

Cyanobacterial Peptides in Anticancer Therapy: A Comprehensive Review of Mechanisms, Clinical Advances, and Biotechnological Innovation


Abstract
1. Introduction
2. Anticancer Peptides from Cyanobacteria: Classes and Examples
2.1. Cyclic Depsipeptides and Lipopeptides
2.2. Peptide–Polyketide Hybrids
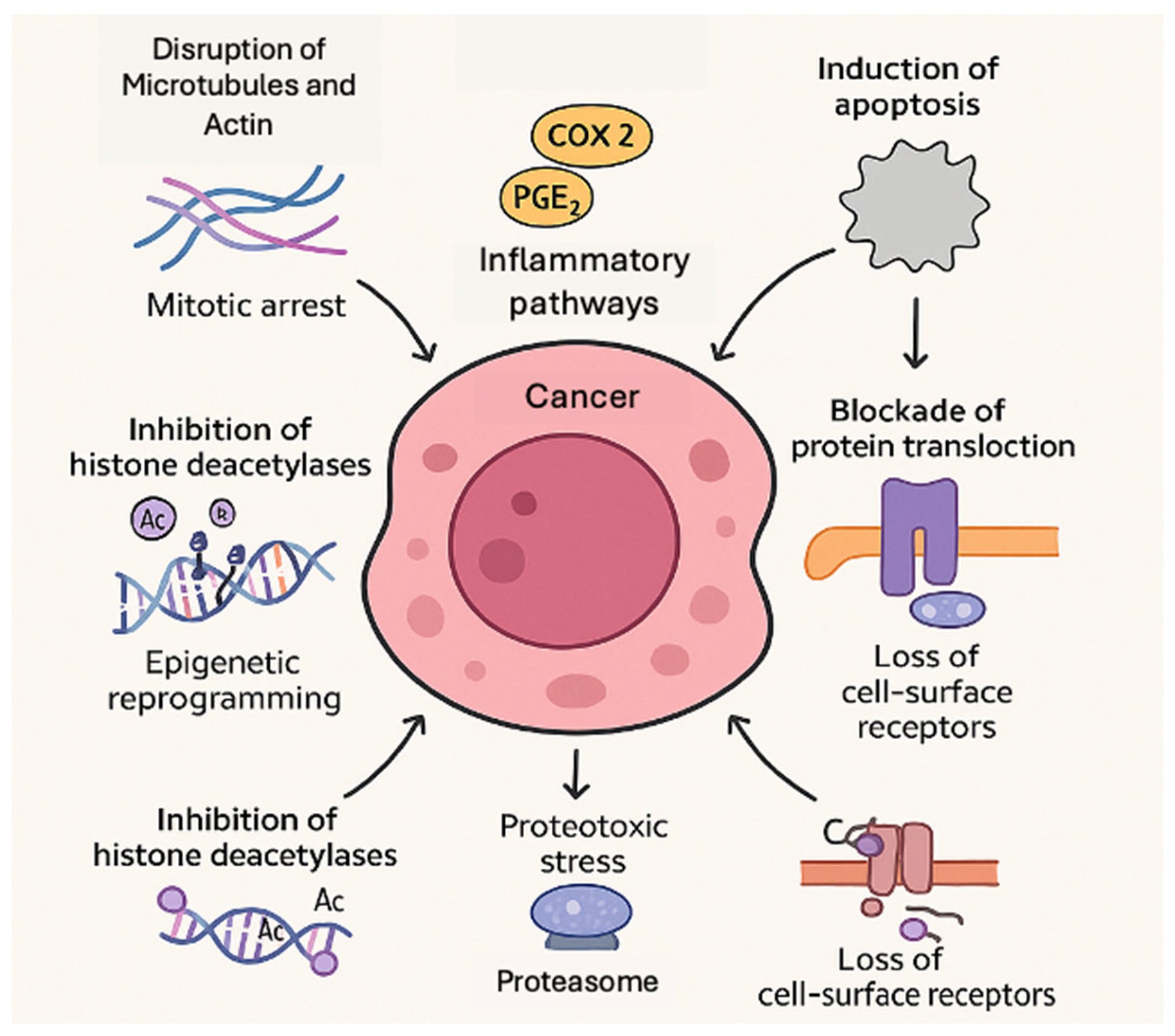
3. Molecular Mechanisms of Action
3.1. Disruption of Microtubules and Actin (Mitotic Arrest)
3.2. Induction of Apoptosis (Intrinsic and Extrinsic Pathways)
3.3. Inhibition of Histone Deacetylases (Epigenetic Reprogramming)
3.4. Inhibition of the Proteasome (Proteostasis Disruption)
3.5. Blockade of Protein Translocation (Sec61 Inhibition)
3.6. Protease Inhibition (Anti-Invasive and Cytotoxic Effects)
3.7. Modulation of COX-2 and Inflammatory Pathways
4. Clinical Development Status of Cyanobacterial Peptides
4.1. Antibody–Drug Conjugates with Auristatin Payloads
- Brentuximab vedotin (Adcetris) was the first such ADC, approved in 2011 for relapsed Hodgkin’s lymphoma and anaplastic large-cell lymphoma (ALCL). It consists of an anti-CD30 antibody linked to MMAE (monomethyl auristatin E). Upon binding to CD30 on lymphoma cells, the ADC is internalized and releases MMAE, which then induces mitotic arrest and apoptosis [69]. Brentuximab vedotin produced significantly improved outcomes in CD30+ lymphomas and is now a standard therapy.
- Polatuzumab vedotin (Polivy) targets CD79b on B-cell tumors and was approved in 2019 for relapsed or refractory diffuse large B-cell lymphoma (DLBCL) in combination with chemotherapy. In the pivotal phase II trial (GO29365), adding polatuzumab vedotin to bendamustine + rituximab significantly improved outcomes versus bendamustine + rituximab alone. Patients receiving the polatuzumab combination had a higher complete response rate (40% vs. 18%, p = 0.026) and superior median overall survival (12.4 vs. 4.7 months) [70].
- Enfortumab vedotin is an anti-Nectin-4 ADC carrying MMAE, granted accelerated approval in 2019 for metastatic urothelial carcinoma after platinum and PD-1/L1 therapy. In a single-arm phase II study (EV-201), enfortumab vedotin achieved a 44% overall response rate (12% complete responses) in heavily pretreated bladder cancer, with a median response duration of 7.6 months (FDA 2019). A phase III trial (EV-301) confirmed its benefit over chemotherapy; enfortumab vedotin monotherapy yielded an objective response rate of ~40% (vs. ~18% with chemo) and significantly prolonged median overall survival (12.9 vs. 9.0 months, HR 0.70, p ~0.001) in this setting [71]. This marked the first therapy to improve survival in post-immunotherapy bladder cancer. Enfortumab vedotin is also being explored in other Nectin-4-expressing tumors; for example, a cohort of patients with head and neck cancer showed a confirmed ~24% response rate on enfortumab vedotin [72], indicating activity beyond urothelial carcinoma.
- Tisotumab vedotin is an ADC against tissue factor, granted accelerated FDA approval in 2021 for recurrent or metastatic cervical cancer after chemotherapy. Approval was based on the phase II innovaTV 204 trial, which demonstrated a 24% objective response rate (7% complete responses), with a median response duration of 8.3 months in a refractory cervical cancer population [73]. A subsequent phase III trial (innovaTV 301) confirmed a clinical benefit over chemotherapy. In that randomized study, tisotumab vedotin improved median overall survival (11.5 vs. 9.5 months, HR 0.70, p = 0.004) and produced higher response rates (18% vs. 5%) than investigator’s choice chemo [74].
- Disitamab vedotin is a HER2-directed ADC (comprising an anti-HER2 antibody attached to MMAE) approved in China in 2021 for HER2-positive advanced gastric cancer, including tumors with low HER2 expression. In a pivotal single-arm phase II trial in patients with HER2-overexpressing gastric/gastroesophageal junction cancer who had failed ≥2 prior regimens, disitamab vedotin achieved a 24.8% objective response rate (95% CI 17.5–33.3%) [75]. Although the ORR was modest, some responses were durable, and median overall survival was ~7.9 months in this late-line setting. Beyond gastric cancer, disitamab vedotin has shown notable activity in HER2-positive urothelial carcinoma. A combined analysis of two phase II studies in advanced bladder cancer reported a confirmed ORR of ~50% with disitamab vedotin monotherapy [76] in patients refractory to standard chemotherapy. This high response rate, along with a manageable safety profile, highlights the promise of disitamab vedotin in HER2-expressing urothelial tumors and supports ongoing trials in these indications.
- Belantamab mafodotin is a B-cell maturation antigen (BCMA)-targeted ADC that received accelerated approval in 2020 for relapsed or refractory multiple myeloma after at least four prior therapies (including a proteasome inhibitor, an immunomodulatory drug, and an anti-CD38 antibody). The approval was driven by the phase II DREAMM-2 study, in which single-agent belantamab mafodotin showed an ~31% overall response rate in heavily pretreated myeloma patients [77]. Notably, a proportion of patients achieved responses lasting ≥6 months, introducing a novel mechanism (delivering MMAF to BCMA-expressing plasma cells). However, belantamab mafodotin was associated with corneal toxicity (keratopathy), necessitating careful eye monitoring and dose adjustments. Importantly, follow-up trials raised questions about its risk–benefit profile. The confirmatory phase III DREAMM-3 trial, comparing belantamab mafodotin to standard pomalidomide–dexamethasone, failed to show an improvement in progression-free survival. Despite a respectable response rate (~41% with belantamab in DREAMM-3), there was no significant PFS or overall survival advantage over standard therapy [78]. Consequently, in November 2022 the manufacturer voluntarily withdrew belantamab mafodotin from the US market due to insufficient efficacy in the confirmatory study. Ongoing trials are now exploring belantamab in combination regimens (e.g., with proteasome inhibitors or immunomodulators) to see if its benefit can be improved in earlier lines of myeloma treatment.
4.2. Clinical-Stage Investigational Agents (Phases I–III)
- Glembatumumab vedotin is an ADC targeting glycoprotein NMB (gpNMB), a protein often overexpressed in triple-negative breast cancer (TNBC) and melanoma. In the phase 2b METRIC trial for metastatic TNBC, glembatumumab did not improve progression-free survival compared to chemotherapy (median 2.9 vs. 2.8 months, p = 0.97), failing to meet its primary endpoint [81]. While some tumor responses occurred, toxicity (notably rash and neutropenia) was significant, and the program was discontinued. Earlier phase II data in melanoma also showed only modest activity [82].
- Depatuxizumab mafodotin (ABT-414) is an EGFR-targeted ADC with an MMAF payload, developed for glioblastoma. Initial trials in EGFR-amplified gliomas showed some promise, but the phase III INTELLANCE-1 trial in newly diagnosed EGFR-amplified glioblastoma was negative. Depatux-M (with chemoradiation) did not improve survival versus standard chemoradiation plus placebo [83,84].
- Ladiratuzumab vedotin is an ADC targeting LIV-1 (a zinc transporter). It is being evaluated in TNBC and other solid tumors. An ongoing phase Ib/II trial in first-line TNBC combines ladiratuzumab vedotin with pembrolizumab. Preliminary results indicate manageable toxicity and evidence of activity in TNBC and other LIV-1–expressing tumors [85]. In a first-line TNBC cohort, the combination showed a preliminary ORR of about 33% in PD-L1^+ patients, supporting further development [85,86].
- Tasidotin (ILX-651) is a synthetic dolastatin-15 analog evaluated in advanced solid tumors. Phase I trials showed dose-limiting neutropenia at higher doses. Although no dramatic tumor regressions were seen, tasidotin did exhibit some anticancer activity. Notably, one melanoma patient achieved a complete response, and several patients had prolonged stable disease in early trials [87].
- Soblidotin (TZT-1027) is another dolastatin derivative (analog of dolastatin-10) that reached clinical trials with limited success. In a phase II study in refractory non-small cell lung cancer, soblidotin produced no objective tumor responses and a short median time to progression (~1.5 months). The trial concluded that soblidotin lacked meaningful anticancer activity in that setting, and further development for NSCLC was not pursued [88].
- Plitidepsin (Aplidin) is a cyclic depsipeptide originally isolated from a marine tunicate (sea squirt) but sometimes produced by a cyanobacterial symbiont. It has been tested in hematologic malignancies, particularly multiple myeloma. In the randomized phase III ADMYRE trial for relapsed/refractory myeloma, plitidepsin plus dexamethasone showed a significant improvement in progression-free survival compared to dexamethasone alone [89]. The combination also achieved a higher response rate (ORR ~13.8% vs. 1.7% with dex alone; p < 0.01) in this heavily pretreated population [89]. Interestingly, plitidepsin was repurposed during 2020–2021 as an antiviral agent against COVID-19 due to activity against SARS-CoV-2. In early 2024, a phase III trial in hospitalized COVID-19 patients showed faster viral clearance with plitidepsin [90]. While that is outside oncology, it highlights the broad potential of marine/cyanobacterial peptides in medicine. For cancer, plitidepsin is approved in Australia for myeloma and continues in trials elsewhere, indicating that such compounds can find niche clinical use [91].
- Spirulina (Arthrospira) and Phycocyanin are nutritional cyanobacteria that are also being evaluated as supportive care agents to mitigate side effects of cancer therapy. A 2019 randomized study (100 patients) found that dietary Spirulina during chemotherapy significantly reduced myelosuppressive toxicity. Patients who took Spirulina had higher post-chemo white blood cell and neutrophil counts and a lower rate of severe neutropenia compared to controls. They also experienced fewer dose delays and showed improved immune indicators (e.g., increased IgM and CD8+ T-cells) after therapy [61]. Meanwhile, phycocyanin (the antioxidant biliprotein from Spirulina) is being tested for preventing chemotherapy-induced peripheral neuropathy. The ongoing PHYCOCARE trial (phase II, NCT05025826) is evaluating oral phycocyanin vs. placebo in gastrointestinal cancer patients receiving oxaliplatin. The hypothesis is that phycocyanin’s ROS-scavenging properties will protect nerves from oxaliplatin neurotoxicity without compromising the anticancer efficacy of the chemotherapy [92]. Results are pending as of 2025.
- OKI-179 (bocodepsin) is an orally bioavailable prodrug analog of largazole (the marine cyanobacterial HDAC inhibitor). OKI-179 entered first-in-human trials in 2019 for advanced solid tumors. In a phase I dose-escalation study, OKI-179 was well tolerated, with manageable class-related toxicity (reversible thrombocytopenia as the dose-limiting toxicity) [93]. The drug showed dose-proportional exposure and robust HDAC target engagement at tolerated doses. Notably, OKI-179 induced histone acetylation in patient cells, confirming on-target activity. As of 2021, phase I results were encouraging, and an expansion phase Ib/II trial (“NAUTILUS”) launched to combine OKI-179 with a MEK inhibitor in RAS-mutant cancers. This represents one of the first HDAC inhibitor prodrugs derived from a marine cyanobacterial peptide to reach clinical testing.
5. Biotechnological Strategies for Production and Optimization
5.1. Genomic Mining and Activation of Silent Pathways
- Heterologous Expression of BGCs: Large cyanobacterial BGCs can be cloned and expressed in more tractable hosts such as E. coli, Anabaena sp., or Synechococcus elongatus. For instance, the cryptomaldamide BGC from Moorea producens yielded high titers only when expressed in an Anabaena host, highlighting that expression can be host-dependent [102]. Similarly, the microginin BGC from Microcystis was expressed in E. coli, resulting in production of both expected and novel halogenated analogs (including variants not detected in the native strain) [52]. Greunke et al. used promoter refactoring in E. coli to enhance anabaenopeptin production from Nostoc by over 100-fold, demonstrating that heterologous expression coupled with synthetic regulatory elements can achieve scalable yields [103].
- Expression in Model Cyanobacteria: While E. coli and yeast are common heterologous hosts, model cyanobacteria like S. elongatus PCC 7942 offer a photosynthetic production platform that utilizes light and CO2. These hosts also naturally provide certain cofactors and chaperones that may be required for proper folding and activity of cyanobacterial enzymes. S. elongatus was able to support cryptomaldamide biosynthesis, providing an example of using a cyanobacterial chassis to express another cyanobacterium’s pathway (“self-compatible” expression system) [102].
- CRISPRa and Stress Induction: CRISPR-based activation (CRISPRa) involves using a catalytically inactive Cas9 (dCas9) fused to a transcriptional activator to upregulate silent gene clusters. Ke et al. applied this in Streptomyces, activating ten silent PKS/NRPS BGCs and uncovering 22 distinct metabolites [104]. While CRISPRa is still in early development for cyanobacteria, it holds promise for systematically accessing cryptic metabolomes [101]. Additionally, traditional elicitation approaches—such as subjecting cultures to UV light, nutrient limitation (e.g., iron starvation), or other stressors—have led to the activation of silent pathways, yielding compounds like cyanochelins and scytonemin. Overexpression of global regulatory genes has also proven effective in awakening latent biosynthetic activity in cyanobacteria [105,106,107].
5.2. Metabolic Engineering of Native Producers
- Deletion of Competing Pathways: In Synechococcus elongatus, Choi et al. showed that CRISPR-Cas9 knockouts of competing central carbon metabolic genes (including those involved in glycolysis and phycobiliprotein biosynthesis) significantly increased production of isoprenoids. In particular, repression of the phycocyanin subunit gene (cpcB) diverted resources like ATP and amino acids toward heterologous product formation [108]. In another study, Synechocystis sp. PCC 6803 was engineered by deleting the shc gene, which encodes hopene cyclase—a key enzyme in hopanoid (triterpene) synthesis. This redirection of farnesyl diphosphate flux led to a marked increase in alternative triterpene accumulation [109]. These examples demonstrate that knocking out or downregulating competing pathways can free up cellular building blocks for the production of desired compounds.
- Amplifying Precursor Supply: For efficient peptide/polyketide biosynthesis, an ample supply of precursor metabolites (such as specific amino acids, malonyl-CoA, methylmalonyl-CoA, etc.) is essential. Roulet et al. boosted polyketide synthesis in S. elongatus by overexpressing enzymes that increase intracellular levels of malonyl-CoA and methylmalonyl-CoA [7]. Usai et al. combined genetic modifications with exogenous feeding of precursor molecules (e.g., 2-phenylethanol) in cyanobacteria, achieving a synergistic increase in final titers [110]. Such strategies can be adapted to NRPS pathways by supplying uncommon amino acid precursors or installing biosynthetic modules for unusual residues (ornithine, homoserine, etc.) to ensure the pathway is not limited by precursor availability.
- Promoter and Regulatory Engineering: Native BGCs are often under tight regulatory control and may only be weakly expressed. To overcome this, promoters within the pathway can be replaced with strong constitutive or inducible promoters. As mentioned, Greunke et al. did this in E. coli by refactoring the anabaenopeptin BGC, leading to >100-fold increase in production [103]. Choi et al. further used CRISPR interference (dCas12a-based repression) to simultaneously silence competing pathways while using synthetic promoters to boost the target pathway, amplifying terpenoid output [108]. Additionally, Leao et al. identified cryptic clusters in Moorea that were silent due to specific repressors [11]; by targeting those regulatory elements or co-expressing transcriptional activators, they were able to awaken those latent pathways.
- Optimizing Chassis Strains: The use of fast-growing and genetically tractable cyanobacterial strains such as S. elongatus UTEX 2973 has opened new possibilities for metabolic engineering. Knoot et al. showed that this strain (notable for its rapid growth) can serve as a high-biomass chassis for complex pathways, successfully producing hapalindole alkaloids after integration of the relevant BGC [111]. The ability of UTEX 2973 to quickly accumulate biomass and maintain high expression levels makes it ideal for large-scale biosynthetic applications where yield is critical.
5.3. Heterologous Expression in Non-Cyanobacterial Hosts
- Escherichia coli: This bacterium is a popular host due to its fast growth, well-characterized genetics, and ease of manipulation. Several cyanobacterial peptides have been successfully produced in E. coli. For instance, the NRPS/PKS genes encoding hapalosin were cloned into an E. coli BAP1 strain, achieving approximately 45% of the native Fischerella yield [112]. Additionally, lyngbyatoxin A was produced in E. coli at the gram scale. E. coli has also been used to generate microcystin analogs by feeding alternative precursor amino acids to the NRPS machinery, enabling structure–activity relationship studies [113]. These successes illustrate E. coli’s versatility for heterologous expression, although it lacks some eukaryotic post-translational modification systems.
- Yeast (Saccharomyces cerevisiae): Yeast has emerged as a promising host for complex cyanobacterial pathways. In one study, a cyanobacterial NRPS-PKS pathway was reconstructed in S. cerevisiae to produce the sunscreen peptide shinorine. Deletion of a competing yeast pathway via CRISPR increased shinorine yield nearly 10-fold [114]. Yeast offers a eukaryotic expression environment that can support proper folding of large enzymatic complexes and provides subcellular compartmentalization, which can be advantageous for certain pathways. While yeast is not photosynthetic, its metabolic flexibility and its capacity to accommodate large DNA constructs, such as those introduced via yeast artificial chromosomes, make it a robust platform for the production of specialized metabolites [115].
- Streptomyces: These filamentous actinomycetes are renowned for producing antibiotics and are well suited for expressing large and complex cyanobacterial BGCs. Streptomyces species have high native expression of phosphopantetheinyl transferases (needed to activate NRPS/PKS enzymes) and abundant precursor pools. For example, Streptomyces venezuelae was used to express the 26 kb barbamide pathway from Moorea producens, resulting in production of the chlorinated metabolite [116]. More recent studies, using tools like bacterial artificial chromosomes and engineered Streptomyces strains, have achieved heterologous production of cyanobacterial compounds such as lyngbyatoxin A and teleocidins [12,117,118].
5.4. Pathway Refactoring and Synthetic Biology
- Domain swapping and Module Editing: Rational swapping of domains in NRPS and PKS enzymes can change substrate specificity and produce new analogs. For example, Calcott et al. replaced adenylation domains in a Pseudomonas NRPS (for pyoverdine synthesis), which generated novel siderophores with altered amino acid composition [119]. In another example, Thong et al. used CRISPR-Cas9 genome editing to modify specificity-conferring domains in Streptomyces, reprogramming the enduracidin lipopeptide biosynthesis. The engineered strains produced new enduracidin analogs. The engineered strains produced new enduracidin analogs, confirming the success of module editing [120].
- Tailoring Enzyme Manipulation: Structural diversification can also be achieved by modifying tailoring enzymes in the pathway, such as methyltransferases or halogenases. For example, in the daptomycin lipopeptide pathway, knockout of the Glu12-specific methyltransferase yielded a desmethyl analog with altered pharmacological properties [121]. Similarly, Bradley et al. incorporated a tryptophan halogenase gene into a biosynthetic cluster, enabling the production of halogenated derivatives of a microbial alkaloid in vivo [122]. Such interventions expand the chemical space of known compounds by creating new analogs with potentially improved bioactivity or pharmacokinetics.
- Plug-and-Play Pathway Assembly: New cloning technologies now allow rapid refactoring and combinatorial assembly of entire biosynthetic pathways. Methods such as DiPaC (Direct Pathway Cloning) and TAR (transformation-associated recombination) facilitate one-step assembly of large biosynthetic gene clusters (BGCs) [123]. PCR-based refactoring of the complete erythromycin BGC in a single step. More recently, Ouyang et al. applied a similar strategy to clone and express a 16.8 kb radicicol biosynthetic pathway in E. coli, resulting in the production of novel analogs [123]. As further proof of concept, Basitta et al. reorganized the novobiocin antibiotic cluster into artificial operons using an assembly method (AGOS), leading to successful heterologous production [124].
- AI-Guided Design and Computational Modeling: Machine learning and computational tools trained on BGC databases can predict optimal engineering strategies. For instance, Kalkreuter et al. used molecular dynamics simulations to redesign acyltransferase domains in a modular PKS, expanding their substrate range [125]. Such in silico approaches increase the predictability and throughput of pathway engineering by highlighting beneficial mutations or domain swaps before laboratory implementation.
5.5. Scale-Up and Production Systems
- Photobioreactors: Engineered cyanobacteria can be grown in controlled photobioreactors at an industrial scale. For example, Synechococcus strains have been cultivated in 100–500 L photobioreactors using industrial flue gas as a CO2 source, successfully producing high-value compounds like squalene at scale [126]. Similar systems are being adapted for peptide production, offering a sustainable, sunlight-driven manufacturing process. Photobioreactors enable dense cultures under optimized light and nutrient conditions, potentially lowering costs for producing complex peptides compared to fermentation in the dark with expensive media [6,23,24].
- Cell-Free Biosynthesis: In vitro enzymatic systems can bypass living cells to produce natural products. By using purified NRPS/PKS enzymes or crude lysates, researchers have synthesized peptides in a cell-free manner. Notably, yields of approximately 30 mg/L have been achieved for certain model compounds, including cyclic dipeptides and the depsipeptide valinomycin, using E. coli lysate-based or two-stage cell-free systems [127,128]. Cell-free biosynthesis allows for precise control over reaction conditions and substrates, and it avoids issues of compound toxicity or metabolic regulation that occur in vivo. While currently used at small scales, advances in cell-free synthetic biology could enable on-demand production of complex cytotoxins by simply mixing the necessary enzymes and substrates in a reactor.
6. Current Limitations and Future Perspectives
6.1. Enhancing Specificity and Delivery
6.2. Combination Therapies
6.3. Expanding Chemical Diversity
6.4. Safety Profiling and Toxicology
6.5. Clinical Trials and Accessibility
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Qamar, H.; Hussain, K.; Soni, A.; Khan, A.; Hussain, T.; Chénais, B. Cyanobacteria as Natural Therapeutics and Pharmaceutical Potential: Role in Antitumor Activity and as Nanovectors. Molecules 2021, 26, 247. [Google Scholar] [CrossRef]
- Perera, R.M.T.D.; Herath, K.H.I.N.M.; Sanjeewa, K.K.A.; Jayawardena, T.U. Recent Reports on Bioactive Compounds from Marine Cyanobacteria in Relation to Human Health Applications. Life 2023, 13, 1411. [Google Scholar] [CrossRef]
- Robles-Bañuelos, B.; Durán-Riveroll, L.M.; Rangel-López, E.; Pérez-López, H.I.; González-Maya, L. Marine Cyanobacteria as Sources of Lead Anticancer Compounds: A Review of Families of Metabolites with Cytotoxic, Antiproliferative, and Antineoplastic Effects. Molecules 2022, 27, 4814. [Google Scholar] [CrossRef] [PubMed]
- Kallifidas, D.; Dhakal, D.; Chen, M.; Chen, Q.-Y.; Kokkaliari, S.; Colon Rosa, N.A.; Ratnayake, R.; Bruner, S.D.; Paul, V.J.; Ding, Y.; et al. Biosynthesis of Dolastatin 10 in Marine Cyanobacteria, a Prototype for Multiple Approved Cancer Drugs. Org. Lett. 2024, 26, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Sitachitta, N.; Rossi, J.V.; Roberts, M.A.; Flatt, P.M.; Jia, J.; Sherman, D.H.; Gerwick, W.H. Biosynthetic Pathway and Gene Cluster Analysis of Curacin A, an Antitubulin Natural Product from the Tropical Marine Cyanobacterium Lyngbya Majuscula. J. Nat. Prod. 2004, 67, 1356–1367. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Ellis, E.K.; Chen, Q.-Y.; Ratnayake, R. Progress in the Discovery and Development of Anticancer Agents from Marine Cyanobacteria. Nat. Prod. Rep. 2025, 42, 208–256. [Google Scholar] [CrossRef]
- Roulet, J.; Taton, A.; Golden, J.W.; Arabolaza, A.; Burkart, M.D.; Gramajo, H. Development of a Cyanobacterial Heterologous Polyketide Production Platform. Metab. Eng. 2018, 49, 94–104. [Google Scholar] [CrossRef]
- Nandagopal, P.; Steven, A.N.; Chan, L.-W.; Rahmat, Z.; Jamaluddin, H.; Mohd Noh, N.I. Bioactive Metabolites Produced by Cyanobacteria for Growth Adaptation and Their Pharmacological Properties. Biology 2021, 10, 1061. [Google Scholar] [CrossRef]
- Wijewickrama, M.; Greene, A.; Cock, I. Therapeutics from Cyanobacteria: A Review of Cyanobacteria-Derived Compounds as Anti-Cancer Drug Leads. Pharmacogn. Rev. 2023, 17, 230–246. [Google Scholar] [CrossRef]
- Mi, Y.; Zhang, J.; He, S.; Yan, X. New Peptides Isolated from Marine Cyanobacteria, an Overview over the Past Decade. Mar. Drugs 2017, 15, 132. [Google Scholar] [CrossRef]
- Leao, T.; Castelão, G.; Korobeynikov, A.; Monroe, E.A.; Podell, S.; Glukhov, E.; Allen, E.E.; Gerwick, W.H.; Gerwick, L. Comparative Genomics Uncovers the Prolific and Distinctive Metabolic Potential of the Cyanobacterial Genus Moorea. Proc. Natl. Acad. Sci. USA 2017, 114, 3198–3203. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, D.; Chen, M.; Luesch, H.; Ding, Y. Heterologous Production of Cyanobacterial Compounds. J. Ind. Microbiol. Biotechnol. 2021, 48, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Phat, C.; Hong, S.-C. Structural Diversity of Marine Cyclic Peptides and Their Molecular Mechanisms for Anticancer, Antibacterial, Antifungal, and Other Clinical Applications. Peptides 2017, 95, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Monks, N.R.; Liu, S.; Xu, Y.; Yu, H.; Bendelow, A.S.; Moscow, J.A. Potent Cytotoxicity of the Phosphatase Inhibitor Microcystin LR and Microcystin Analogues in OATP1B1- and OATP1B3-Expressing HeLa Cells. Mol. Cancer Ther. 2007, 6, 587–598. [Google Scholar] [CrossRef]
- Dias, E.; Paulino, S.; Pereira, P. Cyanotoxins: From Poisoning to Healing—A Possible Pathway? Limnetica 2015, 34, 159–172. [Google Scholar] [CrossRef]
- Kounnis, V.; Chondrogiannis, G.; Mantzaris, M.D.; Tzakos, A.G.; Fokas, D.; Papanikolaou, N.A.; Galani, V.; Sainis, I.; Briasoulis, E. Microcystin LR Shows Cytotoxic Activity Against Pancreatic Cancer Cells Expressing the Membrane OATP1B1 and OATP1B3 Transporters. Anticancer Res. 2015, 35, 5857–5865. [Google Scholar]
- Alvarinõ, R.; Alonso, E.; Bornancin, L.; Bonnard, I.; Inguimbert, N.; Banaigs, B.; Botana, L.M. Biological Activities of Cyclic and Acyclic B-Type Laxaphycins in SH-SY5Y Human Neuroblastoma Cells. Mar. Drugs 2020, 18, 364. [Google Scholar] [CrossRef]
- Nogle, L.M.; Gerwick, W.H. Somocystinamide A, a Novel Cytotoxic Disulfide Dimer from a Fijian Marine Cyanobacterial Mixed Assemblage. Org. Lett. 2002, 4, 1095–1098. [Google Scholar] [CrossRef]
- Wrasidlo, W.; Mielgo, A.; Torres, V.A.; Barbero, S.; Stoletov, K.; Suyama, T.L.; Klemke, R.L.; Gerwick, W.H.; Carson, D.A.; Stupack, D.G. The Marine Lipopeptide Somocystinamide A Triggers Apoptosis via Caspase 8. Proc. Natl. Acad. Sci. USA 2008, 105, 2313–2318. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Y.; Wang, Z.; Xing, X.; Maguire, A.R.; Luesch, H.; Zhang, H.; Xu, Z.; Ye, T. Total Synthesis and Biological Evaluation of Grassypeptolide A. Chem.–A Eur. J. 2013, 19, 6774–6784. [Google Scholar] [CrossRef]
- Kwan, J.C.; Ratnayake, R.; Abboud, K.A.; Paul, V.J.; Luesch, H. Grassypeptolides A−C, Cytotoxic Bis-Thiazoline Containing Marine Cyclodepsipeptides. J. Org. Chem. 2010, 75, 8012–8023. [Google Scholar] [CrossRef] [PubMed]
- Fathoni, I.; Petitbois, J.G.; Alarif, W.M.; Abdel-Lateff, A.; Al-Lihaibi, S.S.; Yoshimura, E.; Nogata, Y.; Vairappan, C.S.; Sholikhah, E.N.; Okino, T. Bioactivities of Lyngbyabellins from Cyanobacteria of Moorea and Okeania Genera. Molecules 2020, 25, 3986. [Google Scholar] [CrossRef]
- Shertzer, A.L.; Vall, H.; Jordan, M.A.; Gerwick, W.; Wilson, L. The Unusual Mechanism of Action of the Microtubule Targeted Drug Curacin, A. Cancer Res. 2004, 64, 1249. [Google Scholar]
- Eren, E.; Watts, N.R.; Sackett, D.L.; Wingfield, P.T. Conformational Changes in Tubulin upon Binding Cryptophycin-52 Reveal Its Mechanism of Action. J. Biol. Chem. 2021, 297, 101138. [Google Scholar] [CrossRef] [PubMed]
- Figueras, E.; Borbély, A.; Ismail, M.; Frese, M.; Sewald, N. Novel Unit B Cryptophycin Analogues as Payloads for Targeted Therapy. Beilstein J. Org. Chem. 2018, 14, 1281–1286. [Google Scholar] [CrossRef]
- Bowers, A.; West, N.; Taunton, J.; Schreiber, S.L.; Bradner, J.E.; Williams, R.M. Total Synthesis and Biological Mode of Action of Largazole: A Potent Class I Histone Deacetylase Inhibitor. J. Am. Chem. Soc. 2008, 130, 11219–11222. [Google Scholar] [CrossRef]
- Liu, Y.; Salvador, L.A.; Byeon, S.; Ying, Y.; Kwan, J.C.; Law, B.K.; Hong, J.; Luesch, H. Anticolon Cancer Activity of Largazole, a Marine-Derived Tunable Histone Deacetylase Inhibitor. J. Pharmacol. Exp. Ther. 2010, 335, 351–361. [Google Scholar] [CrossRef]
- Hong, J.; Luesch, H. Largazole: From Discovery to Broad-Spectrum Therapy. Nat. Prod. Rep. 2012, 29, 449. [Google Scholar] [CrossRef]
- Ying, Y.; Taori, K.; Kim, H.; Hong, J.; Luesch, H. Total Synthesis and Molecular Target of Largazole, a Histone Deacetylase Inhibitor. J. Am. Chem. Soc. 2008, 130, 8455–8459. [Google Scholar] [CrossRef]
- Kim, B.; Park, H.; Salvador, L.A.; Serrano, P.E.; Kwan, J.C.; Zeller, S.L.; Chen, Q.-Y.; Ryu, S.; Liu, Y.; Byeon, S.; et al. Evaluation of Class I HDAC Isoform Selectivity of Largazole Analogues. Bioorg. Med. Chem. Lett. 2014, 24, 3728–3731. [Google Scholar] [CrossRef]
- Pereira, A.R.; Kale, A.J.; Fenley, A.T.; Byrum, T.; Debonsi, H.M.; Gilson, M.K.; Valeriote, F.A.; Moore, B.S.; Gerwick, W.H. The Carmaphycins: New Proteasome Inhibitors Exhibiting an α,Β-Epoxyketone Warhead from a Marine Cyanobacterium. ChemBioChem 2012, 13, 810–817. [Google Scholar] [CrossRef]
- Estrella-Parra, E.A.; Arreola, R.; Álvarez-Sánchez, M.E.; Torres-Romero, J.C.; Rojas-Espinosa, O.; De la Cruz-Santiago, J.A.; Martinez-Benitez, M.B.; López-Camarillo, C.; Lara-Riegos, J.C.; Arana-Argáez, V.E.; et al. Natural Marine Products as Antiprotozoal Agents against Amitochondrial Parasites. Int. J. Parasitol. Drugs Drug Resist. 2022, 19, 40–46. [Google Scholar] [CrossRef]
- Liu, L.J.; Francisco, K.R.; Sun, Y.U.; Serafim, M.S.M.; Amarasinghe, D.K.; Teixeira, T.R.; Lucero, B.; Kronenberger, T.; Elsayed, W.; Elwakeel, H.; et al. Carmaphycin B-Based Proteasome Inhibitors to Treat Human African Trypanosomiasis: Structure–Activity Relationship and In Vivo Efficacy. ACS Infect. Dis. 2024, 10, 4182–4193. [Google Scholar] [CrossRef]
- Almaliti, J.; Miller, B.; Pietraszkiewicz, H.; Glukhov, E.; Naman, C.B.; Kline, T.; Hanson, J.; Li, X.; Zhou, S.; Valeriote, F.A.; et al. Exploration of the Carmaphycins as Payloads in Antibody Drug Conjugate Anticancer Agents. Eur. J. Med. Chem. 2019, 161, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Pohl, M.O.; Martin-Sancho, L.; Ratnayake, R.; White, K.M.; Riva, L.; Chen, Q.-Y.; Lieber, G.; Busnadiego, I.; Yin, X.; Lin, S.; et al. Sec61 Inhibitor Apratoxin S4 Potently Inhibits SARS-CoV-2 and Exhibits Broad-Spectrum Antiviral Activity. ACS Infect. Dis. 2022, 8, 1265–1279. [Google Scholar] [CrossRef]
- Paatero, A.O.; Kellosalo, J.; Dunyak, B.M.; Almaliti, J.; Gestwicki, J.E.; Gerwick, W.H.; Taunton, J.; Paavilainen, V.O. Apratoxin Kills Cells by Direct Blockade of the Sec61 Protein Translocation Channel. Cell Chem. Biol. 2016, 23, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-C.; Chen, Z.; Jiang, Y.; Akare, S.; Kolber-Simonds, D.; Condon, K.; Agoulnik, S.; Tendyke, K.; Shen, Y.; Wu, K.-M.; et al. Apratoxin A Shows Novel Pancreas-Targeting Activity through the Binding of Sec 61. Mol. Cancer Ther. 2016, 15, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Ratnayake, R.; Wang, M.; Chen, Q.-Y.; Raisch, K.P.; Dang, L.H.; Law, B.K.; Luesch, H. Inhibition of Cotranslational Translocation by Apratoxin S4: Effects on Oncogenic Receptor Tyrosine Kinases and the Fate of Transmembrane Proteins Produced in the Cytoplasm. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100053. [Google Scholar] [CrossRef]
- Ahmed, S.; Alam, W.; Aschner, M.; Filosa, R.; Cheang, W.S.; Jeandet, P.; Saso, L.; Khan, H. Marine Cyanobacterial Peptides in Neuroblastoma: Search for Better Therapeutic Options. Cancers 2023, 15, 2515. [Google Scholar] [CrossRef]
- Hau, A.M.; Greenwood, J.A.; Löhr, C.V.; Serrill, J.D.; Proteau, P.J.; Ganley, I.G.; McPhail, K.L.; Ishmael, J.E. Coibamide A Induces MTOR-Independent Autophagy and Cell Death in Human Glioblastoma Cells. PLoS ONE 2013, 8, e65250. [Google Scholar] [CrossRef]
- Serrill, J.D.; Wan, X.; Hau, A.M.; Jang, H.S.; Coleman, D.J.; Indra, A.K.; Alani, A.W.G.; McPhail, K.L.; Ishmael, J.E. Coibamide A, a Natural Lariat Depsipeptide, Inhibits VEGFA/VEGFR2 Expression and Suppresses Tumor Growth in Glioblastoma Xenografts. Investig. New Drugs 2016, 34, 24–40. [Google Scholar] [CrossRef]
- Tranter, D.; Paatero, A.O.; Kawaguchi, S.; Kazemi, S.; Serrill, J.D.; Kellosalo, J.; Vogel, W.K.; Richter, U.; Mattos, D.R.; Wan, X.; et al. Coibamide A Targets Sec61 to Prevent Biogenesis of Secretory and Membrane Proteins. ACS Chem. Biol. 2020, 15, 2125–2136. [Google Scholar] [CrossRef] [PubMed]
- Akaiwa, M.; Dugal-Tessier, J.; Mendelsohn, B.A. Antibody–Drug Conjugate Payloads; Study of Auristatin Derivatives. Chem. Pharm. Bull. 2020, 68, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.P.; Cheung, Y.K.; Shah, D.K. Whole-Body Pharmacokinetics and Physiologically Based Pharmacokinetic Model for Monomethyl Auristatin E (MMAE). J. Clin. Med. 2021, 10, 1332. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.; Teicher, B. Cryptophycins: A Novel Class of Potent Antimitotic Antitumor Depsipeptides. Curr. Pharm. Des. 2001, 7, 1259–1276. [Google Scholar] [CrossRef]
- Wang, X.; Gigant, B.; Zheng, X.; Chen, Q. Microtubule-targeting Agents for Cancer Treatment: Seven Binding Sites and Three Strategies. MedComm–Oncol. 2023, 2, e46. [Google Scholar] [CrossRef]
- Suyama, T.L.; Gerwick, W.H. Stereospecific Total Synthesis of Somocystinamide, A. Org. Lett. 2008, 10, 4449–4452. [Google Scholar] [CrossRef]
- Liu, X.; Nasveschuk, C.G.; Andrew, J.P.; Ungermannova, D.; Christopher, G. Macrocyclic Compounds Useful as Inhibitors of Histone Deacetylases. U.S. Patent No. US20130203681A1, 8 August 2013. [Google Scholar]
- Al-Awadhi, F.H.; Salvador-Reyes, L.A.; Elsadek, L.A.; Ratnayake, R.; Chen, Q.-Y.; Luesch, H. Largazole Is a Brain-Penetrant Class I HDAC Inhibitor with Extended Applicability to Glioblastoma and CNS Diseases. ACS Chem. Neurosci. 2020, 11, 1937–1943. [Google Scholar] [CrossRef]
- Wu, L.-C.; Wen, Z.-S.; Qiu, Y.-T.; Chen, X.-Q.; Chen, H.-B.; Wei, M.-M.; Liu, Z.; Jiang, S.; Zhou, G.-B. Largazole Arrests Cell Cycle at G1 Phase and Triggers Proteasomal Degradation of E2F1 in Lung Cancer Cells. ACS Med. Chem. Lett. 2013, 4, 921–926. [Google Scholar] [CrossRef]
- Pavlik, C.M.; Wong, C.Y.B.; Ononye, S.; Lopez, D.D.; Engene, N.; McPhail, K.L.; Gerwick, W.H.; Balunas, M.J. Santacruzamate A, a Potent and Selective Histone Deacetylase Inhibitor from the Panamanian Marine Cyanobacterium Cf. Symploca Sp. J. Nat. Prod. 2013, 76, 2026–2033. [Google Scholar] [CrossRef]
- Eusébio, N.; Castelo-Branco, R.; Sousa, D.; Preto, M.; D’Agostino, P.; Gulder, T.A.M.; Leão, P.N. Discovery and Heterologous Expression of Microginins from Microcystis Aeruginosa LEGE 91341. ACS Synth. Biol. 2022, 11, 3493–3503. [Google Scholar] [CrossRef]
- Almaliti, J.; Fajtová, P.; Calla, J.; LaMonte, G.M.; Feng, M.; Rocamora, F.; Ottilie, S.; Glukhov, E.; Boura, E.; Suhandynata, R.T.; et al. Development of Potent and Highly Selective Epoxyketone-Based Plasmodium Proteasome Inhibitors. Chem.–A Eur. J. 2023, 29, e202203958. [Google Scholar] [CrossRef] [PubMed]
- Ashhurst, A.S.; Tang, A.H.; Fajtová, P.; Yoon, M.C.; Aggarwal, A.; Bedding, M.J.; Stoye, A.; Beretta, L.; Pwee, D.; Drelich, A.; et al. Potent Anti-SARS-CoV-2 Activity by the Natural Product Gallinamide A and Analogues via Inhibition of Cathepsin, L.J. Med. Chem. 2022, 65, 2956–2970. [Google Scholar] [CrossRef]
- Xiao, L.-X.; Li, X.-J.; Yu, H.-Y.; Qiu, R.-J.; Zhai, Z.-Y.; Ding, W.-F.; Zhu, M.-S.; Zhong, W.; Fang, C.-F.; Yang, J.; et al. Macrophage-Derived Cathepsin L Promotes Epithelial-Mesenchymal Transition and M2 Polarization in Gastric Cancer. World J. Gastroenterol. 2024, 30, 5032–5054. [Google Scholar] [CrossRef] [PubMed]
- Bian, B.; Mongrain, S.; Cagnol, S.; Langlois, M.-J.; Boulanger, J.; Bernatchez, G.; Carrier, J.C.; Boudreau, F.; Rivard, N. Cathepsin B Promotes Colorectal Tumorigenesis, Cell Invasion, and Metastasis. Mol. Carcinog. 2016, 55, 671–687. [Google Scholar] [CrossRef]
- Boudreau, P.D.; Miller, B.W.; McCall, L.-I.; Almaliti, J.; Reher, R.; Hirata, K.; Le, T.; Siqueira-Neto, J.L.; Hook, V.; Gerwick, W.H. Design of Gallinamide A Analogs as Potent Inhibitors of the Cysteine Proteases Human Cathepsin L and Trypanosoma Cruzi Cruzain. J. Med. Chem. 2019, 62, 9026–9044. [Google Scholar] [CrossRef] [PubMed]
- Al-Awadhi, F.; Salvador, L.; Law, B.; Paul, V.; Luesch, H. Kempopeptin C, a Novel Marine-Derived Serine Protease Inhibitor Targeting Invasive Breast Cancer. Mar. Drugs 2017, 15, 290. [Google Scholar] [CrossRef]
- Al-Awadhi, F.H.; Paul, V.J.; Luesch, H. Structural Diversity and Anticancer Activity of Marine-Derived Elastase Inhibitors: Key Features and Mechanisms Mediating the Antimetastatic Effects in Invasive Breast Cancer. ChemBioChem 2018, 19, 815–825. [Google Scholar] [CrossRef]
- Heinilä, L.M.P.; Jokela, J.; Ahmed, M.N.; Wahlsten, M.; Kumar, S.; Hrouzek, P.; Permi, P.; Koistinen, H.; Fewer, D.P.; Sivonen, K. Discovery of Varlaxins, New Aeruginosin-Type Inhibitors of Human Trypsins. Org. Biomol. Chem. 2022, 20, 2681–2692. [Google Scholar] [CrossRef]
- Ge, Y.; Kang, Y.-K.; Dong, L.; Liu, L.-H.; An, G.-Y. The Efficacy of Dietary Spirulina as an Adjunct to Chemotherapy to Improve Immune Function and Reduce Myelosuppression in Patients with Malignant Tumors. Transl. Cancer Res. 2019, 8, 1065–1073. [Google Scholar] [CrossRef]
- Reddy, M.C.; Subhashini, J.; Mahipal, S.V.K.; Bhat, V.B.; Srinivas Reddy, P.; Kiranmai, G.; Madyastha, K.M.; Reddanna, P. C-Phycocyanin, a Selective Cyclooxygenase-2 Inhibitor, Induces Apoptosis in Lipopolysaccharide-Stimulated RAW 264.7 Macrophages. Biochem. Biophys. Res. Commun. 2003, 304, 385–392. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 Promotes Tumor Growth and Suppresses Tumor Immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, Y.; Liu, G.; Liu, H.; Zhu, F.; Ji, H.; Li, B. C-Phycocyanin Exerts Anti-Cancer Effects via the MAPK Signaling Pathway in MDA-MB-231 Cells. Cancer Cell Int. 2018, 18, 12. [Google Scholar] [CrossRef]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in Cancer: A Review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.R.; Jo, S.A.; Lee, H.; Yoon, Y.D.; Kwon, J.-H.; Yang, J.-W.; Choi, B.J.; Park, K.H.; Lee, M.Y.; Lee, C.W.; et al. Inhibition of Skin Inflammation by Scytonemin, an Ultraviolet Sunscreen Pigment. Mar. Drugs 2020, 18, 300. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jiang, M.; Wang, L.; Yu, S. Combined Chemotherapy with Cyclooxygenase-2 (COX-2) Inhibitors in Treating Human Cancers: Recent Advancement. Biomed. Pharmacother. 2020, 129, 110389. [Google Scholar] [CrossRef]
- Kefayat, A.; Ghahremani, F.; Safavi, A.; Hajiaghababa, A.; Moshtaghian, J. C-Phycocyanin: A Natural Product with Radiosensitizing Property for Enhancement of Colon Cancer Radiation Therapy Efficacy through Inhibition of COX-2 Expression. Sci. Rep. 2019, 9, 19161. [Google Scholar] [CrossRef]
- Deng, C.; Pan, B.; O’Connor, O.A. Brentuximab Vedotin. Clin. Cancer Res. 2013, 19, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2020, 38, 155–165. [Google Scholar] [CrossRef]
- Powles, T.; Valderrama, B.P.; Gupta, S.; Bedke, J.; Kikuchi, E.; Hoffman-Censits, J.; Iyer, G.; Vulsteke, C.; Park, S.H.; Shin, S.J.; et al. Enfortumab Vedotin and Pembrolizumab in Untreated Advanced Urothelial Cancer. N. Engl. J. Med. 2024, 390, 875–888. [Google Scholar] [CrossRef]
- Swiecicki, P.L.; Yilmaz, E.; Rosenberg, A.J.; Fujisawa, T.; Bruce, J.Y.; Meng, C.; Wozniak, M.A.; Wang, L.; Gorla, S.; Geiger, J.L. Enfortumab Vedotin (EV) in the Previously Treated Advanced Head and Neck Cancer (HNC) Cohort of EV-202. Int. J. Radiat. Oncol. Biol. Phys. 2024, 118, e18. [Google Scholar] [CrossRef]
- Coleman, R.L.; Lorusso, D.; Gennigens, C.; González-Martín, A.; Randall, L.; Cibula, D.; Lund, B.; Woelber, L.; Pignata, S.; Forget, F.; et al. Efficacy and Safety of Tisotumab Vedotin in Previously Treated Recurrent or Metastatic Cervical Cancer (InnovaTV 204/GOG-3023/ENGOT-Cx6): A Multicentre, Open-Label, Single-Arm, Phase 2 Study. Lancet Oncol. 2021, 22, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Vergote, I.; González-Martín, A.; Fujiwara, K.; Kalbacher, E.; Bagaméri, A.; Ghamande, S.; Lee, J.-Y.; Banerjee, S.; Maluf, F.C.; Lorusso, D.; et al. Tisotumab Vedotin as Second- or Third-Line Therapy for Recurrent Cervical Cancer. N. Engl. J. Med. 2024, 391, 44–55. [Google Scholar] [CrossRef]
- Peng, Z.; Liu, T.; Wei, J.; Wang, A.; He, Y.; Yang, L.; Zhang, X.; Fan, N.; Luo, S.; Li, Z.; et al. Efficacy and Safety of a Novel Anti-HER2 Therapeutic Antibody RC48 in Patients with HER2-overexpressing, Locally Advanced or Metastatic Gastric or Gastroesophageal Junction Cancer: A Single-arm Phase II Study. Cancer Commun. 2021, 41, 1173–1182. [Google Scholar] [CrossRef]
- Sheng, X.; Wang, L.; He, Z.; Shi, Y.; Luo, H.; Han, W.; Yao, X.; Shi, B.; Liu, J.; Hu, C.; et al. Efficacy and Safety of Disitamab Vedotin in Patients With Human Epidermal Growth Factor Receptor 2–Positive Locally Advanced or Metastatic Urothelial Carcinoma: A Combined Analysis of Two Phase II Clinical Trials. J. Clin. Oncol. 2024, 42, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Nooka, A.K.; Cohen, A.D.; Lee, H.C.; Badros, A.; Suvannasankha, A.; Callander, N.; Abdallah, A.; Trudel, S.; Chari, A.; Libby, E.N.; et al. Single-agent Belantamab Mafodotin in Patients with Relapsed/Refractory Multiple Myeloma: Final Analysis of the DREAMM-2 Trial. Cancer 2023, 129, 3746–3760. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Abdullah, H.A.; Opalinska, J.B.; Paka, P.; Richards, E.; Weisel, K.; Trudel, S.; Mateos, M.-V.; Dimopoulos, M.A.; Lonial, S. The Clinical Journey of Belantamab Mafodotin in Relapsed or Refractory Multiple Myeloma: Lessons in Drug Development. Blood Cancer J. 2025, 15, 15. [Google Scholar] [CrossRef]
- Cheng-Sánchez, I.; Moya-Utrera, F.; Porras-Alcalá, C.; López-Romero, J.M.; Sarabia, F. Antibody-Drug Conjugates Containing Payloads from Marine Origin. Mar. Drugs 2022, 20, 494. [Google Scholar] [CrossRef]
- Gogia, P.; Ashraf, H.; Bhasin, S.; Xu, Y. Antibody–Drug Conjugates: A Review of Approved Drugs and Their Clinical Level of Evidence. Cancers 2023, 15, 3886. [Google Scholar] [CrossRef]
- Vahdat, L.T.; Schmid, P.; Forero-Torres, A.; Blackwell, K.; Telli, M.L.; Melisko, M.; Möbus, V.; Cortes, J.; Montero, A.J.; Ma, C.; et al. Glembatumumab Vedotin for Patients with Metastatic, GpNMB Overexpressing, Triple-Negative Breast Cancer (“METRIC”): A Randomized Multicenter Study. NPJ Breast Cancer 2021, 7, 57. [Google Scholar] [CrossRef]
- Ott, P.A.; Pavlick, A.C.; Johnson, D.B.; Hart, L.L.; Infante, J.R.; Luke, J.J.; Lutzky, J.; Rothschild, N.E.; Spitler, L.E.; Cowey, C.L.; et al. A Phase 2 Study of Glembatumumab Vedotin, an Antibody-drug Conjugate Targeting Glycoprotein NMB, in Patients with Advanced Melanoma. Cancer 2019, 125, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bent, M.; Eoli, M.; Sepulveda, J.M.; Smits, M.; Walenkamp, A.; Frenel, J.-S.; Franceschi, E.; Clement, P.M.; Chinot, O.; De Vos, F.; et al. INTELLANCE 2/EORTC 1410 Randomized Phase II Study of Depatux-M Alone and with Temozolomide vs Temozolomide or Lomustine in Recurrent EGFR Amplified Glioblastoma. Neuro Oncol. 2020, 22, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Reardon, D.A.; Lassman, A.B.; Merrell, R.; van den Bent, M.; Butowski, N.; Lwin, Z.; Wheeler, H.; Fichtel, L.; Scott, A.M.; et al. Safety, Pharmacokinetics, and Antitumor Response of Depatuxizumab Mafodotin as Monotherapy or in Combination with Temozolomide in Patients with Glioblastoma. Neuro Oncol. 2018, 20, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Arkenau, H.-T.; Guthrie, T.; Mekhail, T.; Cortinovis, D.; Antonuzzo, L.; Bruce, J.Y.; Gabrail, N.; Anderson, I.; Oh, S.C.; Oh, S.Y.; et al. 643TiP Open-Label, Phase II Study of Ladiratuzumab Vedotin (LV) for Unresectable Locally Advanced or Metastatic Solid Tumors. Ann. Oncol. 2021, 32, S671–S672. [Google Scholar] [CrossRef]
- Meisel, J.L.; Pluard, T.J.; Vinayak, S.; Stringer-Reasor, E.M.; Brown-Glaberman, U.; Dillon, P.M.; Basho, R.K.; Varadarajan, R.; O’Shaughnessy, J.; Han, H.S.; et al. Phase 1b/2 Study of Ladiratuzumab Vedotin (LV) in Combination with Pembrolizumab for First-Line Treatment of Triple-Negative Breast Cancer (SGNLVA-002, Trial in Progress). J. Clin. Oncol. 2022, 40, TPS1127. [Google Scholar] [CrossRef]
- Ebbinghaus, S.; Rubin, E.; Hersh, E.; Cranmer, L.D.; Bonate, P.L.; Fram, R.J.; Jekunen, A.; Weitman, S.; Hammond, L.A. A Phase I Study of the Dolastatin-15 Analogue Tasidotin (ILX651) Administered Intravenously Daily for 5 Consecutive Days Every 3 Weeks in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2005, 11, 7807–7816. [Google Scholar] [CrossRef]
- Riely, G.J.; Gadgeel, S.; Rothman, I.; Saidman, B.; Sabbath, K.; Feit, K.; Kris, M.G.; Rizvi, N.A. A Phase 2 Study of TZT-1027, Administered Weekly to Patients with Advanced Non-Small Cell Lung Cancer Following Treatment with Platinum-Based Chemotherapy. Lung Cancer 2007, 55, 181–185. [Google Scholar] [CrossRef]
- Spicka, I.; Ocio, E.M.; Oakervee, H.E.; Greil, R.; Banh, R.H.; Huang, S.-Y.; D’Rozario, J.M.; Dimopoulos, M.A.; Martínez, S.; Extremera, S.; et al. Randomized Phase III Study (ADMYRE) of Plitidepsin in Combination with Dexamethasone vs. Dexamethasone Alone in Patients with Relapsed/Refractory Multiple Myeloma. Ann. Hematol. 2019, 98, 2139–2150. [Google Scholar] [CrossRef]
- Landete, P.; Caliman-Sturdza, O.-A.; Lopez-Martin, J.A.; Preotescu, L.; Luca, M.-C.; Kotanidou, A.; Villares, P.; Iglesias, S.-P.; Guisado-Vasco, P.; Saiz-Lou, E.-M.; et al. A Phase III Randomized Controlled Trial of Plitidepsin, a Marine-Derived Compound, in Hospitalized Adults With Moderate COVID-19. Clin. Infect. Dis. 2024, 79, 910–919. [Google Scholar] [CrossRef]
- Therapeutic Goods Administration. Australian Public Assessment Report for Plitidepsin About the Therapeutic Goods Administration (TGA); Therapeutic Goods Administration: Woden, Australia, 2019. [Google Scholar]
- Le Gouill-Jaijarat, C.; Péréon, Y.; Leroy, M.; Lépine, O.; Loloum, A.; Peluchon, C.; Volteau, C.; Martineau, A.S.; Korner, S.; Perrault, C.; et al. PROPERTY: Study Protocol for a Randomized, Double-Blind, Multicenter Placebo-Controlled Trial Assessing Neurotoxicity in Patients with Metastatic Gastrointestinal Cancer Taking PHYCOCARE® during Oxaliplatin-Based Chemotherapy. Trials 2023, 24, 50. [Google Scholar] [CrossRef]
- OnKure Therapeutics. OnKure Therapeutics Announces Positive Topline First-in-Human Phase 1 Results for OKI-179. Press Release, 14 December 2021.. Available online: https://onkuretherapeutics.com/onkure-therapeutics-announces-positive-topline-first-in-human-phase-1-results-for-oki-179/ (accessed on 27 May 2025).
- Banerjee, S.; Oza, A.M.; Birrer, M.J.; Hamilton, E.P.; Hasan, J.; Leary, A.; Moore, K.N.; Mackowiak-Matejczyk, B.; Pikiel, J.; Ray-Coquard, I.; et al. Anti-NaPi2b Antibody–Drug Conjugate Lifastuzumab Vedotin (DNIB0600A) Compared with Pegylated Liposomal Doxorubicin in Patients with Platinum-Resistant Ovarian Cancer in a Randomized, Open-Label, Phase II Study. Ann. Oncol. 2018, 29, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; Vaishampayan, U.N.; LoRusso, P.; Barton, J.; Hua, S.; Reich, S.D.; Shazer, R.; Taylor, C.T.; Xuan, D.; Borghaei, H. First-in-Human Trial of an Anti-5T4 Antibody-Monomethylauristatin Conjugate, PF-06263507, in Patients with Advanced Solid Tumors. Investig. New Drugs 2017, 35, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Bar, J.; Horinouchi, H.; Goldman, J.; Moiseenko, F.; Filippova, E.; Cicin, I.; Ciuleanu, T.; Daaboul, N.; Liu, C.; et al. Telisotuzumab Vedotin Monotherapy in Patients With Previously Treated C-Met Protein–Overexpressing Advanced Nonsquamous EGFR -Wildtype Non–Small Cell Lung Cancer in the Phase II LUMINOSITY Trial. J. Clin. Oncol. 2024, 42, 3000–3011. [Google Scholar] [CrossRef] [PubMed]
- Jordyn Sava FDA Grants Telisotuzumab Vedotin Accelerated Approval in C-MET + NSCLC. Available online: https://www.targetedonc.com/view/fda-grants-telisotuzumab-vedotin-accelerated-approval-in-c-met-nsclc (accessed on 19 May 2025).
- Chang, H.W.; Wang, J.; Liu, H.; Xing, C.; Chen, J.; Frey, G.; Boyle, W.J.; Short, J.M. Preclinical Development of Mecbotamab Vedotin (BA3011), a Novel, AXL-Specific Conditional Active Biologic Antibody–Drug Conjugate. Antib. Ther. 2025, 8, tbaf006. [Google Scholar] [CrossRef]
- BioAtla BioAtla Presented Phase 2 Clinical Trial Data at the IASLC 2023 North America Conference on Lung Cancer and Virtual KOL Event. Available online: https://ir.bioatla.com/news-releases/news-release-details/bioatla-presented-phase-2-clinical-trial-data-iaslc-2023-north#:~:text=BioAtla%20Presented%20Phase%202%20Clinical,squamous (accessed on 4 December 2023).
- BioAtla. BioAtla Granted FDA Fast Track Designation for Ozuriftamab Vedotin (CAB-ROR2-ADC) for Treatment of Patients with Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck. News Release, 23 January 2024.. Available online: https://ir.bioatla.com/news-releases/news-release-details/bioatla-granted-fda-fast-track-designation-ozuriftamab-vedotin/ (accessed on 27 May 2025).
- Mao, D.; Okada, B.K.; Wu, Y.; Xu, F.; Seyedsayamdost, M.R. Recent Advances in Activating Silent Biosynthetic Gene Clusters in Bacteria. Curr. Opin. Microbiol. 2018, 45, 156–163. [Google Scholar] [CrossRef]
- Taton, A.; Ecker, A.; Diaz, B.; Moss, N.A.; Anderson, B.; Reher, R.; Leão, T.F.; Simkovsky, R.; Dorrestein, P.C.; Gerwick, L.; et al. Heterologous Expression of Cryptomaldamide in a Cyanobacterial Host. ACS Synth. Biol. 2020, 9, 3364–3376. [Google Scholar] [CrossRef]
- Greunke, C.; Duell, E.R.; D’Agostino, P.M.; Glöckle, A.; Lamm, K.; Gulder, T.A.M. Direct Pathway Cloning (DiPaC) to Unlock Natural Product Biosynthetic Potential. Metab. Eng. 2018, 47, 334–345. [Google Scholar] [CrossRef]
- Ke, J.; Robinson, D.; Wu, Z.-Y.; Kuftin, A.; Louie, K.; Kosina, S.; Northen, T.; Cheng, J.-F.; Yoshikuni, Y. CRAGE-CRISPR Facilitates Rapid Activation of Secondary Metabolite Biosynthetic Gene Clusters in Bacteria. Cell Chem. Biol. 2022, 29, 696–710.e4. [Google Scholar] [CrossRef]
- Galica, T.; Borbone, N.; Mareš, J.; Kust, A.; Caso, A.; Esposito, G.; Saurav, K.; Hájek, J.; Řeháková, K.; Urajová, P.; et al. Cyanochelins, an Overlooked Class of Widely Distributed Cyanobacterial Siderophores, Discovered by Silent Gene Cluster Awakening. Appl. Environ. Microbiol. 2021, 87, e03128-20. [Google Scholar] [CrossRef]
- Sorrels, C.M.; Proteau, P.J.; Gerwick, W.H. Organization, Evolution, and Expression Analysis of the Biosynthetic Gene Cluster for Scytonemin, a Cyanobacterial UV-Absorbing Pigment. Appl. Environ. Microbiol. 2009, 75, 4861–4869. [Google Scholar] [CrossRef]
- Soule, T.; Palmer, K.; Gao, Q.; Potrafka, R.M.; Stout, V.; Garcia-Pichel, F. A Comparative Genomics Approach to Understanding the Biosynthesis of the Sunscreen Scytonemin in Cyanobacteria. BMC Genom. 2009, 10, 336. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Woo, H.M. CRISPRi-DCas12a: A DCas12a-Mediated CRISPR Interference for Repression of Multiple Genes and Metabolic Engineering in Cyanobacteria. ACS Synth. Biol. 2020, 9, 2351–2361. [Google Scholar] [CrossRef]
- Germann, A.T.; Nakielski, A.; Dietsch, M.; Petzel, T.; Moser, D.; Triesch, S.; Westhoff, P.; Axmann, I.M. A Systematic Overexpression Approach Reveals Native Targets to Increase Squalene Production in Synechocystis Sp. PCC 6803. Front. Plant Sci. 2023, 14, 1024981. [Google Scholar] [CrossRef]
- Usai, G.; Cordara, A.; Re, A.; Polli, M.F.; Mannino, G.; Bertea, C.M.; Fino, D.; Pirri, C.F.; Menin, B. Combining Metabolite Doping and Metabolic Engineering to Improve 2-Phenylethanol Production by Engineered Cyanobacteria. Front. Bioeng. Biotechnol. 2022, 10, 1005960. [Google Scholar] [CrossRef]
- Knoot, C.J.; Khatri, Y.; Hohlman, R.M.; Sherman, D.H.; Pakrasi, H.B. Engineered Production of Hapalindole Alkaloids in the Cyanobacterium Synechococcus Sp. UTEX 2973. ACS Synth. Biol. 2019, 8, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, P.M.; Gulder, T.A.M. Direct Pathway Cloning Combined with Sequence- and Ligation-Independent Cloning for Fast Biosynthetic Gene Cluster Refactoring and Heterologous Expression. ACS Synth. Biol. 2018, 7, 1702–1708. [Google Scholar] [CrossRef]
- Niedermeyer, T.H.J.; Daily, A.; Swiatecka-Hagenbruch, M.; Moscow, J.A. Selectivity and Potency of Microcystin Congeners against OATP1B1 and OATP1B3 Expressing Cancer Cells. PLoS ONE 2014, 9, e91476. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Lee, K.; Jang, J.W.; Hahn, J.-S. Metabolic Engineering of Saccharomyces Cerevisiae for Production of Shinorine, a Sunscreen Material, from Xylose. ACS Synth. Biol. 2019, 8, 346–357. [Google Scholar] [CrossRef]
- Zhao, M.; Ma, J.; Zhang, L.; Qi, H. Engineering Strategies for Enhanced Heterologous Protein Production by Saccharomyces Cerevisiae. Microb. Cell Fact. 2024, 23, 32. [Google Scholar] [CrossRef]
- Kim, E.J.; Lee, J.H.; Choi, H.; Pereira, A.R.; Ban, Y.H.; Yoo, Y.J.; Kim, E.; Park, J.W.; Sherman, D.H.; Gerwick, W.H.; et al. Heterologous Production of 4-O-Demethylbarbamide, a Marine Cyanobacterial Natural Product. Org. Lett. 2012, 14, 5824–5827. [Google Scholar] [CrossRef]
- Ongley, S.E.; Bian, X.; Zhang, Y.; Chau, R.; Gerwick, W.H.; Müller, R.; Neilan, B.A. High Titer Heterologous Production of Lyngbyatoxin in E. Coli, a Protein Kinase C Activator from an Uncultured Marine Cyanobacterium. ACS Chem. Biol. 2013, 8, 1888–1893. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Lee, Y.; Kim, J.H.; Kim, G.; Kim, H.; Kim, W.; Cho, S.; Palsson, B.O.; Cho, B.-K. Streptomyces as Microbial Chassis for Heterologous Protein Expression. Front. Bioeng. Biotechnol. 2021, 9, 804295. [Google Scholar] [CrossRef] [PubMed]
- Calcott, M.J.; Owen, J.G.; Lamont, I.L.; Ackerley, D.F. Biosynthesis of Novel Pyoverdines by Domain Substitution in a Nonribosomal Peptide Synthetase of Pseudomonas Aeruginosa. Appl. Environ. Microbiol. 2014, 80, 5723–5731. [Google Scholar] [CrossRef]
- Thong, W.L.; Zhang, Y.; Zhuo, Y.; Robins, K.J.; Fyans, J.K.; Herbert, A.J.; Law, B.J.C.; Micklefield, J. Gene Editing Enables Rapid Engineering of Complex Antibiotic Assembly Lines. Nat. Commun. 2021, 12, 6872. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.T.; Ritz, D.; Gu, J.-Q.; Alexander, D.; Chu, M.; Miao, V.; Brian, P.; Baltz, R.H. Combinatorial Biosynthesis of Novel Antibiotics Related to Daptomycin. Proc. Natl. Acad. Sci. USA 2006, 103, 17462–17467. [Google Scholar] [CrossRef]
- Bradley, S.A.; Zhang, J.; Jensen, M.K. Deploying Microbial Synthesis for Halogenating and Diversifying Medicinal Alkaloid Scaffolds. Front. Bioeng. Biotechnol. 2020, 8, 594126. [Google Scholar] [CrossRef]
- Ouyang, X.; D’Agostino, P.M.; Wahlsten, M.; Delbaje, E.; Jokela, J.; Permi, P.; Gaiani, G.; Poso, A.; Bartos, P.; Gulder, T.A.M.; et al. Direct Pathway Cloning and Expression of the Radiosumin Biosynthetic Gene Cluster. Org. Biomol. Chem. 2023, 21, 4893–4908. [Google Scholar] [CrossRef]
- Basitta, P.; Westrich, L.; Rösch, M.; Kulik, A.; Gust, B.; Apel, A.K. AGOS: A Plug-and-Play Method for the Assembly of Artificial Gene Operons into Functional Biosynthetic Gene Clusters. ACS Synth. Biol. 2017, 6, 817–825. [Google Scholar] [CrossRef]
- Kalkreuter, E.; Bingham, K.S.; Keeler, A.M.; Lowell, A.N.; Schmidt, J.J.; Sherman, D.H.; Williams, G.J. Computationally-Guided Exchange of Substrate Selectivity Motifs in a Modular Polyketide Synthase Acyltransferase. Nat. Commun. 2021, 12, 2193. [Google Scholar] [CrossRef]
- Choi, S.Y.; Sim, S.J.; Ko, S.C.; Son, J.; Lee, J.S.; Lee, H.J.; Chang, W.S.; Woo, H.M. Scalable Cultivation of Engineered Cyanobacteria for Squalene Production from Industrial Flue Gas in a Closed Photobioreactor. J. Agric. Food Chem. 2020, 68, 10050–10055. [Google Scholar] [CrossRef]
- Goering, A.W.; Li, J.; McClure, R.A.; Thomson, R.J.; Jewett, M.C.; Kelleher, N.L. In Vitro Reconstruction of Nonribosomal Peptide Biosynthesis Directly from DNA Using Cell-Free Protein Synthesis. ACS Synth. Biol. 2017, 6, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Huang, S.; Liu, W.-Q.; Karim, A.S.; Jewett, M.C.; Li, J. Total in Vitro Biosynthesis of the Nonribosomal Macrolactone Peptide Valinomycin. Metab. Eng. 2020, 60, 37–44. [Google Scholar] [CrossRef]
- Lai, Q.; Wu, M.; Wang, R.; Lai, W.; Tao, Y.; Lu, Y.; Wang, Y.; Yu, L.; Zhang, R.; Peng, Y.; et al. Cryptophycin-55/52 Based Antibody-Drug Conjugates: Synthesis, Efficacy, and Mode of Action Studies. Eur. J. Med. Chem. 2020, 199, 112364. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.B.; Corcoran, R.B. Therapeutic Strategies to Target RAS-Mutant Cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.B.; Fece de la Cruz, F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRASG12C Inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef]
- Popin, R.V.; Alvarenga, D.O.; Castelo-Branco, R.; Fewer, D.P.; Sivonen, K. Mining of Cyanobacterial Genomes Indicates Natural Product Biosynthetic Gene Clusters Located in Conjugative Plasmids. Front. Microbiol. 2021, 12, 684565. [Google Scholar] [CrossRef]
- Castelo-Branco, R.; Pereira, J.P.; Freitas, S.; Preto, M.; Vieira, A.R.; Morais, J.; Leão, P.N. Genome-Informed Discovery of Monchicamides A–K: Cyanobactins from the Microcoleaceae Cyanobacterium LEGE 16532. J. Nat. Prod. 2025, 88, 86–93. [Google Scholar] [CrossRef]
- Schmidt, J.J.; Khatri, Y.; Brody, S.I.; Zhu, C.; Pietraszkiewicz, H.; Valeriote, F.A.; Sherman, D.H. A Versatile Chemoenzymatic Synthesis for the Discovery of Potent Cryptophycin Analogs. ACS Chem. Biol. 2020, 15, 524–532. [Google Scholar] [CrossRef]
- Bubik, A.; Frangež, R.; Žužek, M.C.; Gutiérrez-Aguirre, I.; Lah, T.T.; Sedmak, B. Cyanobacterial Cyclic Peptides Can Disrupt Cytoskeleton Organization in Human Astrocytes—A Contribution to the Understanding of the Systemic Toxicity of Cyanotoxins. Toxins 2024, 16, 374. [Google Scholar] [CrossRef]
- Frachet, S.; Danigo, A.; Duchesne, M.; Richard, L.; Sturtz, F.; Magy, L.; Demiot, C. A Mouse Model of Sensory Neuropathy Induced by a Long Course of Monomethyl-Auristatin E Treatment. Toxicol. Appl. Pharmacol. 2023, 474, 116624. [Google Scholar] [CrossRef]
- Chernoff, N.; Hill, D.; Lang, J.; Schmid, J.; Le, T.; Farthing, A.; Huang, H. The Comparative Toxicity of 10 Microcystin Congeners Administered Orally to Mice: Clinical Effects and Organ Toxicity. Toxins 2020, 12, 403. [Google Scholar] [CrossRef] [PubMed]
- Samuel, J.N.; Booth, C.M.; Eisenhauer, E.; Brundage, M.; Berry, S.R.; Gyawali, B. Association of Quality-of-Life Outcomes in Cancer Drug Trials with Survival Outcomes and Drug Class. JAMA Oncol. 2022, 8, 879. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.; Sridhar, S.S.; Zhang, J.; Smith, D.; Ruether, D.; Flaig, T.W.; Baranda, J.; Lang, J.; Plimack, E.R.; Sangha, R.; et al. EV-101: A Phase I Study of Single-Agent Enfortumab Vedotin in Patients with Nectin-4–Positive Solid Tumors, Including Metastatic Urothelial Carcinoma. J. Clin. Oncol. 2020, 38, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Radford, J.; Connors, J.M.; Długosz-Danecka, M.; Kim, W.-S.; Gallamini, A.; Ramchandren, R.; Friedberg, J.W.; Advani, R.; Hutchings, M.; et al. Overall Survival with Brentuximab Vedotin in Stage III or IV Hodgkin’s Lymphoma. N. Engl. J. Med. 2022, 387, 310–320. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Peptide (Source) | Structural Class | Primary Mechanisms |
| Microcystin-LR (Microcystis) | Cyclic heptapeptide (depsipeptide) | Inhibits serine/threonine phosphatases PP1/PP2A; triggers rapid cytotoxicity |
| Laxaphycin A/B (Anabaena) | Cyclic lipopeptides | Disrupt cellular membranes (synergistic cytotoxic action) |
| Somocystinamide A (Lyngbya) | Dimeric cyclic lipopeptide | Induces extrinsic apoptosis (caspase-8 activation); anti-angiogenic (targets tumor vasculature) |
| Grassypeptolide A (Lyngbya) | Cyclic depsipeptide | Binds to actin filaments; destabilizes cytoskeleton (mitotic arrest and apoptosis) |
| Lyngbyabellin A (Lyngbya) | Cyclic depsipeptide | Binds to actin (some analogs bind tubulin); disrupts cytoskeletal dynamics |
| Dolastatin 10 (Symploca) | Linear pentapeptide (NRPS-derived) | Binds β-tubulin (vinca domain); prevents microtubule assembly (mitotic arrest) |
| Symplostatin 1 (Symploca) | Linear peptide (dolastatin analog) | Binds β-tubulin similar to dolastatin 10; precursor to auristatin analogs used in ADCs |
| Cryptophycin-52 (Nostoc) | Cyclic depsipeptide | Binds tubulin (distinct site); causes microtubule depolymerization (mitotic collapse) |
| Curacin A (Lyngbya) | Hybrid polyketide–peptide | Binds β-tubulin (colchicine site); inhibits microtubule polymerization (mitotic arrest) |
| Largazole (Symploca/Caldora) | Cyclic depsipeptide | Inhibits class I histone deacetylases (HDAC1/2/3); causes hyperacetylation of histones (epigenetic reprogramming) |
| Carmaphycin A/B ( Symploca) | Linear peptide (epoxyketone) | Irreversibly inhibits the proteasome (via epoxyketone warhead); blocks protein degradation, inducing cell death |
| Apratoxin A (Moorea/Lyngbya) | Cyclic depsipeptide (PKS-NRPS hybrid) | Blocks Sec61 translocon (inhibits co-translational protein translocation into ER); downregulates growth factor receptors |
| Coibamide A (Leptolyngbya) | Cyclic depsipeptide (PKS-NRPS hybrid) | Blocks Sec61 translocon; induces caspase-dependent apoptosis; disrupts mTOR signaling and angiogenesis |
| Agent (Type) | Indication | Regulatory Status | Key Efficacy/Safety Findings |
|---|---|---|---|
| Brentuximab vedotin (ADC, MMAE from dolastatin) | Hodgkin lymphoma, ALCL (CD30⁺ lymphomas) | Approved (US/EU 2011–12; expanded 2018+) | Improves PFS/OS in CD30⁺ lymphomas; first auristatin ADC to reach market/Common tox: peripheral neuropathy. |
| Polatuzumab vedotin (ADC, MMAE) | R/R DLBCL (with BR chemo) | Approved (FDA 2019; EU 2020) | +BR improved CR 40% vs. 18% and median OS 12.4 vs. 4.7 mo over BR alone. Priority review granted due to 58% lower risk of death/Notable tox: cytopenias, neuropathy. |
| Enfortumab vedotin (ADC, MMAE) | Metastatic urothelial carcinoma | Approved (FDA 2019; EU 2022) | ORR 44% (12% CR) in post-platinum/IO bladder cancer; confirmed OS benefit vs. chemo in Phase III/Tox: neuropathy, rash; rare serious hyperglycemia. |
| Tisotumab vedotin (ADC, MMAE) | Recurrent/metastatic cervical CA | Approved (FDA 2021) | ORR 24% (7% CR); median DOR 8.3 mo in refractory cervical cancer (single-arm Phase II); OS benefit vs. chemo (Phase III) /Tox: ocular (boxed warning for conjunctival/corneal injury). |
| Belantamab mafodotin (ADC, MMAF) | Relapsed multiple myeloma (BCMA-targeted) | Approved (FDA/EMA 2020); Withdrawn (US 2022) | ~31% ORR in heavily pretreated myeloma (monotherapy) with some durable responses/Notable toxicity: corneal damage (keratopathy). Approval withdrawn after Phase III trial showed no PFS benefit over standard therapy. |
| Disitamab vedotin (ADC, MMAE) | HER2+ gastric/GEJ adenocarcinoma | Approved (China 2021) | ~30% ORR in HER2 IHC 2+ or 3+ gastric cancer after 2+ lines (conditional approval). Showing activity in HER2-low tumors as well; Phase II completed (ORR ~25% gastric; ~50% bladder)/Key tox: nausea, marrow suppression, liver enzyme elevations. |
| Agent (Type) | Target | Indications | Phase | Status/Key Outcomes |
|---|---|---|---|---|
| Glembatumumab vedotin (ADC, MMAE payload) | gpNMB | Metastatic triple-negative breast cancer (TNBC); also tested in melanoma | Phase 2b (METRIC, NCT01997333) | No benefit over chemo in TNBC (median PFS 2.9 vs. 2.8 mo); significant toxicity (rash, neutropenia); discontinued after failing primary endpoint. Minimal activity seen in melanoma as well. |
| Depatuxizumab mafodotin (ADC, MMAF payload) | EGFR | EGFR-amplified glioblastoma | Phase 3 (INTELLANCE-1, NCT02573324) | No survival improvement when added to standard chemoradiation; Phase III trial in newly diagnosed GBM was negative, leading to termination of the program. |
| Ladiratuzumab vedotin (ADC, MMAE) | LIV-1 | Triple-negative breast cancer; LIV-1–expressing solid tumors | Phase 1b/2 (NCT03310957) | Ongoing. Manageable toxicity and preliminary efficacy in TNBC. In first-line TNBC (PD-L1+), ladiratuzumab + pembrolizumab showed ~33% ORR, warranting further development. |
| Tasidotin (ILX-651; synthetic peptide) | Tubulin | Advanced solid tumors (refractory) | Phase 1 | Dose-limiting neutropenia at higher doses. No dramatic responses; some anti-tumor activity with stable disease observed. Notably, melanoma CR reported, but no further development beyond Phase I. |
| Soblidotin (TZT-1027) | Tubulin | Non–small cell lung cancer (refractory NSCLC) | Phase 2 | No objective responses in Phase II; median time to progression ~1.5 months. Showed minimal efficacy, and development was halted for NSCLC. |
| Lifastuzumab vedotin (DNIB0600A, ADC) | NaPi2b | Non-sq NSCLC; Platinum-resistant ovarian cancer | Phase 2 (NCT01991210) | LIFA achieved higher ORR (34% vs. 15%) than chemo, but PFS benefit was modest (5.3 vs. 3.1 mo) and not statistically significant. Consequently, the ADC was discontinued for lack of clear superiority. |
| PF-06263507 (anti-5T4 ADC) | 5T4 | Advanced solid tumors (5T4-expressing; lung, breast, ovarian) | Phase 1 (NCT01891669) | First-in-human dose escalation completed; ocular toxicities (e.g., photophobia, conjunctivitis) were dose-limiting. Demonstrated insufficient clinical activity; the program was discontinued after Phase I. |
| Telisotuzumab vedotin (“Teliso-V”, ADC) | c-MET | c-MET–overexpressing NSCLC (EGFR wild-type) | Phase 2 (LUMINOSITY, NCT03539536) | Achieved durable responses in c-MET high non-squamous NSCLC. In c-MET high patients, ORR ~53% with prolonged benefit. Received FDA accelerated approval in 2023 for advanced NSCLC with high c-MET expression. Common toxicities: fatigue, peripheral neuropathy (manageable). |
| Mecbotamab vedotin (BA3011, CAB-ADC) | AXL | Solid tumors with AXL expression | Phase 2 (NCT03425279) | Conditionally active ADC (pH-dependent tumor targeting). Interim Phase II results in refractory NSCLC show promising efficacy (objective responses in heavily pretreated patients). Development ongoing; well-tolerated with limited off-tumor toxicity due to CAB activation mechanism. |
| Ozuriftamab vedotin (BA3021, CAB-ADC) | ROR2 | ROR2-positive cancers (melanoma; head and neck SCC; NSCLC) | Phase 2 (NCT03504488) | Conditionally active (CAB) ADC targeting ROR2. Early Phase II data in metastatic head and neck cancer showed encouraging response signals, leading to FDA Fast Track designation. Trials ongoing to confirm efficacy in ROR2-expressing tumors; toxicities so far manageable and mostly mild. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.; Nihan, K.; Kwon, Y.R. Cyanobacterial Peptides in Anticancer Therapy: A Comprehensive Review of Mechanisms, Clinical Advances, and Biotechnological Innovation. Mar. Drugs 2025, 23, 233. https://doi.org/10.3390/md23060233
Lee H, Nihan K, Kwon YR. Cyanobacterial Peptides in Anticancer Therapy: A Comprehensive Review of Mechanisms, Clinical Advances, and Biotechnological Innovation. Marine Drugs. 2025; 23(6):233. https://doi.org/10.3390/md23060233
Chicago/Turabian StyleLee, Heayyean, Khuld Nihan, and Yale Ryan Kwon. 2025. "Cyanobacterial Peptides in Anticancer Therapy: A Comprehensive Review of Mechanisms, Clinical Advances, and Biotechnological Innovation" Marine Drugs 23, no. 6: 233. https://doi.org/10.3390/md23060233
APA StyleLee, H., Nihan, K., & Kwon, Y. R. (2025). Cyanobacterial Peptides in Anticancer Therapy: A Comprehensive Review of Mechanisms, Clinical Advances, and Biotechnological Innovation. Marine Drugs, 23(6), 233. https://doi.org/10.3390/md23060233