

Targeting Ferroptosis in Tumors: Novel Marine-Derived Compounds as Regulators of Lipid Peroxidation and GPX4 Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Biology of Ferroptosis
1.2. Marine Natural Products in Drug Discovery
2. Marine Compounds and Ferroptosis
2.1. Molecular Mechanisms of Marine Compounds Targeting Ferroptosis Pathways
2.2. Clinical Translational Challenges of Targeting Ferroptosis Pathways with Marine Compounds
3. Mechanisms, Strategies, and Challenges of Marine Compounds in Regulating Ferroptosis
3.1. Core Signaling Pathways and Therapeutic Targets of Ferroptosis
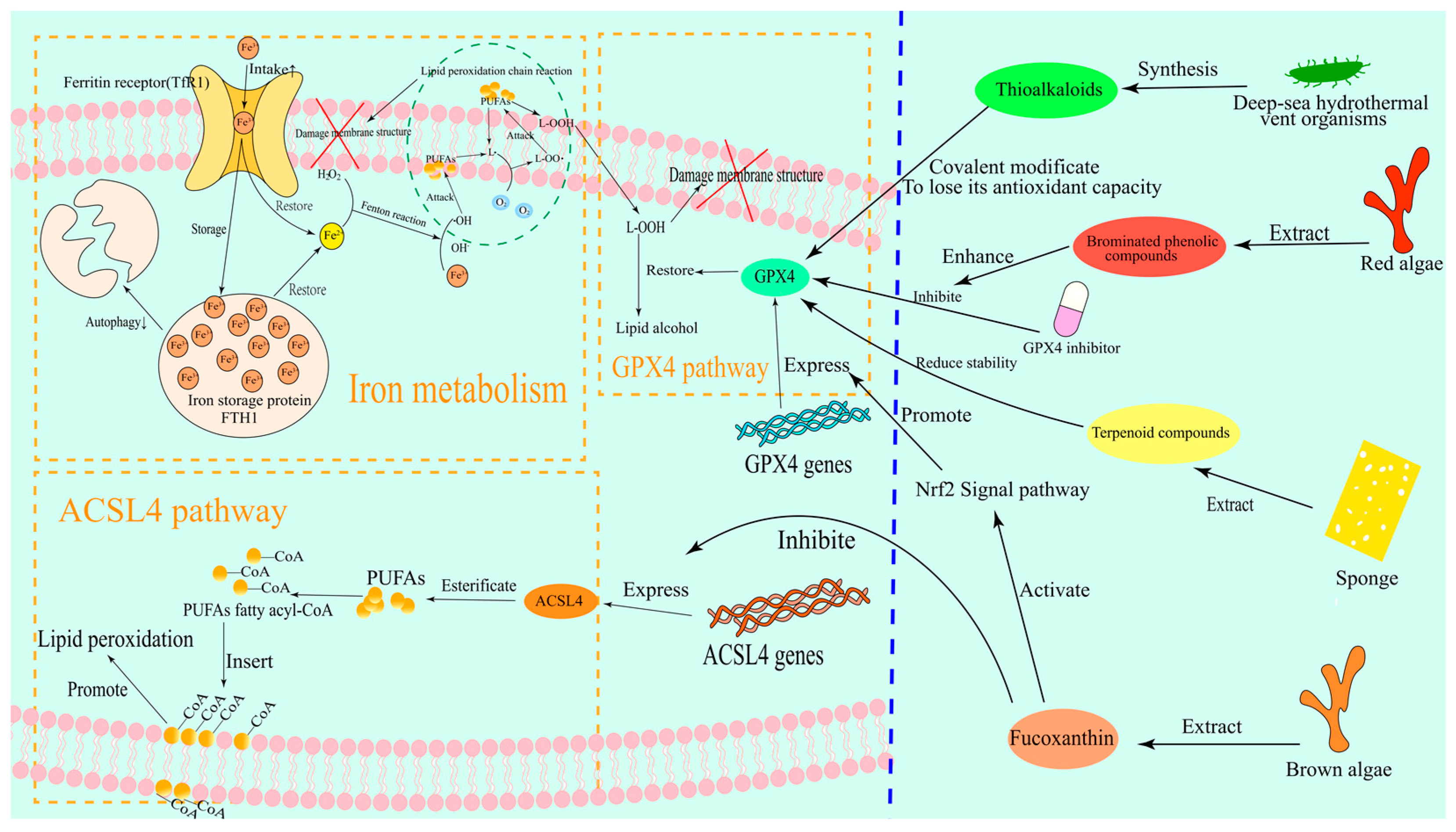
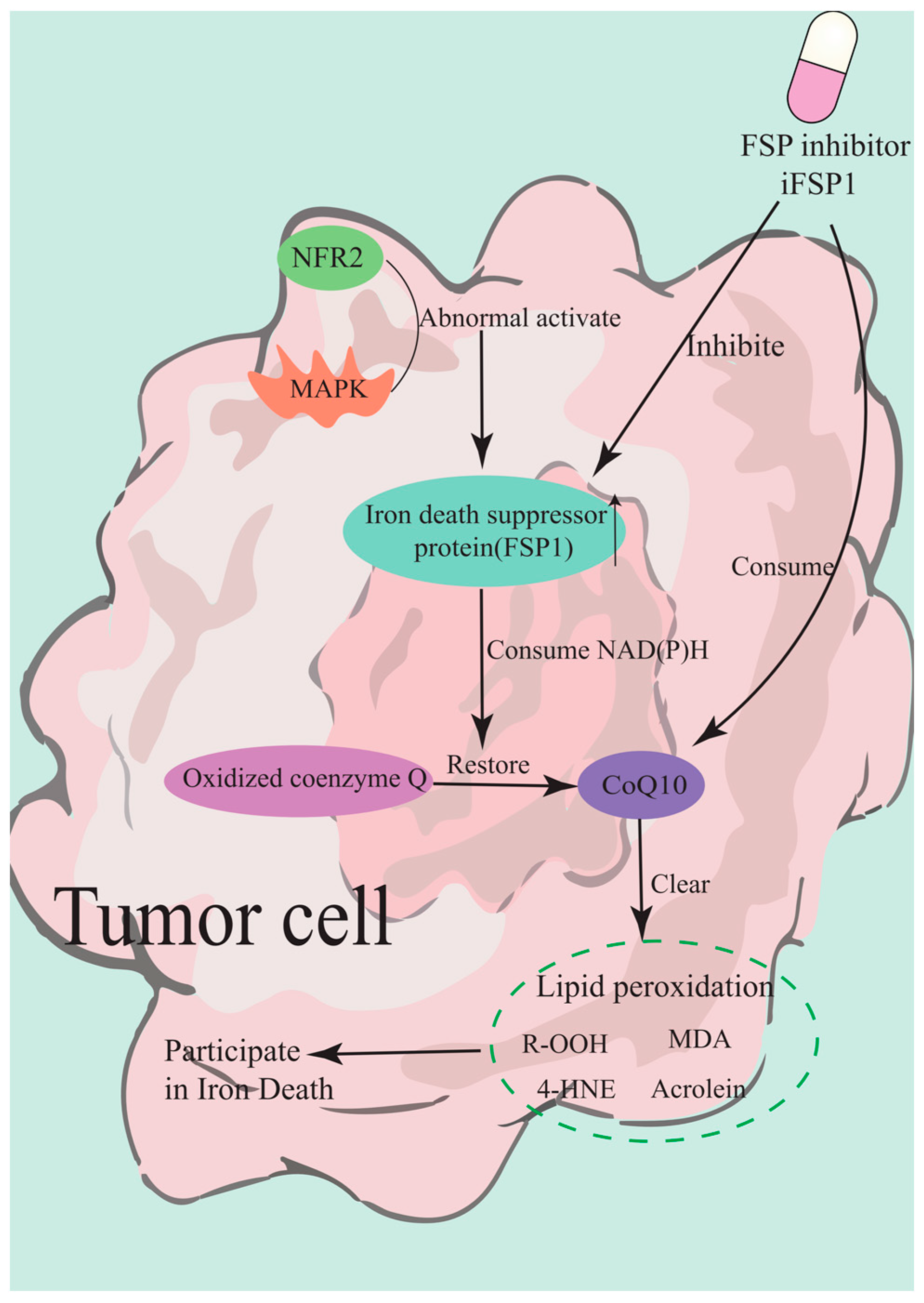
3.1.1. GPX4-Dependent Pathways
3.1.2. ACSL4(1)-Driven Lipid Peroxidation
3.1.3. Iron Metabolism Network
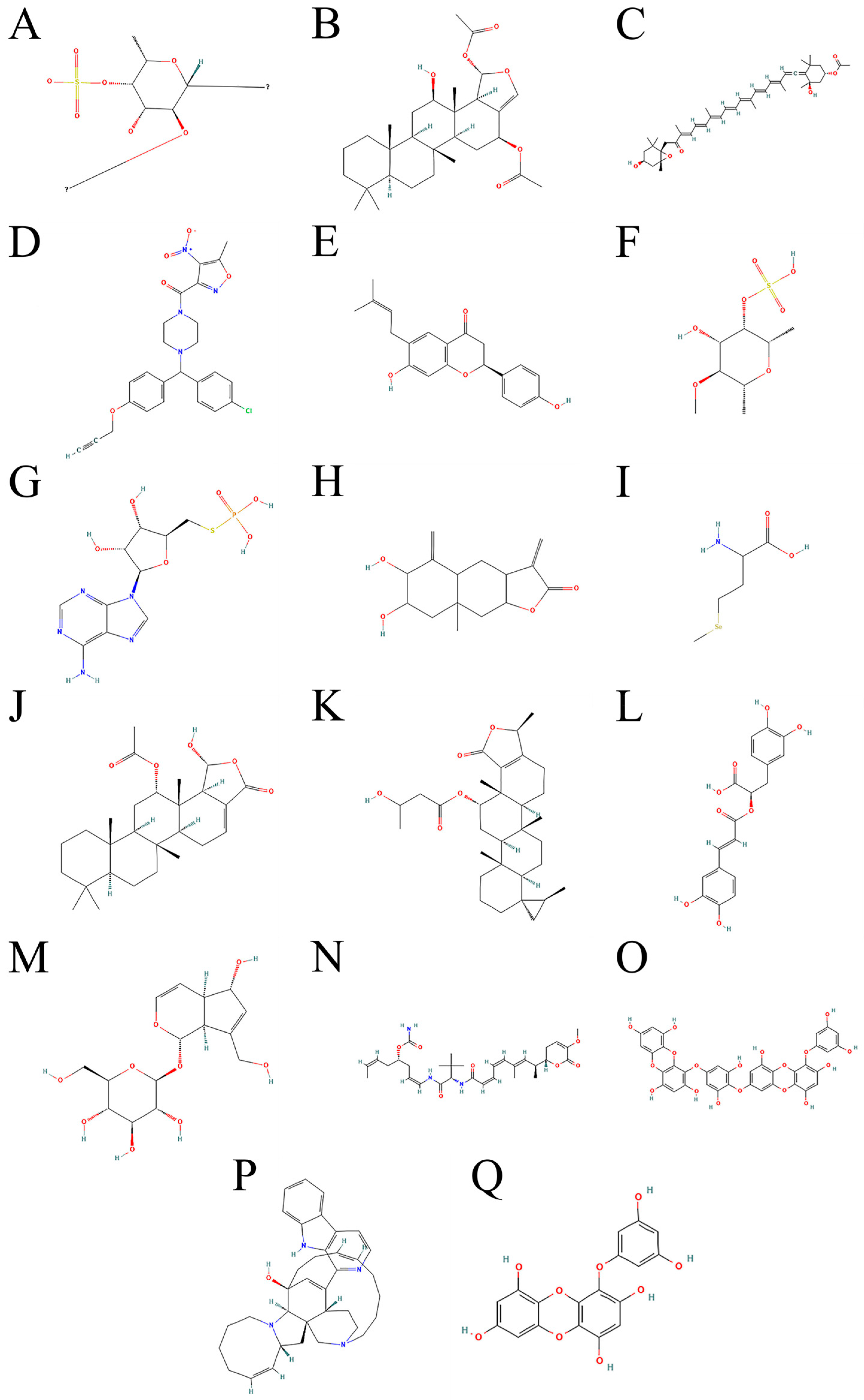
3.2. Selection and Mechanism of Marine Natural Products
3.2.1. Marine Ferroptosis Inducers
3.2.2. Marine-Derived Ferroptosis Inhibitors
3.2.3. Technology-Driven Screening Strategies
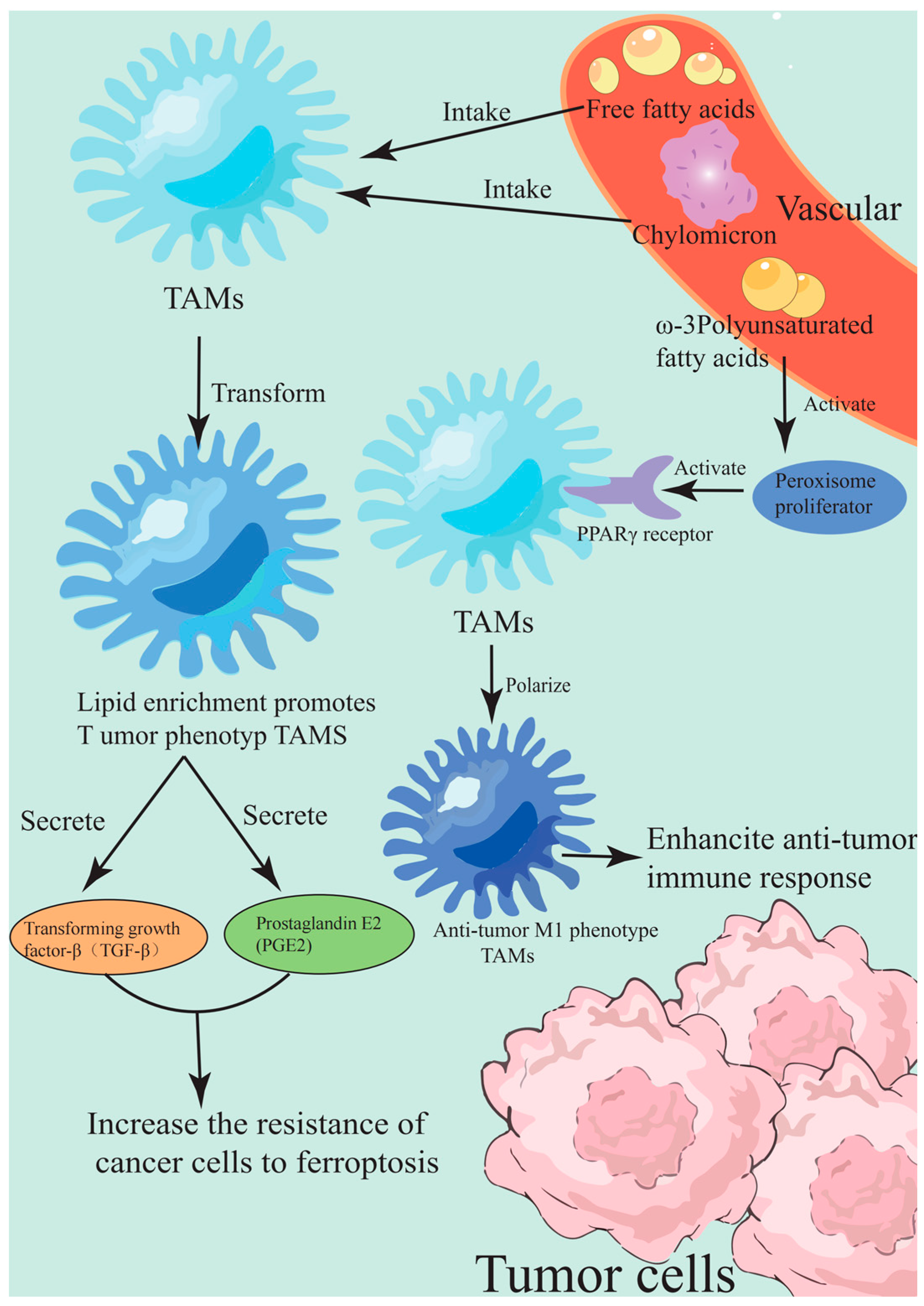
3.3. Remodeling Strategies of TME
3.3.1. Reprogramming of Lipid Metabolism
3.3.2. Oxidative Stress and Immune Regulation
4. Challenges and Solutions
4.1. Improve Bioavailability
4.2. Mechanisms of Drug Resistance
4.3. Clinical Translation
5. Future Prospects
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 4-HNE | 4-Hydroxynonenal |
| AA | Arachidonic acid |
| ACSL3 | Long-chain acyl-CoA synthetase 3 |
| ACSL4(1) | Long-chain acyl-CoA synthetase 4 |
| AdA | Adrenal acid |
| ATGL | Adipose triglyceride lipase |
| ASMP(7) | Actin-derived small molecular peptide |
| BRD4 | Bromodomain-containing protein 4 |
| CFAs | Cyclic fatty acids |
| CoQ | Coenzyme Q |
| CRISPR-Cas9 | Clustered Regularly Interspaced Short Palindromic Repeats-Cas9 |
| CSCs) | Cancer stem cells |
| DAMPs | Damage-associated molecular patterns |
| DHA | Docosahexaenoic acid |
| DUBs | Deubiquitinizing enzymes |
| eIF | Eukaryotic translation initiation factor |
| EMT | Epithelial-mesenchymal transition |
| EPR | Enhanced permeability and retention |
| ETC | Electron transport chain |
| EPA | Eicosapentaenoic acid |
| FG-CDs@Cu | Functionalized carbon dots loaded with copper ions |
| FSP1 | Ferroptosis suppressor protein 1 |
| FTH1 | Ferritin heavy chain |
| FXR | Farnesoid X receptor |
| GPX4 | Glutathione peroxidase 4 |
| GSH | glutathione |
| HDAC | Histone deacetylase |
| HMGB1 | High mobility group box 1 |
| iFSP1 | Inhibitor of FSP1 |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| LIP | Labile iron pool |
| LPO | Lipid peroxides |
| MAPK | Mitogen-activated protein kinase |
| MALT1 | Mucosa-associated lymphoid tissue lymphoma translocation protein 1 |
| MDA | Malondialdehyde |
| mtROS | Mitochondrial reactive oxygen species |
| NPC | Nasopharyngeal carcinoma |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| NSCLC | Non-small cell lung cancer |
| PD-1 | Programmed Cell Death Protein 1 |
| PD-L1 | Programmed Death-Ligand 1 |
| PDO | Patient-derived organoids |
| PGE2 | Prostaglandin E2 |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PSCs | Pancreatic stellate cells |
| PUFAs | Polyunsaturated fatty acids |
| RCD | Regulatory cell death |
| ROS | Reactive oxygen species |
| SAR | Structure-activity relationship |
| SCD1 | Stearoyl-CoA desaturase 1 |
| SDH | Succinate dehydrogenase |
| Sec | Selenocysteine |
| SPs | Sulfated polysaccharides |
| STING | Stimulator of interferon genes |
| TAMs | Tumor-associated macrophages |
| TfR1 | Transferrin receptor 1 |
| TGF-β | Transforming growth factor-β |
| TME | Tumor microenvironment |
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Yue, H.; Yuan, L.; Zhang, W.; Zhang, S.; Wei, W.; Ma, G. Macrophage responses to the physical burden of cell-sized particles. J. Mater. Chem. B 2018, 6, 393–400. [Google Scholar] [CrossRef]
- Xue, D.; Zhou, C.; Shi, Y.; Lu, H.; Xu, R.; He, X. Nuclear transcription factor Nrf2 suppresses prostate cancer cells growth and migration through upregulating ferroportin. Oncotarget 2016, 7, 78804–78812. [Google Scholar] [CrossRef]
- Yuan, H.; Pratte, J.; Giardina, C. Ferroptosis and its potential as a therapeutic target. Biochem. Pharmacol. 2021, 186, 114486. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, Y.; Zhao, X.; Shao, L.; Liu, G.; Sun, C.; Xu, R.; Zhang, Z. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain. Behav. Immun. 2021, 93, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, Z.; Pan, K.; Li, J.; Chen, Q. The function and mechanism of ferroptosis in cancer. Apoptosis 2020, 25, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Tian, R.L.; Wang, T.X.; Huang, Z.X.; Yang, Z.; Guan, K.L.; Xiong, Y.; Wang, P.; Ye, D. Temsirolimus inhibits FSP1 enzyme activity to induce ferroptosis and restrain liver cancer progression. J. Mol. Cell. Biol. 2025, 16, mjae036. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Zeng, K.; Li, W.; Wang, Y.; Zhang, Z.; Zhang, L.; Zhang, W.; Xing, Y.; Zhou, C. Inhibition of CDK1 Overcomes Oxaliplatin Resistance by Regulating ACSL4-mediated Ferroptosis in Colorectal Cancer. Adv. Sci. 2023, 10, e2301088. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Wang, W.; Wang, W.; Kryczek, I.; Li, X.; Bian, Y.; Sell, A.; Wei, S.; Grove, S.; Johnson, J.K.; et al. CD8+ T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell 2022, 40, 365–378.e6. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, S. Ferroptosis Signaling Pathways: Alzheimer’s Disease. Horm. Metab. Res. 2023, 55, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Simmons, G., Jr.; Vale, G.; Deng, Y.; Kim, J.; Kim, H.; Zhang, R.; McDonald, J.G.; Ye, J.; Stockwell, E.B.; et al. FAF1 blocks ferroptosis by inhibiting peroxidation of polyunsaturated fatty acids. Proc. Natl. Acad. Sci. USA 2022, 119, e2107189119. [Google Scholar] [CrossRef]
- Song, X.; Long, D. Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front. Neurosci. 2020, 14, 267. [Google Scholar] [CrossRef]
- Zhang, W.; Wen, W.; Tan, R.; Zhang, M.; Zhong, T.; Wang, J.; Chen, H.; Fang, X. Ferroptosis: Potential therapeutic targets and prognostic predictions for acute myeloid leukemia (Review). Oncol. Lett. 2024, 28, 574. [Google Scholar] [CrossRef]
- Hanke, N.; Rami, A. Inhibition of autophagy rescues HT22 hippocampal neurons from erastin-induced ferroptosis. Neural Regen. Res. 2023, 18, 1548. [Google Scholar] [CrossRef]
- Zhu, J.; Berisa, M.; Schwörer, S.; Qin, W.; Cross, J.R.; Thompson, C.B. Transsulfuration Activity Can Support Cell Growth upon Extracellular Cysteine Limitation. Cell Metab. 2019, 30, 865–876.e5. [Google Scholar] [CrossRef]
- Kose, T.; Sharp, P.A.; Latunde-Dada, G.O. Upregulation of Nrf2 Signalling and the Inhibition of Erastin-Induced Ferroptosis by Ferulic Acid in MIN6 Cells. Int. J. Mol. Sci. 2022, 23, 15886. [Google Scholar] [CrossRef]
- Ghosal, J.; Sinchana, V.K.; Chakrabarty, S. Ferroptosis meets microRNAs: A new frontier in anti-cancer therapy. Free Radic. Biol. Med. 2025, 226, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Cheff, D.M.; Huang, C.; Scholzen, K.C.; Gencheva, R.; Ronzetti, M.H.; Cheng, Q.; Hall, M.D.; Arnér, E.S.J. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol. 2023, 62, 102703. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.; Son, S.; Min, S.; Hong, H.; Kim, C.; An, J.; Kim, J.S.; Kang, H. Stimuli-responsive ferroptosis for cancer therapy. Chem. Soc. Rev. 2023, 52, 3955–3972. [Google Scholar] [CrossRef]
- Adzavon, K.P.; Zhao, W.; He, X.; Sheng, W. Ferroptosis resistance in cancer cells: Nanoparticles for combination therapy as a solution. Front. Pharmacol. 2024, 15, 1416382. [Google Scholar] [CrossRef]
- Ren, Y. Ferroptosis and EMT: Key targets for combating cancer progression and therapy resistance. Cell Mol. Life Sci. 2023, 80, 263. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, G.; Chen, X. Mechanism of ferroptosis resistance in cancer cells. Cancer Drug Resist. 2024, 7, 47. [Google Scholar] [CrossRef]
- Xu, S. Insights into emerging mechanisms of ferroptosis: New regulators for cancer therapeutics. Cell Biol. Toxicol. 2025, 41, 63. [Google Scholar] [CrossRef]
- Fan, R.; Deng, A.; Lin, R.; Zhang, S.; Cheng, C.; Zhuang, J.; Hai, Y.; Zhao, M.; Yang, L.; Wei, G. A platinum(IV)–artesunate complex triggers ferroptosis by boosting cytoplasmic and mitochondrial lipid peroxidation to enhance tumor immunotherapy. MedComm 2024, 5, e570. [Google Scholar] [CrossRef]
- Pontel, L.B.; Bueno-Costa, A.; Morellato, A.E.; Carvalho Santos, J.; Roué, G.; Esteller, M. Acute lymphoblastic leukemia necessitates GSH-dependent ferroptosis defenses to overcome FSP1-epigenetic silencing. Redox Biol. 2022, 55, 102408. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, S.; Hao, H.; Deng, X.; Fu, J.; Guo, Y.; Yuan, Y.; Jiao, Y.; Han, S. Targeting Ferroptosis in Cancer by Natural Products: An Updated Review. Am. J. Chin. Med. 2023, 51, 547–574. [Google Scholar] [CrossRef]
- Kaftan, G.; Erdoğan, M.A.; El-Shazly, M.; Lu, M.-C.; Shih, S.-P.; Lin, H.-Y.; Saso, L.; Armagan, G. Heteronemin Promotes Iron-Dependent Cell Death in Pancreatic Cancer. Naunyn. Schmiedebergs. Arch. Pharmacol. 2024, 397, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-T.; Bow, Y.-D.; Fu, P.-J.; Li, C.-Y.; Wu, C.-Y.; Chang, Y.-H.; Teng, Y.-N.; Li, R.-N.; Lu, M.-C.; Liu, Y.-C.; et al. A Marine Terpenoid, Heteronemin, Induces Both the Apoptosis and Ferroptosis of Hepatocellular Carcinoma Cells and Involves the ROS and MAPK Pathways. Oxid. Med. Cell. Longev. 2021, 2021, 7689045. [Google Scholar] [CrossRef]
- Zuo, H.-L.; Huang, H.-Y.; Lin, Y.-C.-D.; Liu, K.-M.; Lin, T.-S.; Wang, Y.-B.; Huang, H.-D. Effects of Natural Products on Enzymes Involved in Ferroptosis: Regulation and Implications. Molecules 2023, 28, 7929. [Google Scholar] [CrossRef]
- Yang, J.; Liu, J.; Kuang, W.; Lin, Y.; Zhong, S.; Kraithong, S.; Zhang, X.; Wong, I.N.; Huang, R. Structural characterization and ferroptosis-related immunomodulatory of a novel exopolysaccharide isolated from marine fungus Aspergillus medius. Int. J. Biol. Macromol. 2024, 265, 130703. [Google Scholar] [CrossRef] [PubMed]
- Du, H.-F.; Wu, J.-W.; Zhu, Y.-S.; Hua, Z.-H.; Jin, S.-Z.; Ji, J.-C.; Wang, C.-S.; Qian, G.-Y.; Jin, X.-D.; Ding, H.-M. Fucoxanthin Induces Ferroptosis in Cancer Cells via Downregulation of the Nrf2/HO−1/GPX4 Pathway. Molecules 2024, 29, 2832. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Zhao, R.; Ostermann, L.B.; Li, Z.; Tcheng, M.; Yazdani, S.J.; Moayed, A.; Pryor, M.L.; Slngh, S.; Baran, N.; et al. Mitochondrial regulation of GPX4 inhibition–mediated ferroptosis in acute myeloid leukemia. Leukemia 2024, 38, 729–740. [Google Scholar] [CrossRef]
- Wang, H.; Wang, C.; Li, B.; Zheng, C.; Liu, G.; Liu, Z.; Zhang, L.; Xu, P. Discovery of ML210-Based glutathione peroxidase 4 (GPX4) degrader inducing ferroptosis of human cancer cells. Eur. J. Med. Chem. 2023, 254, 115343. [Google Scholar] [CrossRef]
- Costa, I.; Barbosa, D.J.; Benfeito, S.; Silva, V.; Chavarria, D.; Borges, F.; Remião, F.; Silva, R. Molecular mechanisms of ferroptosis and their involvement in brain diseases. Pharmacol. Ther. 2023, 244, 108373. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, G.; Tian, M.; Yin, Y.; Bao, Y.; Guan, X.; Ding, C.; Yu, S. Brucella rough RB51 infection activates P53-Slc7a11-Gpx4/GSH pathway to induce ferroptosis to attenuate the intracellular survival on macrophages. Vet. Microbiol. 2024, 298, 110224. [Google Scholar] [CrossRef]
- Lei, M.; Zhang, Y.-L.; Huang, F.-Y.; Chen, H.-Y.; Chen, M.-H.; Wu, R.-H.; Dai, S.-Z.; He, G.-S.; Tan, G.-H.; Zheng, W.-P. Gankyrin inhibits ferroptosis through the p53/SLC7A11/GPX4 axis in triple-negative breast cancer cells. Sci. Rep. 2023, 13, 21916. [Google Scholar] [CrossRef]
- Yang, Y.; Ma, Y.; Li, Q.; Ling, Y.; Zhou, Y.; Chu, K.; Xue, L.; Tao, S. STAT6 inhibits ferroptosis and alleviates acute lung injury via regulating P53/SLC7A11 pathway. Cell Death Dis. 2022, 13, 530. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Gao, X.; Zou, L.; Lei, M.; Feng, J.; Hu, Z. Bavachin Induces Ferroptosis through the STAT3/P53/SLC7A11 Axis in Osteosarcoma Cells. Oxid. Med. Cell. Longev. 2021, 2021, 1783485. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Yamada, Y.; Taguchi, S.; Kojima, R.; Masumoto, H.; Kimura, S.; Niijima, T.; Toyama, T.; Kise, R.; Sato, E.; et al. Conjugated fatty acids drive ferroptosis through chaperone-mediated autophagic degradation of GPX4 by targeting mitochondria. Cell Death Dis. 2024, 15, 884. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Wang, C.; Liu, G.; Bi, C.; Wang, X.; Zhou, Q.; Jin, H. SLC7A11/xCT in cancer: Biological functions and therapeutic implications. Am. J. Cancer Res. 2020, 10, 3106–3126. [Google Scholar]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef]
- Trombetti, S.; Iaccarino, N.; Riccio, P.; Sessa, R.; Catapano, R.; Salvatore, M.; Luka, S.; De Nicola, S.; Izzo, P.; Roperto, S.; et al. Over-Expressed GATA-1S, the Short Isoform of the Hematopoietic Transcriptional Factor GATA-1, Inhibits Ferroptosis in K562 Myeloid Leukemia Cells by Preventing Lipid Peroxidation. Antioxidants 2023, 12, 537. [Google Scholar] [CrossRef]
- Xie, Y.; Kang, R.; Klionsky, D.J.; Tang, D. GPX4 in cell death, autophagy, and disease. Autophagy 2023, 19, 2621–2638. [Google Scholar] [CrossRef]
- Bao, Y.; Li, G.; Li, S.; Zhang, H.; Wu, X.; Yan, R.; Wang, Z.; Guo, C.; Jin, Y. Multifunctional Tumor-Targeting Carbon Dots for Tumor Microenvironment Activated Ferroptosis and Immunotherapy in Cancer Treatment. ACS Appl. Mater. Interfaces 2023, 15, 56834–56845. [Google Scholar] [CrossRef]
- Hao, W.; Sun, N.; Fan, Y.; Chen, M.; Liu, Q.; Yang, M.; Yang, Y.; Gao, C. Targeted Ferroptosis-Immunotherapy Synergy: Enhanced Antiglioma Efficacy with Hybrid Nanovesicles Comprising NK Cell-Derived Exosomes and RSL3-Loaded Liposomes. ACS Appl. Mater. Interfaces 2024, 16, 28193–28208. [Google Scholar] [CrossRef]
- Wiernicki, B.; Maschalidi, S.; Pinney, J.; Adjemian, S.; Vanden Berghe, T.; Ravichandran, K.S.; Vandenabeele, P. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat. Commun. 2022, 13, 3676. [Google Scholar] [CrossRef]
- Yang, M.; Liu, K.; Chen, P.; Zhu, H.; Wang, J.; Huang, J. Bromodomain-containing protein 4 (BRD4) as an epigenetic regulator of fatty acid metabolism genes and ferroptosis. Cell Death Dis. 2022, 13, 912. [Google Scholar] [CrossRef] [PubMed]
- Tschuck, J.; Theilacker, L.; Rothenaigner, I.; Weiß, S.A.I.; Akdogan, B.; Lam, V.T.; Müller, C.; Graf, R.; Brandner, S.; Pütz, C.; et al. Farnesoid X receptor activation by bile acids suppresses lipid peroxidation and ferroptosis. Nat. Commun. 2023, 14, 6908. [Google Scholar] [CrossRef]
- Bhat, K.P.; Vijay, J.; Vilas, C.K.; Asundi, J.; Zou, J.; Lau, T.; Cai, X.; Ahmed, M.; Kabza, M.; Weng, J.; et al. CRISPR activation screens identify the SWI/SNF ATPases as suppressors of ferroptosis. Cell Rep. 2024, 43, 114345. [Google Scholar] [CrossRef]
- Ma, T.; Du, J.; Zhang, Y.; Wang, Y.; Wang, B.; Zhang, T. GPX4-independent ferroptosis—A new strategy in disease’s therapy. Cell Death Discov. 2022, 8, 434. [Google Scholar] [CrossRef]
- Liu, S.; Wang, J. Recent Progress of Glutathione Peroxidase 4 Inhibitors in Cancer Therapy. Mini-Rev. Med. Chem. 2025, 25, 42–57. [Google Scholar] [CrossRef]
- Zheng, C.; Wang, C.; Sun, D.; Wang, H.; Li, B.; Liu, G.; Liu, Z.; Zhang, L.; Xu, P. Structure-activity relationship study of RSL3-based GPX4 degraders and its potential noncovalent optimization. Eur. J. Med. Chem. 2023, 255, 115393. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Min, D.W.; Kim, D.; Kim, J.; Kim, M.J.; Lim, H.; Lee, J.-Y. GPX4 overexpressed non-small cell lung cancer cells are sensitive to RSL3-induced ferroptosis. Sci. Rep. 2023, 13, 8872. [Google Scholar] [CrossRef]
- Kumada, H.; Itoh, M.; Tohda, S. Effect of Ferroptosis Inducers and Inhibitors on Cell Proliferation in Acute Leukemia. Anticancer Res. 2024, 44, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Liu, J.; Lu, Z.; Li, J.; Zhang, S.; Li, Q.; Geng, F.; Pan, Y. Role of ferroptosis in Porphyromonas gingivalis- induced impairment of epithelial junction. J. Oral Microbiol. 2024, 16, 2334578. [Google Scholar] [CrossRef]
- Tokunaga, F. BACH to the ferroptosis. J. Biochem. 2024, 176, 423–426. [Google Scholar] [CrossRef]
- Dong, J.; Li, M.; Peng, R.; Zhang, Y.; Qiao, Z.; Sun, N. ACACA reduces lipid accumulation through dual regulation of lipid metabolism and mitochondrial function via AMPK- PPARα- CPT1A axis. J. Transl. Med. 2024, 22, 196. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wu, L.; Xu, S. An overview of GPX4-targeting TPDs for cancer therapy. Bioorg. Med. Chem. 2025, 118, 118046. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef]
- Chen, G.Y.; O’Leary, B.R.; Du, J.; Carroll, R.S.; Steers, G.J.; Buettner, G.R.; Cullen, J.J. Pharmacologic Ascorbate Radiosensitizes Pancreatic Cancer but Radioprotects Normal Tissue: The Role of Oxidative Stress-Induced Lipid Peroxidation. Antioxidants 2024, 13, 361. [Google Scholar] [CrossRef]
- Cheng, K.; Yang, G.; Huang, M.; Huang, Y.; Wang, C. Exogenous 1,25(OH)2D3/VD3 counteracts RSL3-Induced ferroptosis by enhancing antioxidant capacity and regulating iron ion transport: Using zebrafish as a model. Chem. Biol. Interact. 2024, 387, 110828. [Google Scholar] [CrossRef] [PubMed]
- Weaver, K.; Skouta, R. The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities. Biomedicines 2022, 10, 891. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Yang, F.; Li, Q. Post-Translational Modification of GPX4 is a Promising Target for Treating Ferroptosis-Related Diseases. Front. Mol. Biosci. 2022, 9, 901565. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Roh, J.-L. Targeting GPX4 in human cancer: Implications of ferroptosis induction for tackling cancer resilience. Cancer Lett. 2023, 559, 216119. [Google Scholar] [CrossRef]
- Lin, J.; Deng, L.; Qi, A.; Jiang, H.; Xu, D.; Zheng, Y.; Zhang, Z.; Guo, X.; Hu, B.; Li, P. Catalpol alleviates hypoxia ischemia-induced brain damage by inhibiting ferroptosis through the PI3K/NRF2/system Xc-/GPX4 axis in neonatal rats. Eur. J. Pharmacol. 2024, 968, 176406. [Google Scholar] [CrossRef]
- Zhu, L.; Hu, S.; Yan, X.; Zeng, Q.; Zhang, B.; Jiang, L.; Yao, S.Q.; Ge, J. Ugi reaction-assisted assembly of covalent PROTACs against glutathione peroxidase 4. Bioorganic Chem. 2023, 134, 106461. [Google Scholar] [CrossRef]
- Cai, M.; Ma, F.; Hu, C.; Li, H.; Cao, F.; Li, Y.; Dong, J.; Qin, J.-J. Design and synthesis of proteolysis-targeting chimeras (PROTACs) as degraders of glutathione peroxidase 4. Bioorg. Med. Chem. 2023, 90, 117352. [Google Scholar] [CrossRef]
- Zhuo, B.; Qin, C.; Deng, S.; Jiang, H.; Si, S.; Tao, F.; Cai, F.; Meng, Z. The role of ACSL4 in stroke: Mechanisms and potential therapeutic target. Mol. Cell. Biochem. 2025, 480, 2223–2246. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Zhang, X.; Yang, M.; Dong, X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 2019, 31, 1904197. [Google Scholar] [CrossRef]
- Miao, Z.; Tian, W.; Ye, Y.; Gu, W.; Bao, Z.; Xu, L.; Sun, G.; Li, C.; Tu, Y.; Chao, H.; et al. Hsp90 induces Acsl4-dependent glioma ferroptosis via dephosphorylating Ser637 at Drp1. Cell Death Dis. 2022, 13, 548. [Google Scholar] [CrossRef]
- Chen, P.; Wang, D.; Xiao, T.; Gu, W.; Yang, H.; Yang, M.; Wang, H. ACSL4 promotes ferroptosis and M1 macrophage polarization to regulate the tumorigenesis of nasopharyngeal carcinoma. Int. Immunopharmacol. 2023, 122, 110629. [Google Scholar] [CrossRef] [PubMed]
- Rousselle, A.; Lodka, D.; Sonnemann, J.; Kling, L.; Kettritz, R.; Schreiber, A. Endothelial but not systemic ferroptosis inhibition protects from antineutrophil cytoplasmic antibody–induced crescentic glomerulonephritis. Kidney Int. 2025, 107, 1037–1050. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, C.; Wu, B.; Li, C.; Lin, J.; Huang, P. Thermoresponsive Ozone-Enriched Spray Gel for Postsurgical Treatment of Hepatocellular Carcinoma. ACS Nano 2023, 17, 3518–3527. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Yan, Y.; Long, T.; Xu, J.; Chang, C.; Kang, M.; Wang, X.; Chen, Y.; Qiu, J. Ferroptosis: A potential therapeutic target in cardio-cerebrovascular diseases. Mol. Cell. Biochem. 2025. [Google Scholar] [CrossRef]
- Gao, X.; Li, Y.; Shen, J.; Huang, Y.; Wang, Y.; Niu, X. LC-MS untargeted metabolomics reveals metabolic disturbance and ferroptosis in MWCNTs-induced hepatotoxicity of Cyprinus carpio. Aquat. Toxicol. 2024, 275, 107078. [Google Scholar] [CrossRef]
- Zuo, Y.-B.; Zhang, Y.-F.; Zhang, R.; Tian, J.-W.; Lv, X.-B.; Li, R.; Li, S.-P.; Cheng, M.-D.; Shan, J.; Zhao, Z.; et al. Ferroptosis in Cancer Progression: Role of Noncoding RNAs. Int. J. Biol. Sci. 2022, 18, 1829–1843. [Google Scholar] [CrossRef]
- Jia, C.; Gou, Y.; Gao, Y.; Pei, X.; Jin, X.; Li, B.; Zhang, Z.; He, Y.; Ji, E.-S.; Zhao, Y. Rosmarinic acid liposomes suppress ferroptosis in ischemic brain via inhibition of TfR1 in BMECs. Phytomedicine 2024, 132, 155835. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423.e7. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-X.; Zhang, Z.-S.; Qin, J.; Zhang, M.-Z.; Cao, J.-L.; Li, Y.-Y.; Wang, M.-Q.; Hou, L.-L.; Fang, D.; Xie, S.-Q. Aucubin enhances the antitumor activity of cisplatin through the inhibition of PD-L1 expression in hepatocellular carcinoma. Phytomedicine 2023, 112, 154715. [Google Scholar] [CrossRef]
- Zhao, P.; Yuan, Q.; Liang, C.; Ma, Y.; Zhu, X.; Hao, X.; Li, X.; Shi, J.; Fu, Q.; Fan, H.; et al. GPX4 degradation contributes to fluoride-induced neuronal ferroptosis and cognitive impairment via mtROS-chaperone-mediated autophagy. Sci. Total Environ. 2024, 927, 172069. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Qi, F.; Qie, H.; Du, S.; Li, L.; Zhang, Y.; Xu, K.; Li, D.; Xu, Y. Oleic Acid Inhibits SDC4 and Promotes Ferroptosis in Lung Cancer Through GPX4/ACSL4. Clin. Respir. J. 2024, 18, e70014. [Google Scholar] [CrossRef]
- Dong, Y.; Li, Y.; Tang, W.; Chen, Q.; Kong, C. Increased Trophoblast Cell Ferroptosis via HMGB1/ACSL4 Pathway Is Associated with Spontaneous Abortion. Reprod. Sci. 2025, 32, 1713–1722. [Google Scholar] [CrossRef]
- Turrini, E.; Maffei, F.; Fimognari, C. Effect of the Marine Polyketide Plocabulin on Tumor Progression. Mar Drugs. 2022, 21, 38. [Google Scholar] [CrossRef]
- Ou, Z.; Deng, Y.; Wu, Y.; Wang, Y.; Zhao, Y.; Liu, C.; Wang, Z.; Liu, M.; Hu, X.; Fang, L.; et al. Tongqiao Huoxue Decoction inhibits ferroptosis by facilitating ACSL4 ubiquitination degradation for neuroprotection against cerebral ischemia-reperfusion injury. Phytomedicine 2024, 130, 155701. [Google Scholar] [CrossRef]
- Li, W.; Yu, J.; Wang, J.; Fan, X.; Xu, X.; Wang, H.; Xiong, Y.; Li, X.; Zhang, X.; Zhang, Q.; et al. How does ferrocene correlate with ferroptosis? Multiple approaches to explore ferrocene-appended GPX4 inhibitors as anticancer agents. Chem. Sci. 2024, 15, 10477–10490. [Google Scholar] [CrossRef]
- Lomartire, S.; Gonçalves, A.M.M. Marine Macroalgae Polyphenols as Potential Neuroprotective Antioxidants in Neurodegenerative Diseases. Mar. Drugs 2023, 21, 261. [Google Scholar] [CrossRef]
- Sathishkumar, K.; Sathuvan, M. Brown algal bioactive molecules: A new frontier in oral cancer treatment. Nat. Prod. Res. 2025, 39, 3005–3007. [Google Scholar] [CrossRef]
- Scarpellini, C.; Klejborowska, G.; Lanthier, C.; Toye, A.; Musiałek, K.; Van San, E.; Walravens, M.; Berg, M.; Hassannia, B.; Van Der Veken, P.; et al. Oxazole-Based Ferroptosis Inhibitors with Promising Properties to Treat Central Nervous System Diseases. J. Med. Chem. 2025, 68, 4908–4928. [Google Scholar] [CrossRef]
- Han, X.; Choi, S.-I.; Men, X.; Lee, S.-J.; Oh, G.; Jin, H.; Oh, H.-J.; Kim, E.; Kim, J.; Lee, B.-Y.; et al. Radical Scavenging-Linked Anti-Obesity Effect of Standardized Ecklonia stolonifera Extract on 3T3-L1 Preadipocytes and High-Fat Diet-Fed ICR Mice. J. Med. Food 2023, 26, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Ke, K.; Li, L.; Lu, C.; Zhu, Q.; Wang, Y.; Mou, Y.; Wang, H.; Jin, W. The crosstalk effect between ferrous and other ions metabolism in ferroptosis for therapy of cancer. Front. Oncol. 2022, 12, 916082. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Zhao, L.; Yoo, S.; Lin, Z.; Zhang, Y.; Yang, W.; Piao, J. Emodin induces ferroptosis in colorectal cancer through NCOA4-mediated ferritinophagy and NF-κb pathway inactivation. Apoptosis 2024, 29, 1810–1823. [Google Scholar] [CrossRef]
- Yi, X.; Wang, Q.; Zhang, M.; Shu, Q.; Zhu, J. Ferroptosis: A novel therapeutic target of natural products against doxorubicin-induced cardiotoxicity. Biomed. Pharmacother. 2024, 178, 117217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, J. Induction of ferroptosis by natural phenols: A promising strategy for cancer therapy. Phytother. Res. 2024, 38, 2041–2076. [Google Scholar] [CrossRef]
- Wang, B.; Wang, J.; Liu, C.; Li, C.; Meng, T.; Chen, J.; Liu, Q.; He, W.; Liu, Z.; Zhou, Y. Ferroptosis: Latest evidence and perspectives on plant-derived natural active compounds mitigating doxorubicin-induced cardiotoxicity. J. Appl. Toxicol. 2025, 45, 135–158. [Google Scholar] [CrossRef]
- Tschuck, J.; Tonnus, W.; Gavali, S.; Kolak, A.; Mallais, M.; Maremonti, F.; Sato, M.; Rothenaigner, I.; Friedmann Angeli, J.P.; Pratt, D.A.; et al. Seratrodast inhibits ferroptosis by suppressing lipid peroxidation. Cell Death Dis. 2024, 15, 853. [Google Scholar] [CrossRef]
- Lv, Y.; Liang, C.; Sun, Q.; Zhu, J.; Xu, H.; Li, X.; Li, Y.; Wang, Q.; Yuan, H.; Chu, B.; et al. Structural insights into FSP1 catalysis and ferroptosis inhibition. Nat. Commun. 2023, 14, 5933. [Google Scholar] [CrossRef]
- Lee, J.; Roh, J.-L. Lipid metabolism in ferroptosis: Unraveling key mechanisms and therapeutic potential in cancer. Biochim. Biophys. Acta BBA—Rev. Cancer 2025, 1880, 189258. [Google Scholar] [CrossRef]
- Vermonden, P.; Martin, M.; Glowacka, K.; Neefs, I.; Ecker, J.; Höring, M.; Liebisch, G.; Debier, C.; Feron, O.; Larondelle, Y. Phospholipase PLA2G7 is complementary to GPX4 in mitigating punicic-acid-induced ferroptosis in prostate cancer cells. iScience 2024, 27, 109774. [Google Scholar] [CrossRef]
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 2022, 82, 2215–2227. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, X.; Chen, Y.; Tang, Q.; He, C.; Ding, X.; Hu, J.; Cai, Z.; Li, X.; Qiao, H.; et al. Targeting PAX8 sensitizes ovarian cancer cells to ferroptosis by inhibiting glutathione synthesis. Apoptosis 2024, 29, 1499–1514. [Google Scholar] [CrossRef]
- Wang, J.; Liao, L.; Miao, B.; Yang, B.; Li, B.; Ma, X.; Fitz, A.; Wu, S.; He, J.; Zhang, Q.; et al. Deciphering the role of the MALT1–RC3H1 axis in regulating GPX4 protein stability. Proc. Natl. Acad. Sci. USA 2025, 122, e2419625121. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, Q.; Zhao, M.; Wang, X.; Zhang, Y.; Gan, B.; Zhang, P. The deubiquitinase ZRANB1 is an E3 ubiquitin ligase for SLC7A11 and regulates ferroptotic resistance. J. Cell Biol. 2023, 222, e202212072. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Dimou, J.; Watt, M.J. Lipid droplets and ferroptosis as new players in brain cancer glioblastoma progression and therapeutic resistance. Front. Oncol. 2022, 12, 1085034. [Google Scholar] [CrossRef] [PubMed]
- Petan, T. Lipid Droplets in Cancer. In Organelles in Disease; Pedersen, S.H.F., Barber, D.L., Eds.; Reviews of Physiology, Biochemistry and Pharmacology; Springer International Publishing: Cham, Switzerland, 2020; Volume 185, pp. 53–86. ISBN 978-3-031-22594-9. [Google Scholar]
- Yorek, M.; Jiang, X.; Liu, S.; Hao, J.; Yu, J.; Avellino, A.; Liu, Z.; Curry, M.; Keen, H.; Shao, J.; et al. FABP4-mediated lipid accumulation and lipolysis in tumor-associated macrophages promote breast cancer metastasis. eLife 2024, 13, RP101221. [Google Scholar] [CrossRef]
- Yang, X.; Deng, B.; Zhao, W.; Guo, Y.; Wan, Y.; Wu, Z.; Su, S.; Gu, J.; Hu, X.; Feng, W.; et al. FABP5+ lipid-loaded macrophages process tumour-derived unsaturated fatty acid signal to suppress T-cell antitumour immunity. J. Hepatol. 2025, 82, 676–689. [Google Scholar] [CrossRef]
- Lorito, N.; Subbiani, A.; Smiriglia, A.; Bacci, M.; Bonechi, F.; Tronci, L.; Romano, E.; Corrado, A.; Longo, D.L.; Iozzo, M.; et al. FADS1/2 control lipid metabolism and ferroptosis susceptibility in triple-negative breast cancer. EMBO Mol. Med. 2024, 16, 1533–1559. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, M.; Cao, J.; Wang, F.; Han, J.R.; Wu, T.W.; Li, L.; Yu, J.; Fan, Y.; Xie, G.; et al. ACSL4 and polyunsaturated lipids support metastatic extravasation and colonization. Cell 2025, 188, 412–429.e27. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, P.; Xu, J.; Lv, G.; Li, Y. Lipid metabolism in tumor microenvironment: Novel therapeutic targets. Cancer Cell Int. 2022, 22, 224. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Zhu, J.; Wang, Y.; Chen, W.; Fang, S.; Mao, W.; Xu, Z.; Yang, Y.; Weng, Q.; Zhao, Z.; et al. Targeted xCT-mediated Ferroptosis and Protumoral Polarization of Macrophages Is Effective against HCC and Enhances the Efficacy of the Anti-PD-1/L1 Response. Adv. Sci. 2023, 10, 3973. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Han, X.; Egeblad, M. Isolation of mouse mammary carcinoma-derived macrophages and cancer cells for co-culture assays. STAR Protoc. 2022, 3, 101833. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Cai, D.; Gou, S.; Bai, Y.; Lei, H.; Li, Y.; Chen, Y.; Zhao, Y.; Shen, J.; Wu, X.; et al. The dynamic role of ferroptosis in cancer immunoediting: Implications for immunotherapy. Pharmacol. Res. 2025, 214, 107674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, T.; Zhang, X.; Xu, Y.; Ming, J.; Wang, X.; Liu, Z.; Li, J.; Su, X. Synchronously Delivering Melittin and Evoking Ferroptosis via Tumor Microenvironment-Triggered Self-Destructive Metal–Organic Frameworks to Boost Cancer Immunotherapy. Adv. Healthc. Mater. 2025, 14, 2500003. [Google Scholar] [CrossRef]
- Rothe, T.; Gruber, F.; Uderhardt, S.; Ipseiz, N.; Rössner, S.; Oskolkova, O.; Blüml, S.; Leitinger, N.; Bicker, W.; Bochkov, V.N.; et al. 12/15-lipoxygenase–mediated enzymatic lipid oxidation regulates DC maturation and function. J. Clin. Investig. 2015, 125, 1944–1954. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Kang, R.; Tang, D. Cell type-specific induction of ferroptosis to boost antitumor immunity. OncoImmunology 2023, 12, 2282252. [Google Scholar] [CrossRef]
- Zheng, Y.; Sun, L.; Guo, J.; Ma, J. The crosstalk between ferroptosis and anti-tumor immunity in the tumor microenvironment: Molecular mechanisms and therapeutic controversy. Cancer Commun. 2023, 43, 1071–1096. [Google Scholar] [CrossRef]
- Xu, X.; Tian, M.; Deng, L.; Jiang, H.; Han, J.; Zhen, C.; Huang, L.; Liu, W. Structural degradation and uptake of resveratrol-encapsulated liposomes using an in vitro digestion combined with Caco-2 cell absorption model. Food Chem. 2023, 403, 133943. [Google Scholar] [CrossRef]
- Nguyen-Huu, A.-M.; Le, N.T.T.; Vo Do, M.H.; Dong Yen, P.N.; Nguyen-Dinh, T.-D.; Nguyen, N.H.; Nguyen, D.H. Development and Characterization of Quercetin-Loaded Polymeric Liposomes with Gelatin–Poly(ethylene glycol)–Folic Acid Coating to Increase Their Long-Circulating and Anticancer Activity. ACS Appl. Bio Mater. 2024, 7, 4454–4470. [Google Scholar] [CrossRef] [PubMed]
- Amiri, H.; Shabanpour, B.; Pourashouri, P.; Kashiri, M. Preparation of functional supplement powder using nanoliposome-containing marine bioactive compounds. J. Food Sci. 2024, 89, 8658–8672. [Google Scholar] [CrossRef] [PubMed]
- Dhanisha, S.S.; Drishya, S.; Guruvayoorappan, C. Encapsulating Naringenin in biomimetic proteolipid vesicles abrogates cancer metastasis by targeting apoptotic signaling axis. Food Chem. 2024, 434, 137445. [Google Scholar] [CrossRef]
- Ettoumi, F.; Zhang, R.; Xu, Y.; Li, L.; Huang, H.; Luo, Z. Synthesis and characterization of fucoidan/chitosan-coated nanoliposomes for enhanced stability and oral bioavailability of hydrophilic catechin and hydrophobic juglone. Food Chem. 2023, 423, 136330. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Youssef, M.; Liang, H.; Li, J.; Li, B. Sea Buckthorn Flavonoid Extract Co-Loaded Zein/Gum Arabic Nanoparticles: Evaluating Cellular Absorption in Caco-2 Cells and Antioxidant Activity in Hepg2 Cells. 2024. Available online: https://ssrn.com/abstract=4946419 (accessed on 17 June 2025).
- Menchinskaya, E.S.; Gorbach, V.I.; Pislyagin, E.A.; Gorpenchenko, T.Y.; Pimenova, E.A.; Guzhova, I.V.; Aminin, D.L.; Yermak, I.M. Interaction of Liposomes Containing the Carrageenan/Echinochrome Complex with Human HaCaT Keratinocytes In Vitro. Mar. Drugs 2024, 22, 561. [Google Scholar] [CrossRef]
- Hadian, K. Ferroptosis Suppressor Protein 1 (FSP1) and Coenzyme Q10 Cooperatively Suppress Ferroptosis. Biochemistry 2020, 59, 637–638. [Google Scholar] [CrossRef]
- Hendricks, J.M.; Doubravsky, C.E.; Wehri, E.; Li, Z.; Roberts, M.A.; Deol, K.K.; Lange, M.; Lasheras-Otero, I.; Momper, J.D.; Dixon, S.J.; et al. Identification of structurally diverse FSP1 inhibitors that sensitize cancer cells to ferroptosis. Cell Chem. Biol. 2023, 30, 1090–1103.e7. [Google Scholar] [CrossRef]
- Li, W.; Liang, L.; Liu, S.; Yi, H.; Zhou, Y. FSP1: A key regulator of ferroptosis. Trends Mol. Med. 2023, 29, 753–764. [Google Scholar] [CrossRef]
- Kim, J.W.; Kim, M.-J.; Han, T.-H.; Lee, J.-Y.; Kim, S.; Kim, H.; Oh, K.-J.; Kim, W.K.; Han, B.-S.; Bae, K.-H.; et al. FSP1 confers ferroptosis resistance in KEAP1 mutant non-small cell lung carcinoma in NRF2-dependent and -independent manner. Cell Death Dis. 2023, 14, 567. [Google Scholar] [CrossRef]
- Müller, F.; Lim, J.K.M.; Bebber, C.M.; Seidel, E.; Tishina, S.; Dahlhaus, A.; Stroh, J.; Beck, J.; Yapici, F.I.; Nakayama, K.; et al. Elevated FSP1 protects KRAS-mutated cells from ferroptosis during tumor initiation. Cell Death Differ. 2023, 30, 442–456. [Google Scholar] [CrossRef]
- Chen, J.; Zhan, Q.; Li, L.; Xi, S.; Cai, L.; Liu, R.; Chen, L. Cell-membrane targeting sonodynamic therapy combination with FSP1 inhibition for ferroptosis-boosted immunotherapy. Mater. Today Bio 2025, 30, 101407. [Google Scholar] [CrossRef]
- Peng, Z.; Ding, Y.-N.; Yang, Z.-M.; Li, X.-J.; Zhuang, Z.; Lu, Y.; Tang, Q.-S.; Hang, C.-H.; Li, W. Neuron-targeted liposomal coenzyme Q10 attenuates neuronal ferroptosis after subarachnoid hemorrhage by activating the ferroptosis suppressor protein 1/coenzyme Q10 system. Acta Biomater. 2024, 179, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Chen, C.; Wang, C.; Guo, Y.; Sun, B.; Tian, J.; Yan, J.; Li, D.; Chen, G. Targeting GPX4-mediated ferroptosis protection sensitizes BRCA1-deficient cancer cells to PARP inhibitors. Redox Biol. 2024, 76, 103350. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, J.; Li, L.; Ye, Z.; Liu, J.; Chen, Y.; Hu, B.; Tang, J.; Feng, G.; Li, Z.; et al. Susceptibility of Mitophagy-Deficient Tumors to Ferroptosis Induction by Relieving the Suppression of Lipid Peroxidation. Adv. Sci. 2025, 12, 2412593. [Google Scholar] [CrossRef]
- Shen, Q.; Zhu, X.; Huo, M.; Lin, Y.; Zhang, W.; Yang, M.; Zhang, Y.; Zhang, L.; Gai, Y. A hollow nanozyme-based multifunctional platform enhances sonodynamic–chemodynamic-induced ferroptosis for cancer therapy. RSC Adv. 2025, 15, 9408–9419. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Shi, Z.; Sun, Y.; Ning, H.; Gu, X.; Zhang, L. Prospects for Anti-Tumor Mechanism and Potential Clinical Application Based on Glutathione Peroxidase 4 Mediated Ferroptosis. Int. J. Mol. Sci. 2023, 24, 1607. [Google Scholar] [CrossRef]
- Li, P.; Chu, D.; Ding, G.; Qin, D.; Bu, Y.; Tian, B. IGF2BP3 suppresses ferroptosis in lung adenocarcinoma by m6A-dependent regulation of TFAP2A to transcriptionally activate SLC7A11/GPX4. Mol. Cell. Biochem. 2025, 480, 2361–2375. [Google Scholar] [CrossRef]
- Song, H.; Park, J.Y.; Kim, J.-H.; Shin, T.-S.; Hong, S.A.; Huda, M.N.; Kim, B.J.; Kim, J.G. Establishment of Patient-Derived Gastric Cancer Organoid Model From Tissue Obtained by Endoscopic Biopsies. J. Korean Med. Sci. 2022, 37, e220. [Google Scholar] [CrossRef]
- Li, G.; Lu, X.; Zhang, S.; Zhang, J.; Fu, X.; Zhang, M.; Teng, L.; Sun, F. Multi-Enzyme Cascade-Triggered Nitric Oxide Release Nanoplatform Combined with Chemo Starvation-like Therapy for Multidrug-Resistant Cancers. ACS Appl. Mater. Interfaces 2023, 15, 31285–31299. [Google Scholar] [CrossRef]
- Chen, Y.; Li, X.; Wang, S.; Miao, R.; Zhong, J. Targeting Iron Metabolism and Ferroptosis as Novel Therapeutic Approaches in Cardiovascular Diseases. Nutrients 2023, 15, 591. [Google Scholar] [CrossRef]
- Wu, D.; Wang, Z.; Zhang, Y.; Yang, Y.; Yang, Y.; Zu, G.; Yu, X.; Chen, W.; Qin, Y.; Xu, X.; et al. IL15RA-STAT3-GPX4/ACSL3 signaling leads to ferroptosis resistance in pancreatic cancer. Acta Biochim. Biophys. Sin. 2025, 57, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, R.; Cai, X.; Zhang, L.; Wu, B.; Tan, H.; Zhou, K.; Wang, H.; Liu, Y.; Luo, Y.; et al. Acceptor Elongation Boosted Intersystem Crossing Affords Efficient NIR Type-I and AIE-Active Photosensitizers for Targeting Ferroptosis-Based Cancer Therapy. Adv. Healthc. Mater. 2025, 14, 2404505. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Wang, Q.; Zhang, Q.; Cai, M.; Liu, S.; Zhang, W. Molecular mechanisms of ferroptosis and its antitumor applications in natural products. Acta Biochim. Biophys. Sin. 2023, 55, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Cui, L.; Zhang, Y.; Guo, C.; Deng, L.; Wen, Z.; Lu, Z.; Shi, X.; Xing, H.; Liu, Y.; et al. Screening for Potential Therapeutic Agents for Non-Small Cell Lung Cancer by Targeting Ferroptosis. Front. Mol. Biosci. 2022, 9, 917602. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Chen, X.; Chen, Z.; Ma, Y. Targeting Ferroptosis in Tumors: Novel Marine-Derived Compounds as Regulators of Lipid Peroxidation and GPX4 Signaling. Mar. Drugs 2025, 23, 258. https://doi.org/10.3390/md23060258
Wu Y, Chen X, Chen Z, Ma Y. Targeting Ferroptosis in Tumors: Novel Marine-Derived Compounds as Regulators of Lipid Peroxidation and GPX4 Signaling. Marine Drugs. 2025; 23(6):258. https://doi.org/10.3390/md23060258
Chicago/Turabian StyleWu, Yimao, Xiaoyan Chen, Zichang Chen, and Yunqi Ma. 2025. "Targeting Ferroptosis in Tumors: Novel Marine-Derived Compounds as Regulators of Lipid Peroxidation and GPX4 Signaling" Marine Drugs 23, no. 6: 258. https://doi.org/10.3390/md23060258
APA StyleWu, Y., Chen, X., Chen, Z., & Ma, Y. (2025). Targeting Ferroptosis in Tumors: Novel Marine-Derived Compounds as Regulators of Lipid Peroxidation and GPX4 Signaling. Marine Drugs, 23(6), 258. https://doi.org/10.3390/md23060258