
Exploring the Biosynthetic Potential of Microorganisms from the South China Sea Cold Seep Using Culture-Dependent and Culture-Independent Approaches


 ,
,  , and
, and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
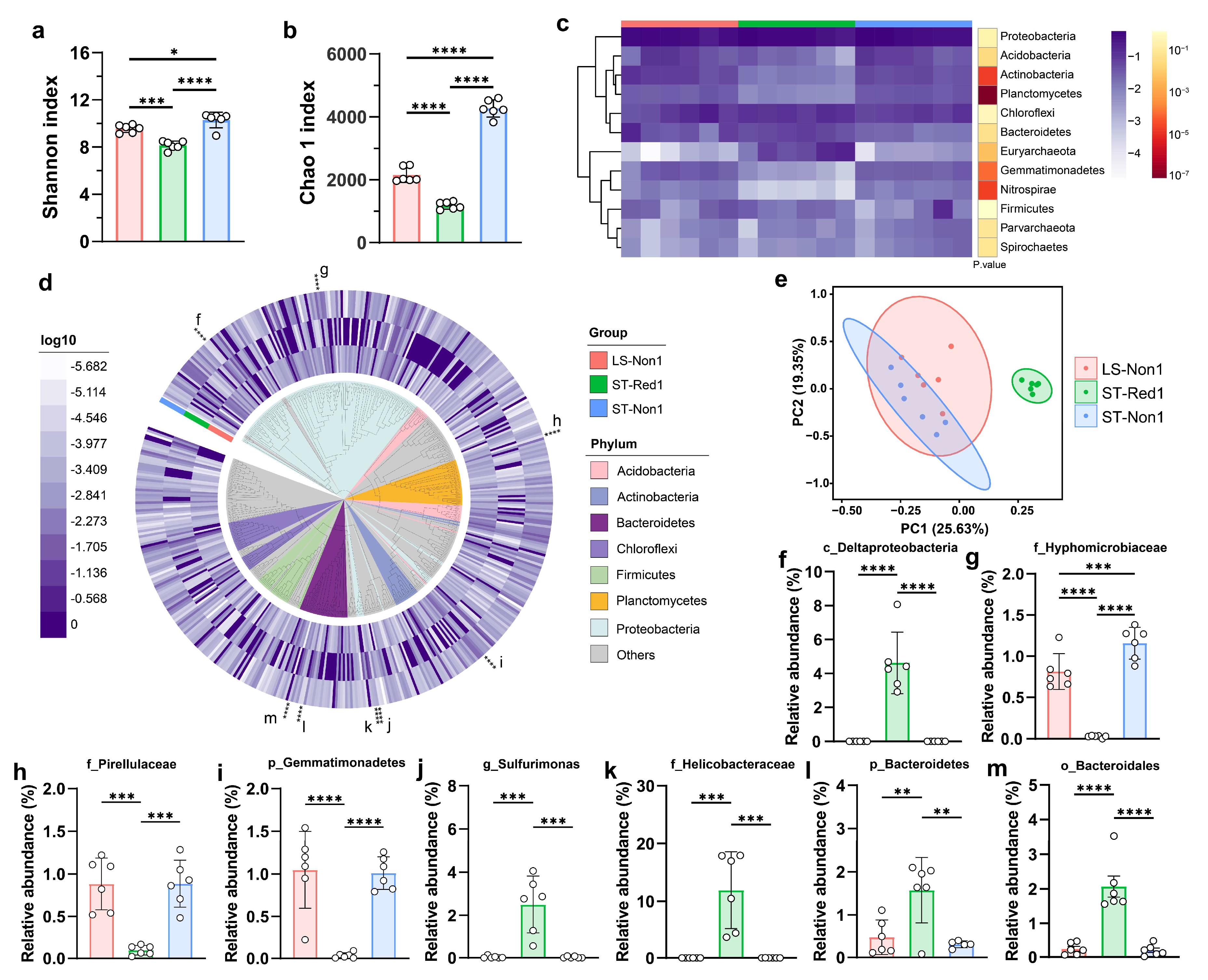
2.1. 16S rRNA Sequencing Reveals the Bacterial Diversity and Metabolic Potential in Cold Seeps
2.2. Enrichment and Selective Culturing Yield 518 Bacterial Isolations from Cold Seep
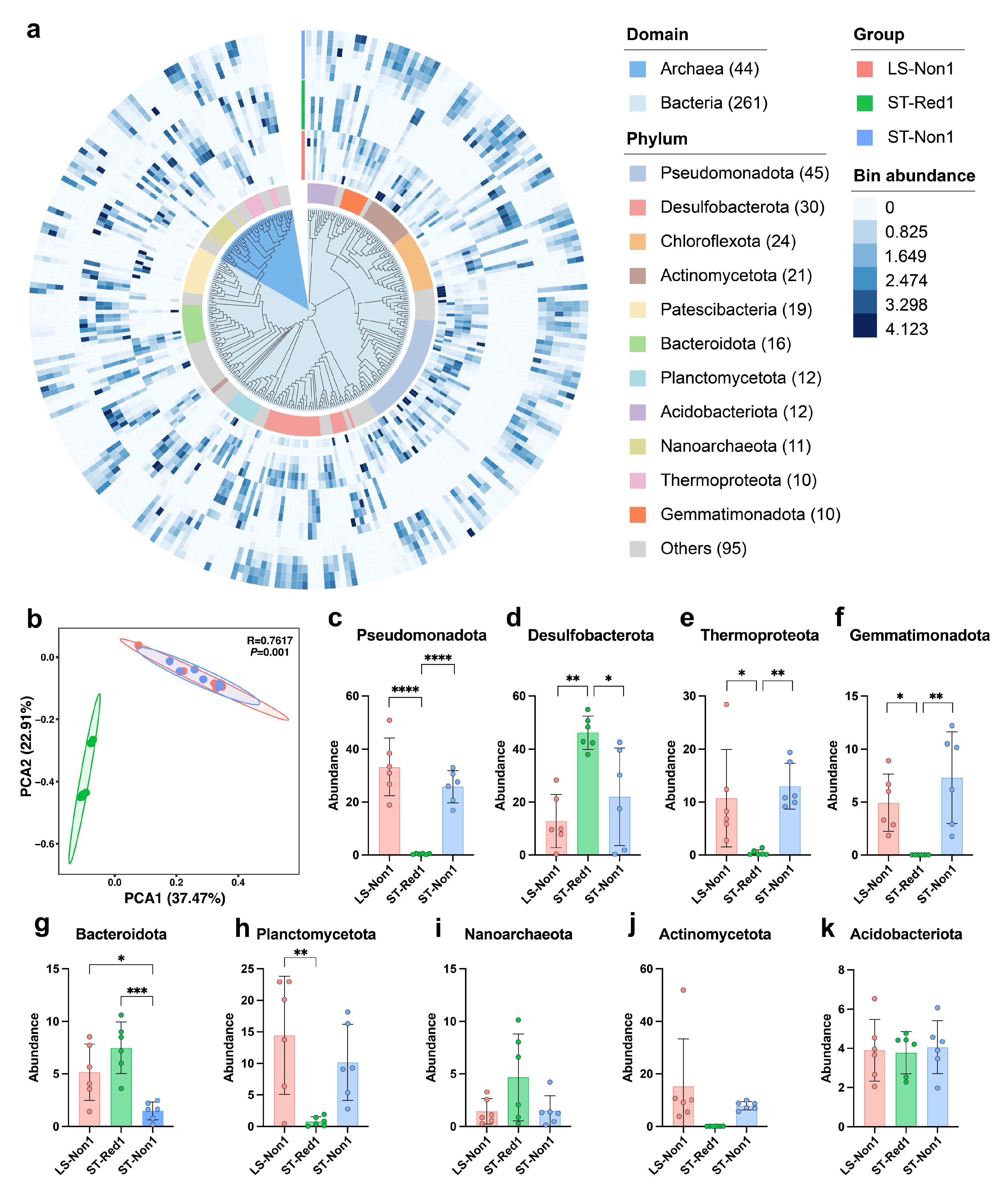
2.3. Metagenomic Study Confirms Microbial Differences Across Cold Seeps
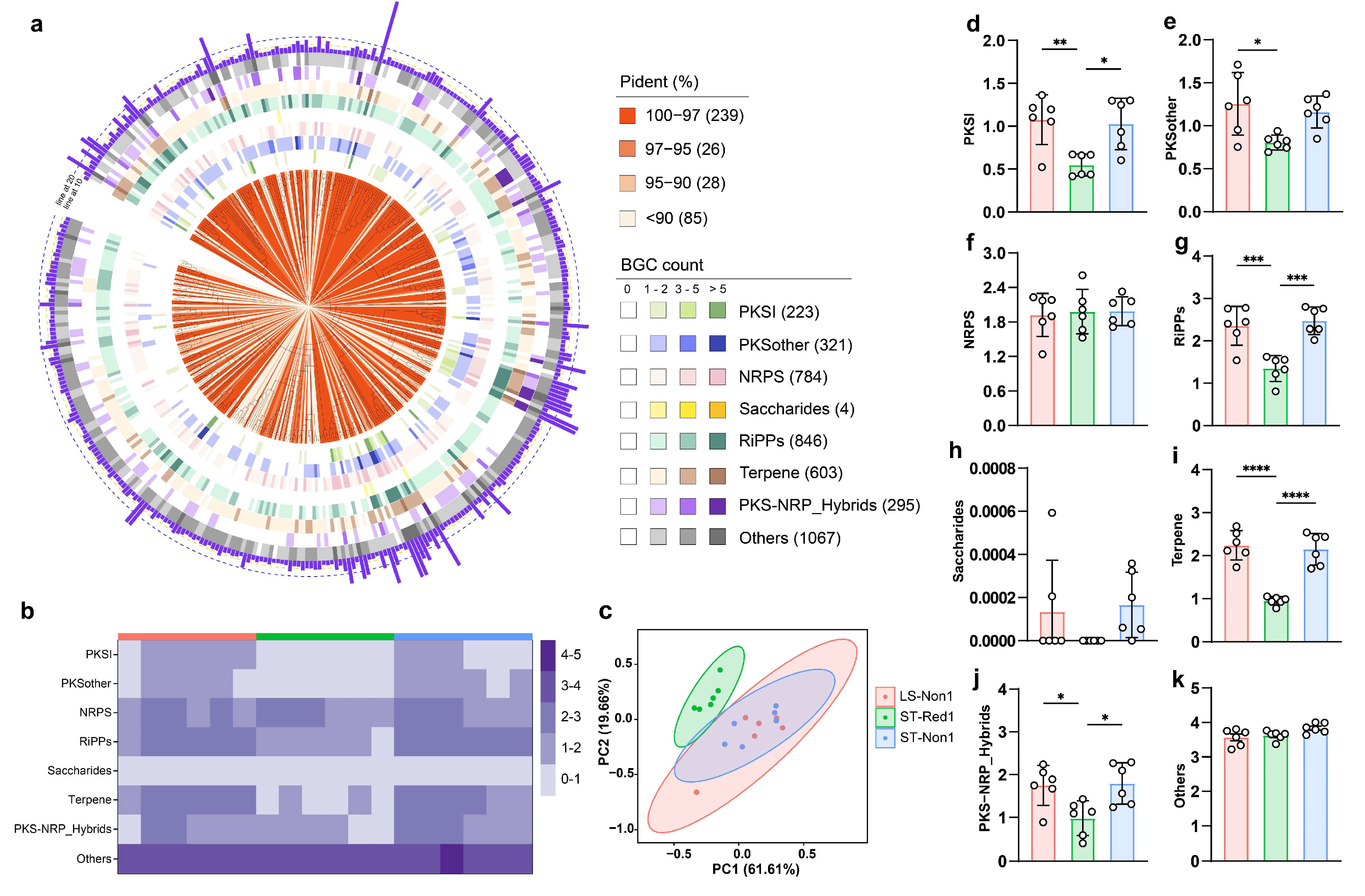
2.4. Cold Seep Microbes Harbor Diverse and Site-Specific BGCs
2.5. Depth-Dependent Distribution and Function of Biosynthetic Gene Clusters in Cold Seeps
3. Discussion
4. Materials and Methods
4.1. Cold Seep Sample Source and Collection
4.2. Isolation and Culture of Strains
4.3. DNA Extraction, Amplification, and 16S rRNA Sequencing
4.4. Metagenomic Sequencing, MAG Construction, and Bioinformatics Analysis
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Joye, S.B. The Geology and Biogeochemistry of Hydrocarbon Seeps. Annu. Rev. Earth Planet. Sci. 2020, 48, 205–231. [Google Scholar] [CrossRef]
- Orsi, W.D. Ecology and Evolution of Seafloor and Subseafloor Microbial Communities. Nat. Rev. Microbiol. 2018, 16, 671–683. [Google Scholar] [CrossRef]
- Cong, M.; Pang, X.; Zhao, K.; Song, Y.; Liu, Y.; Wang, J. Deep-Sea Natural Products from Extreme Environments: Cold Seeps and Hydrothermal Vents. Mar. Drugs 2022, 20, 404. [Google Scholar] [CrossRef]
- Åström, E.K.L.; Carroll, M.L.; Ambrose, W.G., Jr.; Sen, A.; Silyakova, A.; Carroll, J. Methane Cold Seeps as Biological Oases in the High-Arctic Deep Sea. Limnol. Oceanogr. 2018, 63, S209–S231. [Google Scholar] [CrossRef]
- Lee, O.O.; Wang, Y.; Tian, R.; Zhang, W.; Shek, C.S.; Bougouffa, S.; Al-Suwailem, A.; Batang, Z.B.; Xu, W.; Wang, G.C.; et al. In Situ Environment Rather than Substrate Type Dictates Microbial Community Structure of Biofilms in a Cold Seep System. Sci. Rep. 2014, 4, 3587. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-L.; Wu, Y.-Z.; Zhou, G.; Huang, H.; Wang, Y. Metabolic Diversification of Anaerobic Methanotrophic Archaea in a Deep-Sea Cold Seep. Mar. Life Sci. Technol. 2020, 2, 431–441. [Google Scholar] [CrossRef]
- Li, W.-L.; Dong, X.; Lu, R.; Zhou, Y.-L.; Zheng, P.-F.; Feng, D.; Wang, Y. Microbial Ecology of Sulfur Cycling near the Sulfate–Methane Transition of Deep-Sea Cold Seep Sediments. Environ. Microbiol. 2021, 23, 6844–6858. [Google Scholar] [CrossRef] [PubMed]
- Ruppel, C.D.; Kessler, J.D. The Interaction of Climate Change and Methane Hydrates. Rev. Geophys. 2017, 55, 126–168. [Google Scholar] [CrossRef]
- Cho, J.-C.; Lee, S.-H.; Oh, H.-R.; Lee, J.-H.; Kim, S.-J. Cold-Seep Sediment Harbors Phylogenetically Diverse Uncultured Bacteria. J. Microbiol. Biotechnol. 2004, 14, 906–913. [Google Scholar]
- Li, L.; Zhang, W.; Zhang, S.; Song, L.; Sun, Q.; Zhang, H.; Xiang, H.; Dong, X. Bacteria and Archaea Synergistically Convert Glycine Betaine to Biogenic Methane in the Formosa Cold Seep of the South China Sea. mSystems 2021, 6, e0070321. [Google Scholar] [CrossRef]
- Nogi, Y.; Mori, K.; Uchida, H.; Hatada, Y. Shimia sagamensis Sp. Nov., a Marine Bacterium Isolated from Cold-Seep Sediment. Int. J. Syst. Evol. Microbiol. 2015, 65, 2786–2790. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhou, D.; Zhang, B.; Zhang, J. Fontisubflavum oceani Gen. Nov., Sp. Nov., Isolated from the Deep-Sea Cold Seep Water of South China Sea. Int. J. Syst. Evol. Microbiol. 2024, 74, 006256. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Kato, C.; Horikoshi, K. Microbial Diversity in Sediments Collected from the Deepest Cold-Seep Area, the Japan Trench. Mar. Biotechnol. 1999, 1, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Peng, Y.; Wang, M.; Woods, L.; Wu, W.; Wang, Y.; Xiao, X.; Li, J.; Jia, K.; Greening, C.; et al. Evolutionary Ecology of Microbial Populations Inhabiting Deep Sea Sediments Associated with Cold Seeps. Nat. Commun. 2023, 14, 1127. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, M.; Zhang, H.; He, W.; Cao, L.; Lian, C.; Zhong, Z.; Wang, H.; Fu, L.; Zhang, X.; et al. Metabolism Interactions Promote the Overall Functioning of the Episymbiotic Chemosynthetic Community of Shinkaia crosnieri of Cold Seeps. mSystems 2022, 7, e0032022. [Google Scholar] [CrossRef]
- Dong, X.; Zhang, T.; Wu, W.; Peng, Y.; Liu, X.; Han, Y.; Chen, X.; Gao, Z.; Xia, J.; Shao, Z.; et al. A Vast Repertoire of Secondary Metabolites Potentially Influences Community Dynamics and Biogeochemical Processes in Cold Seeps. Sci. Adv. 2024, 10, eadl2281. [Google Scholar] [CrossRef]
- Giordano, D. Bioactive Molecules from Extreme Environments II. Mar. Drugs 2021, 19, 642. [Google Scholar] [CrossRef]
- Wright, E.S.; Vetsigian, K.H. Inhibitory Interactions Promote Frequent Bistability among Competing Bacteria. Nat. Commun. 2016, 7, 11274. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Lu, W.-Y.; Li, H.-J.; Li, Q.-Y.; Wu, Y.-C. Application of Marine Natural Products in Drug Research. Bioorg. Med. Chem. 2021, 35, 116058. [Google Scholar] [CrossRef]
- Ghareeb, M.A.; Tammam, M.A.; El-Demerdash, A.; Atanasov, A.G. Insights about Clinically Approved and Preclinically Investigated Marine Natural Products. Curr. Res. Biotechnol. 2020, 2, 88–102. [Google Scholar] [CrossRef]
- Dilshad, R.; Jamil, N.; Batool, R. Biosynthetic Gene Clusters in Bacteria: A Review: Bacterial Secondary Metabolites Producing Genes. Proc. Pak. Acad. Sci. B Life Environ. Sci. 2021, 58, 29–42. [Google Scholar] [CrossRef]
- Wei, B.; Hu, G.-A.; Zhou, Z.-Y.; Yu, W.-C.; Du, A.-Q.; Yang, C.-L.; Yu, Y.-L.; Chen, J.-W.; Zhang, H.-W.; Wu, Q.; et al. Global Analysis of the Biosynthetic Chemical Space of Marine Prokaryotes. Microbiome 2023, 11, 144. [Google Scholar] [CrossRef]
- Kumar, M.; Ji, B.; Zengler, K.; Nielsen, J. Modelling Approaches for Studying the Microbiome. Nat. Microbiol. 2019, 4, 1253–1267. [Google Scholar] [CrossRef]
- Medema, M.H.; Fischbach, M.A. Computational Approaches to Natural Product Discovery. Nat. Chem. Biol. 2015, 11, 639–648. [Google Scholar] [CrossRef]
- Paoli, L.; Ruscheweyh, H.-J.; Forneris, C.C.; Hubrich, F.; Kautsar, S.; Bhushan, A.; Lotti, A.; Clayssen, Q.; Salazar, G.; Milanese, A.; et al. Biosynthetic Potential of the Global Ocean Microbiome. Nature 2022, 607, 111–118. [Google Scholar] [CrossRef]
- Klemetsen, T.; Raknes, I.A.; Fu, J.; Agafonov, A.; Balasundaram, S.V.; Tartari, G.; Robertsen, E.; Willassen, N.P. The MAR Databases: Development and Implementation of Databases Specific for Marine Metagenomics. Nucleic Acids Res. 2018, 46, D692–D699. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Luo, Y.; Tan, X.; Zhao, D.; Bi, X.; Li, C.; Zheng, Y.; Xiang, H.; Hu, S. Global Marine Cold Seep Metagenomes Reveal Diversity of Taxonomy, Metabolic Function, and Natural Products. Genom. Proteom. Bioinform. 2024, 22, qzad006. [Google Scholar] [CrossRef]
- Ma, B.; Lu, C.; Wang, Y.; Yu, J.; Zhao, K.; Xue, R.; Ren, H.; Lv, X.; Pan, R.; Zhang, J.; et al. A Genomic Catalogue of Soil Microbiomes Boosts Mining of Biodiversity and Genetic Resources. Nat. Commun. 2023, 14, 7318. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Páez-Espino, D.; Call, L.; Low, S.J.; Sberro, H.; Ivanova, N.N.; Proal, A.D.; Fischbach, M.A.; Bhatt, A.S.; Hugenholtz, P.; et al. Metagenomic Compendium of 189,680 DNA Viruses from the Human Gut Microbiome. Nat. Microbiol. 2021, 6, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xia, J.; Jiang, L.; Tan, Y.; An, Y.; Zhu, X.; Ruan, J.; Chen, Z.; Zhen, H.; Ma, Y.; et al. Characterization of the Human Skin Resistome and Identification of Two Microbiota Cutotypes. Microbiome 2021, 9, 47. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.N.; et al. antiSMASH 7.0: New and Improved Predictions for Detection, Regulation, Chemical Structures and Visualisation. Nucleic Acids Res. 2023, 51, W46–W50. [Google Scholar] [CrossRef]
- Hannigan, G.D.; Prihoda, D.; Palicka, A.; Soukup, J.; Klempir, O.; Rampula, L.; Durcak, J.; Wurst, M.; Kotowski, J.; Chang, D.; et al. A Deep Learning Genome-Mining Strategy for Biosynthetic Gene Cluster Prediction. Nucleic Acids Res. 2019, 47, e110. [Google Scholar] [CrossRef]
- Skinnider, M.A.; Johnston, C.W.; Gunabalasingam, M.; Merwin, N.J.; Kieliszek, A.M.; MacLellan, R.J.; Li, H.; Ranieri, M.R.M.; Webster, A.L.H.; Cao, M.P.T.; et al. Comprehensive Prediction of Secondary Metabolite Structure and Biological Activity from Microbial Genome Sequences. Nat. Commun. 2020, 11, 6058. [Google Scholar] [CrossRef]
- Wei, B.; Zhou, Z.-Y.; Lai, C.; Du, A.-Q.; Hu, G.-A.; Yu, W.-C.; Yu, Y.-L.; Chen, J.-W.; Zhang, H.-W.; Wu, Q.-H.; et al. Predicting the Secondary Metabolic Potential of Microbiomes from Marker Genes Using PSMPA. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum Information about a Single Amplified Genome (MISAG) and a Metagenome-Assembled Genome (MIMAG) of Bacteria and Archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef]
- Waschulin, V.; Borsetto, C.; James, R.; Newsham, K.K.; Donadio, S.; Corre, C.; Wellington, E. Biosynthetic Potential of Uncultured Antarctic Soil Bacteria Revealed through Long-Read Metagenomic Sequencing. ISME J. 2022, 16, 101–111. [Google Scholar] [CrossRef]
- Pascal Andreu, V.; Augustijn, H.E.; van den Berg, K.; van der Hooft, J.J.J.; Fischbach, M.A.; Medema, M.H. BiG-MAP: An Automated Pipeline to Profile Metabolic Gene Cluster Abundance and Expression in Microbiomes. mSystems 2021, 6, e0093721. [Google Scholar] [CrossRef]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Navarro-Munoz, J.C.; Terlouw, B.R.; van der Hooft, J.J.J.; van Santen, J.A.; Tracanna, V.; Duran, H.G.S.; Andreu, V.P.; et al. MIBiG 2.0: A Repository for Biosynthetic Gene Clusters of Known Function. Nucleic Acids Res. 2020, 48, D454–D458. [Google Scholar] [CrossRef]
- Navarro-Munoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A Computational Framework to Explore Large-Scale Biosynthetic Diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef]
- Brickman, T.J.; Armstrong, S.K. Essential Role of the Iron-Regulated Outer Membrane Receptor FauA in Alcaligin Siderophore-Mediated Iron Uptake inBordetella Species. J. Bacteriol. 1999, 181, 5958–5966. [Google Scholar] [CrossRef]
- Garibaldi, J.A.; Neilands, J.B. Isolation and Properties of Ferrichrome a. J. Am. Chem. Soc. 1955, 77, 2429–2430. [Google Scholar] [CrossRef]
- Müller, G.; Raymond, K.N. Specificity and Mechanism of Ferrioxamine-Mediated Iron Transport in Streptomyces pilosus. J. Bacteriol. 1984, 160, 304–312. [Google Scholar] [CrossRef]
- Ellermann, M.; Arthur, J.C. Siderophore-Mediated Iron Acquisition and Modulation of Host-Bacterial Interactions. Free Radic. Biol. Med. 2017, 105, 68–78. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Shi, L.-D.; Li, J.; Xiao, X.; Shao, Z.; Dong, X. Unexpected Genetic and Microbial Diversity for Arsenic Cycling in Deep Sea Cold Seep Sediments. npj Biofilms Microbiomes 2023, 9, 13. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, M.; Wang, H.; Chen, H.; Cao, L.; Zhong, Z.; Lian, C.; Zhou, L.; Li, C. Metagenome Sequencing and 768 Microbial Genomes from Cold Seep in South China Sea. Sci. Data 2022, 9, 480. [Google Scholar] [CrossRef]
- Poretsky, R.; Rodriguez-R, L.M.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and Limitations of 16S rRNA Gene Amplicon Sequencing in Revealing Temporal Microbial Community Dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef]
- Mande, S.S.; Mohammed, M.H.; Ghosh, T.S. Classification of Metagenomic Sequences: Methods and Challenges. Brief. Bioinform. 2012, 13, 669–681. [Google Scholar] [CrossRef]
- Alain, K.; Querellou, J. Cultivating the Uncultured: Limits, Advances and Future Challenges. Extremophiles 2009, 13, 583–594. [Google Scholar] [CrossRef]
- Zhan, M.; Zeng, W.; Liu, H.; Li, J.; Meng, Q.; Peng, Y. Simultaneous Nitrogen and Sulfur Removal through Synergy of Sulfammox, Anammox and Sulfur-Driven Autotrophic Denitrification in a Modified Bioreactor Enhanced by Activated Carbon. Environ. Res. 2023, 232, 116341. [Google Scholar] [CrossRef]
- Suess, E. Marine Cold Seeps and Their Manifestations: Geological Control, Biogeochemical Criteria and Environmental Conditions. Int. J. Earth. Sci. 2014, 103, 1889–1916. [Google Scholar] [CrossRef]
- Sun, Q.; Xu, K.; Cao, L.; Du, Z.; Wang, M.; Sun, L. Nitrogen and Sulfur Cycling Driven by Campylobacterota in the Sediment–Water Interface of Deep-Sea Cold Seep: A Case in the South China Sea. mBio 2023, 14, e00117-23. [Google Scholar] [CrossRef]
- Kleindienst, S.; Ramette, A.; Amann, R.; Knittel, K. Distribution and in Situ Abundance of Sulfate-Reducing Bacteria in Diverse Marine Hydrocarbon Seep Sediments. Environ. Microbiol. 2012, 14, 2689–2710. [Google Scholar] [CrossRef]
- Lin, T.J.; Ver Eecke, H.C.; Breves, E.A.; Dyar, M.D.; Jamieson, J.W.; Hannington, M.D.; Dahle, H.; Bishop, J.L.; Lane, M.D.; Butterfield, D.A.; et al. Linkages between Mineralogy, Fluid Chemistry, and Microbial Communities within Hydrothermal Chimneys from the Endeavour Segment, Juan de Fuca Ridge. Geochem. Geophys. Geosyst. 2016, 17, 300–323. [Google Scholar] [CrossRef] [PubMed]
- Kumar, U.; Panneerselvam, P.; Gupta, V.V.S.R.; Manjunath, M.; Priyadarshinee, P.; Sahoo, A.; Dash, S.R.; Kaviraj, M.; Annapurna, K. Diversity of Sulfur-Oxidizing and Sulfur-Reducing Microbes in Diverse Ecosystems. In Advances in Soil Microbiology: Recent Trends and Future Prospects: Volume 1: Soil-Microbe Interaction; Adhya, T.K., Lal, B., Mohapatra, B., Paul, D., Das, S., Eds.; Springer: Singapore, 2018; pp. 65–89. ISBN 978-981-10-6178-3. [Google Scholar]
- Krom, M.D.; Mortimer, R.J.G.; Poulton, S.W.; Hayes, P.; Davies, I.M.; Davison, W.; Zhang, H. In-Situ Determination of Dissolved Iron Production in Recent Marine Sediments. Aquat. Sci. 2002, 64, 282–291. [Google Scholar] [CrossRef]
- Sandy, M.; Butler, A. Microbial Iron Acquisition: Marine and Terrestrial Siderophores. Chem. Rev. 2009, 109, 4580–4595. [Google Scholar] [CrossRef] [PubMed]
- López-Maury, L.; Marguerat, S.; Bähler, J. Tuning Gene Expression to Changing Environments: From Rapid Responses to Evolutionary Adaptation. Nat. Rev. Genet. 2008, 9, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Neilands, J.B. Chapter 1-IRON AND ITS ROLE IN MICROBIAL PHYSIOLOGY. In Microbial Iron Metabolism; Neilands, J.B., Ed.; Academic Press: Cambridge, MA, USA, 1974; pp. 3–34. ISBN 978-0-12-515250-1. [Google Scholar]
- Høvik Hansen, G.; Sørheim, R. Improved Method for Phenotypical Characterization of Marine Bacteria. J. Microbiol. Methods 1991, 13, 231–241. [Google Scholar] [CrossRef]
- Ramasamy, V.; Durairaj, T.; Alagappan, C.; Sudalaimuthu, R.S.S. Cultivation Techniques of Rare Actinobacteria. In Methods in Actinobacteriology; Dharumadurai, D., Ed.; Springer US: New York, NY, USA, 2022; pp. 169–179. ISBN 978-1-0716-1728-1. [Google Scholar]
- El-Sayed, M.H.; Abdellatif, M.M.; Mostafa, H.M.; Elsehemy, I.A.; Kobisi, A.E.-N.A. Biodegradation and Antimicrobial Capability-Induced Heavy Metal Resistance of the Marine-Derived Actinomycetes Nocardia Harenae JJB5 and Amycolatopsis Marina JJB11. World J. Microbiol. Biotechnol. 2024, 40, 202. [Google Scholar] [CrossRef]
- McDonald, D.; Jiang, Y.; Balaban, M.; Cantrell, K.; Zhu, Q.; Gonzalez, A.; Morton, J.T.; Nicolaou, G.; Parks, D.H.; Karst, S.M.; et al. Greengenes2 Unifies Microbial Data in a Single Reference Tree. Nat. Biotechnol. 2024, 42, 715–718. [Google Scholar] [CrossRef]
- Lassmann, T.; Sonnhammer, E.L. Kalign–an Accurate and Fast Multiple Sequence Alignment Algorithm. BMC Bioinform. 2005, 6, 298. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent Updates and New Developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A Flexible Pipeline for Genome-Resolved Metagenomic Data Analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Na, S.-I.; Kim, Y.O.; Yoon, S.-H.; Ha, S.; Baek, I.; Chun, J. UBCG: Up-to-Date Bacterial Core Gene Set and Pipeline for Phylogenomic Tree Reconstruction. J. Microbiol. 2018, 56, 280–285. [Google Scholar] [CrossRef]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & Clustermap.Js: Automatic Generation of Gene Cluster Comparison Figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological Network Exploration with Cytoscape 3. Curr. Protoc. Bioinform. 2014, 47, 1–24. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, G.-A.; Sun, H.-Y.; Yin, Q.-J.; Wang, H.; Liu, S.-Y.; Wang, B.-G.; Wang, H.; Li, X.; Wei, B. Exploring the Biosynthetic Potential of Microorganisms from the South China Sea Cold Seep Using Culture-Dependent and Culture-Independent Approaches. Mar. Drugs 2025, 23, 313. https://doi.org/10.3390/md23080313
Hu G-A, Sun H-Y, Yin Q-J, Wang H, Liu S-Y, Wang B-G, Wang H, Li X, Wei B. Exploring the Biosynthetic Potential of Microorganisms from the South China Sea Cold Seep Using Culture-Dependent and Culture-Independent Approaches. Marine Drugs. 2025; 23(8):313. https://doi.org/10.3390/md23080313
Chicago/Turabian StyleHu, Gang-Ao, Huai-Ying Sun, Qun-Jian Yin, He Wang, Shi-Yi Liu, Bin-Gui Wang, Hong Wang, Xin Li, and Bin Wei. 2025. "Exploring the Biosynthetic Potential of Microorganisms from the South China Sea Cold Seep Using Culture-Dependent and Culture-Independent Approaches" Marine Drugs 23, no. 8: 313. https://doi.org/10.3390/md23080313
APA StyleHu, G.-A., Sun, H.-Y., Yin, Q.-J., Wang, H., Liu, S.-Y., Wang, B.-G., Wang, H., Li, X., & Wei, B. (2025). Exploring the Biosynthetic Potential of Microorganisms from the South China Sea Cold Seep Using Culture-Dependent and Culture-Independent Approaches. Marine Drugs, 23(8), 313. https://doi.org/10.3390/md23080313