67 (1H, d, J = 9.0 Hz), 2.28 (2H, m), 1.91 (1H, m), 1.85 (3H, s), 1.81–1.59 (3H, m), 1.53 (3H, s), 1.48 (3H, br s), 1.47 (3H, s), 1.33 (1H, m), 1.22 (1H, m), 1.19 (3H, s); 13C-NMR (150 MHz, CDCl3) δ ppm: 212.1, 170.0, 139.5, 137.0, 134.4, 128.8, 128.5, 128.2, 127.2, 119.0, 118.7, 117.8, 82.6, 79.0, 56.7, 54.7, 37.5, 31.9, 29.7, 28.9, 25.6, 24.8, 22.5, 17.8, 16.6, 16.5; HRESIMS (m/z) calcd. for C28H36O4Na (M+Na)+ 459.2511, found 459.2501.
3.22. Conversion of 12-epi-21 to 21
To a solution of 12-epi-21 (1.7 mg, 0.006 mmol) in THF (59 μL) were added Ph3P (2.3 mg, 0.009 mmol) and p-NO2BzOH (1.5 mg, 0.009 mmol) at r.t. After stirring for 10 min, DIAD (1.8 mg, 0.009 mmol) was added and the mixture was stirred for an additional 2 hr. The crude mixture was diluted with Et2O and filtered through a silica gel pad. The filtrate was then concentrated to afford the crude ester. To a solution of the crude ester in MeOH (200 μL) was added K2CO3 (12.2 mg, 0.088 mmol) at r.t. After stirring for 30 min at the same temperature, the mixture was diluted with Et2O and then filtered through a silica gel pad. Removal of the solvent gave a residue which was then purified by silica gel column chromatography (hexane/AcOEt = 2:1) to generate xestenone (21, 1.6 mg, 95% yield).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}