

Chondroitin Sulfate as a Lysosomal Enhancer Attenuates Lipid-Driven Inflammation via Lipophagy and Mitophagy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Establishment of a NaOl-Induced NASH Cell Model and Ameliorative Efficacy
2.1.1. NaOl-Induced Lipid Accumulation and Oxidative Stress in HepG2 Cells
2.1.2. CS Restores Lipid Metabolism and Alleviates Oxidative Damage in NaOl-Induced HepG2 Cells
2.2. Subcellular Regulatory Mechanisms of CS in Ameliorating the NASH Cell Model
2.2.1. CS Reduces Intracellular LDs and ROS Overproduction
2.2.2. CS Enhances Lysosomal Acidification to Promote Lipophagy
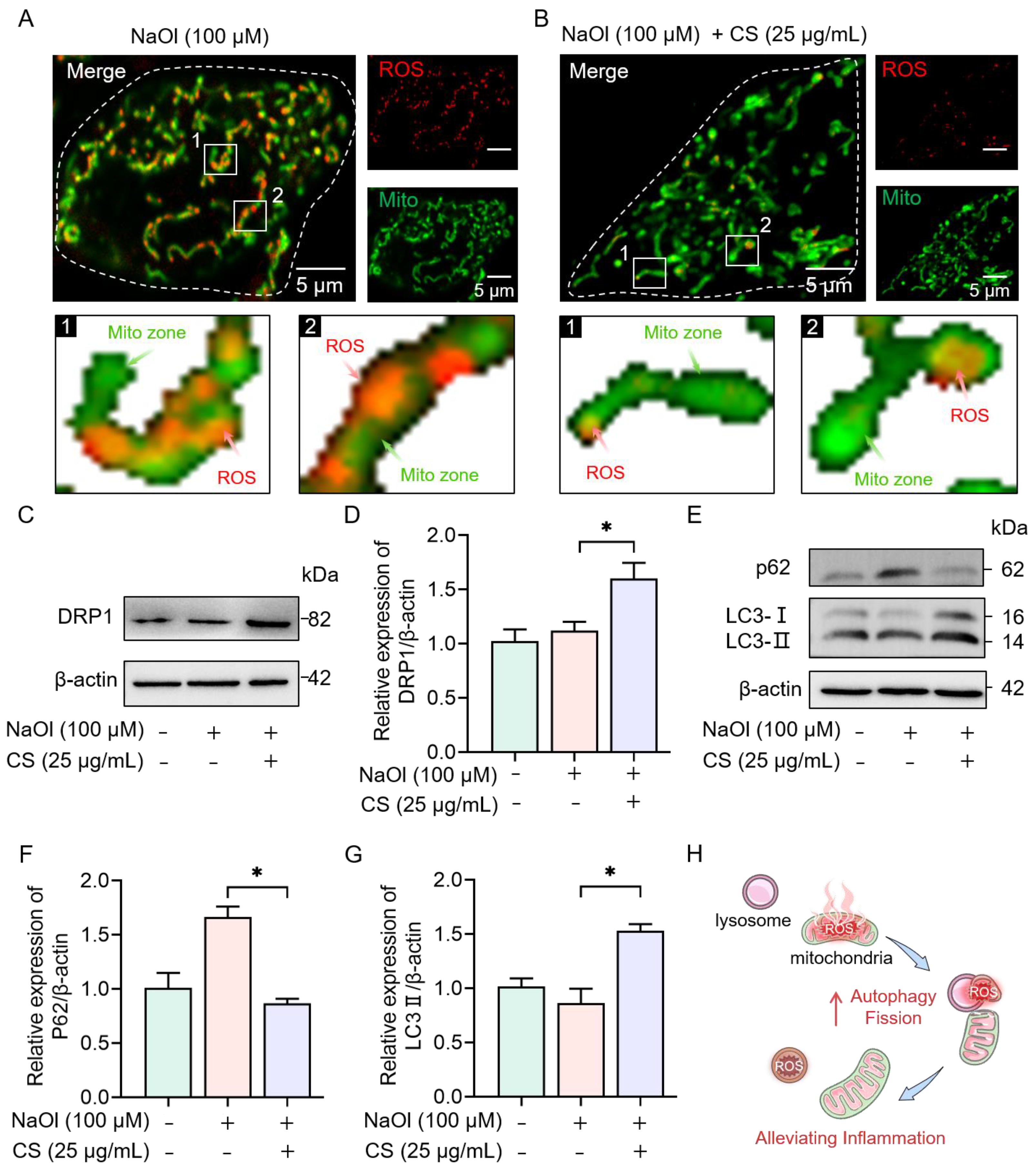
2.2.3. CS Activates Autophagy and Mitochondrial Fission to Eliminate ROS
2.3. Essential Role of Lysosomal Activity in CS-Mediated Regulation of the NASH Cell Model
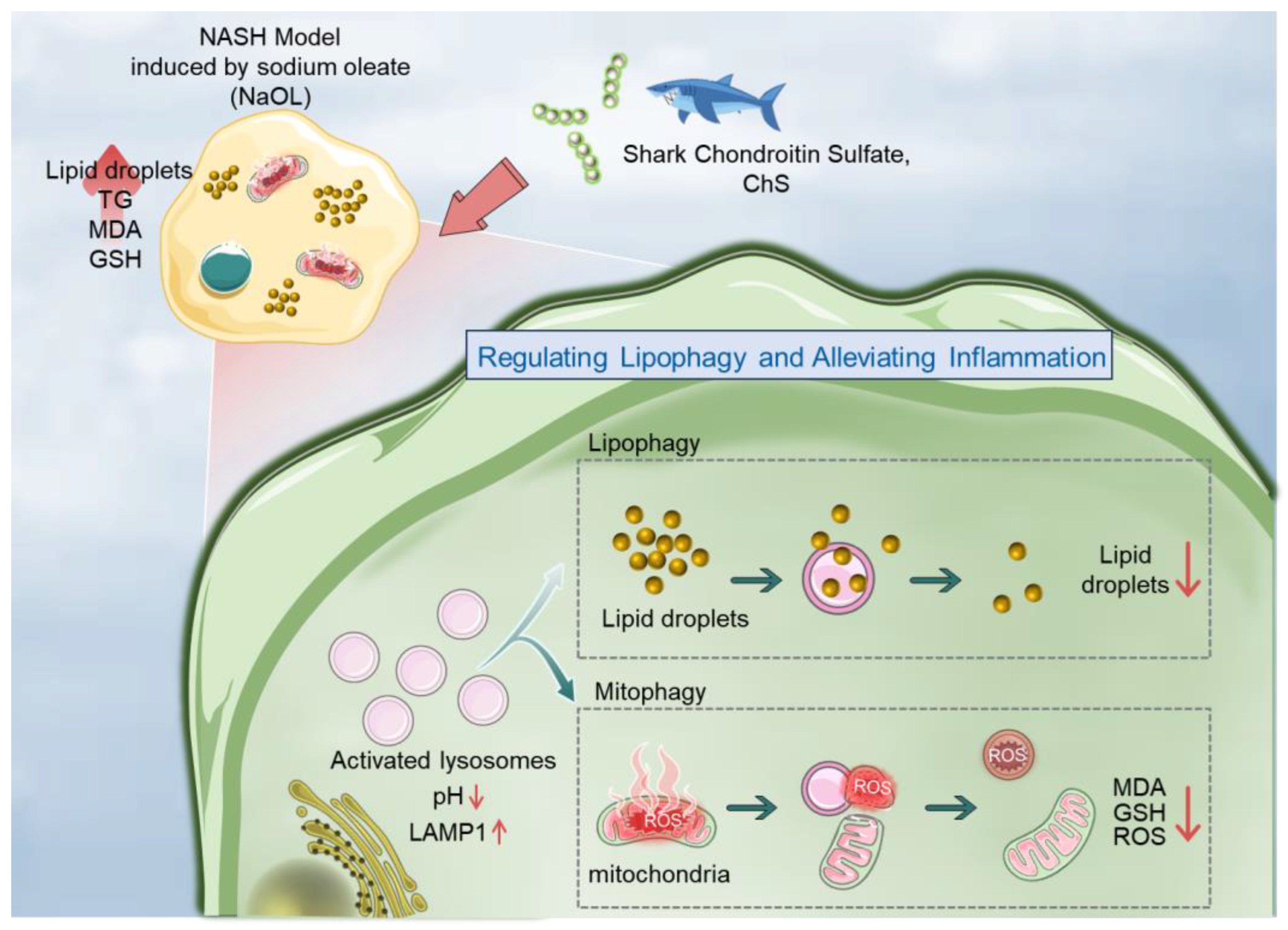
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Treatment
4.3. Structural Characterization of CS
4.3.1. FTIR Spectroscopic Analysis
4.3.2. NMR Spectroscopy
4.3.3. Molecular Weight Measurement
4.3.4. Determination of Sulfate Group Content
4.3.5. Monosaccharide Composition Analysis
4.4. Determination of TG Content
4.5. Determination of MDA and GSH Levels
4.6. ORO Staining Assay
4.7. Fluorescence Imaging of Cells
4.8. Detection of Lysosomal pH
4.9. Quantitative Detection of LDs and ROS Level
4.10. Western Blot Analysis
4.11. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NASH | Non-alcoholic steatohepatitis |
| NAFLD | Non-alcoholic fatty liver disease |
| CS | Chondroitin sulfate |
| NaOl | Sodium oleate |
| MDA | Malondialdehyde |
| GSH | Glutathione |
| ORO | Oil Red O |
| FTIR | Fourier transform infrared spectroscop |
| NMR | Nuclear magnetic resonance |
| GPC-RI-MALLS | Chromatography-refractive index-multiangle laser light scattering |
| DRP1 | Dynamin-related protein 1 |
| ROS | Reactive oxygen species |
| LDs | Lipid droplets |
| LAMP1 | Lysosomal membrane-associated protein 1 |
| CQ | Chloroquine |
| Baf A1 | Bafilomycin A1 |
| CCK-8 | Cell Counting Kit-8 |
References
- Rinella, M.E.; Sanyal, A.J. Management of NAFLD: A stage-based approach. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Cholankeril, G.; Kramer, J.R.; Chu, J.; Yu, X.; Balakrishnan, M.; Li, L.; El-Serag, H.B.; Kanwal, F. Longitudinal changes in fibrosis markers are associated with risk of cirrhosis and hepatocellular carcinoma in non- alcoholic fatty liver disease. J. Hepatol. 2023, 78, 493–500. [Google Scholar] [CrossRef]
- Quek, J.; Chan, K.E.; Wong, Z.Y.; Tan, C.; Tan, B.; Lim, W.H.; Tan, D.; Tang, A.; Tay, P.; Xiao, J.; et al. Global prevalence of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in the overweight and obese population: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2023, 8, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Huby, T.; Gautier, E.L. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat. Rev. Immunol. 2022, 22, 429–443. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhao, S.; Yu, B. Intracellular role of exchangeable apolipoproteins in energy homeostasis, obesity and non-alcoholic fatty liver disease. Biol. Rev. 2015, 90, 367–376. [Google Scholar] [CrossRef]
- Katsagoni, C.N.; Georgoulis, M.; Papatheodoridis, G.V.; Panagiotakos, D.B.; Kontogianni, M.D. Effects of lifestyle interventions on clinical characteristics of patients with non-alcoholic fatty liver disease: A meta-analysis. Metabolism 2017, 68, 119–132. [Google Scholar] [CrossRef]
- Keam, S.J. Resmetirom: First Approval. Drugs 2024, 84, 729–735. [Google Scholar] [CrossRef]
- Huang, J.; Dai, C.; Huang, C.; Tsai, P.; Yeh, M.; Hsu, P.; Huang, S.; Bair, M.; Hou, N.; Huang, C.; et al. First-in-Asian double-blind randomized trial to assess the efficacy and safety of insulin sensitizer in nonalcoholic steatohepatitis patients. Hepatol. Int. 2021, 15, 1136–1147. [Google Scholar] [CrossRef]
- Armstrong, M.; Hull, D.; Guo, K.; Barton, D.; Yu, J.; Tomlinson, J.; Newsome, P. Effect of liraglutide on adipose insulin resistance and hepatic de-novo lipogenesis in non-alcoholic steatohepatitis: Substudy of a phase 2, randomised place-bo-controlled trial. Lancet 2014, 383, 21. [Google Scholar] [CrossRef]
- Ayada, I.; van Kleef, L.A.; Zhang, H.; Liu, K.; Li, P.; Abozaid, Y.J.; Lavrijsen, M.; Janssen, H.L.A.; van der Laan, L.J.W.; Ghanbari, M.; et al. Dissecting the multifaceted impact of statin use on fatty liver disease: A multidimensional study. EBioMedicine 2023, 87, 104392. [Google Scholar] [CrossRef] [PubMed]
- Scorletti, E.; Carr, R.M.M. A new perspective on NAFLD: Focusing on lipid droplets. J. Hepatol. 2022, 76, 934–945. [Google Scholar] [CrossRef]
- Li, Y.; Xie, Z.; Song, Q.; Li, J. Mitochondria homeostasis: Biology and involvement in hepatic steatosis to NASH. Acta Pharmacol. Sin. 2022, 43, 1141–1155. [Google Scholar] [CrossRef]
- Du, J.; Ji, Y.; Qiao, L.; Liu, Y.; Lin, J. Cellular endo-lysosomal dysfunction in the pathogenesis of non-alcoholic fatty liver disease. Liver Int. 2020, 40, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Talari, N.K.; Mattam, U.; Meher, N.K.; Paripati, A.K.; Mahadev, K.; Krishnamoorthy, T.; Sepuri, N. Lipid-droplet associated mitochondria promote fatty-acid oxidation through a distinct bioenergetic pattern in male Wistar rats. Nat. Commun. 2023, 14, 766. [Google Scholar] [CrossRef]
- Ma, X.; Qian, H.; Chen, A.; Ni, H.; Ding, W. Perspectives on mitochondria-ER and mitochondria-lipid droplet contact in hepatocytes and hepatic lipid metabolism. Cells 2021, 10, 2273. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tan, S.; Nicolas, V.; Bauvy, C.; Yang, N.; Zhang, J.; Xue, Y.; Codogno, P.; Shen, H. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013, 23, 508–523. [Google Scholar] [CrossRef]
- Guo, Y.; Li, D.; Zhang, S.; Yang, Y.; Liu, J.; Wang, X.; Liu, C.; Milkie, D.E.; Moore, R.P.; Tulu, U.S.; et al. Visualizing intracellular organelle and cytoskeletal interactions at nanoscale resolution on millisecond timescales. Cell 2018, 175, 1430. [Google Scholar] [CrossRef]
- Tian, M.; Zhan, J.; Lin, W. Single fluorescent probes enabling simultaneous visualization of duple organelles: Design principles, mechanisms, and applications. Coord. Chem. Rev. 2022, 451, 214266. [Google Scholar] [CrossRef]
- Sun, T.; Shao, H.; Wang, R.; Zeng, Q.; Shao, X.; Liu, F.; Li, G.; Ling, P. Single-organelle visualization tracking natural glycosaminoglycans within mitochondria-lysosome crosstalk for inflammatory homeostasis. Int. J. Biol. Macromol. 2025, 303, 140362. [Google Scholar] [CrossRef]
- Melgar-Lesmes, P.; Garcia-Polite, F.; Del-Rey-Puech, P.; Rosas, E.; Dreyfuss, J.L.; Montell, E.; Verges, J.; Edelman, E.R.; Balcells, M. Treatment with chondroitin sulfate to modulate inflammation and atherogenesis in obesity. Atherosclerosis 2016, 245, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; Kwan, T.S.; Pelletier, J.P. Effects of chondroitin sulfate in the pathophysiology of the osteoarthritic joint: A narrative review. Osteoarthr. Cartil. 2010, 18, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.; Lu, H.; Liu, P.; Zhang, Y.; Zhang, L.; Ren, Q. A chondroitin sulfate purified from shark cartilage and bovine serum albumin interaction activity. Int. J. Biol. Macromol. 2024, 260, 129499. [Google Scholar] [CrossRef]
- Lopez-Senra, E.; Casal-Beiroa, P.; Lopez-Alvarez, M.; Serra, J.; Gonzalez, P.; Valcarcel, J.; Vazquez, J.A.; Burguera, E.F.; Blanco, F.J.; Magalhaes, J. Impact of prevalence ratios of chondroitin sulfate (CS)-4 and -6 isomers derived from marine sources in cell proliferation and chondrogenic differentiation processes. Mar. Drugs 2020, 18, 94. [Google Scholar] [CrossRef]
- Melgar-Lesmes, P.; Sanchez-Herrero, A.; Lozano-Juan, F.; de la Torre, H.J.; Montell, E.; Jimenez, W.; Edelman, E.R.; Balcells, M. Chondroitin sulphate attenuates atherosclerosis in apoE knockout mice involving cellular regulation of the inflammatory response. Thromb. Haemost. 2018, 118, 1329–1339. [Google Scholar] [CrossRef]
- Seol, B.G.; Kim, J.H.; Woo, M.; Song, Y.O.; Choi, Y.H.; Noh, J.S.; Cho, E.J. Skate cartilage extracts containing chondroitin sulfate ameliorates hyperlipidemia-induced inflammation and oxidative stress in high cholesterol diet-fed LDL receptor knockout mice in comparison with shark chondroitin sulfate. Nutr. Res. Pract. 2020, 14, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.O.; Kim, M.; Woo, M.; Baek, J.; Kang, K.; Kim, S.; Roh, S.; Park, C.H.; Jeong, K.; Noh, J. Chondroitin sulfate-rich extract of skate cartilage attenuates lipopolysaccharide-induced liver damage in mice. Mar. Drugs 2017, 15, 178. [Google Scholar] [CrossRef]
- Li, X.; Zhou, L.; Zheng, Y.; He, T.; Guo, H.; Li, J.; Zhang, J. Establishment of a non-alcoholic fatty liver disease model by high fat diet in adult zebrafish. Animal Model. Exp. Med. 2024, 7, 904–913. [Google Scholar] [CrossRef]
- Pang, H.L.; Zhang, L.T.; Zhang, Y.T.; Ren, Q. Separation and purification of bovine nasal cartilage-derived chondroitin sulfat and evaluation of its binding to bovine serum albumin. Int. J. Biol. Macromol. 2024, 277, 134501. [Google Scholar] [CrossRef]
- Wu, R.; Li, P.; Wang, Y.; Su, N.; Xiao, M.; Li, X.; Shang, N. Structural analysis and anti-cancer activity of low-molecular-weight chondroitin sulfate from hybrid sturgeon cartilage. Carbohydr. Polym. 2022, 275, 118700. [Google Scholar] [CrossRef]
- Bedini, E.; De Castro, C.; De Rosa, M.; Di Nola, A.; Iadonisi, A.; Restaino, O.F.; Schiraldi, C.; Parrilli, M. A microbio-logical-chemical strategy to produce chondroitin sulfate A, C. Angew. Chem. Int. Ed. 2011, 50, 6160–6163. [Google Scholar] [CrossRef] [PubMed]
- Garnjanagoonchorn, W.; Wongekalak, L.; Engkagul, A. Determination of chondroitin sulfate from different sources of cartilage. Chem. Eng. Process. 2007, 46, 465–471. [Google Scholar] [CrossRef]
- Liang, J.; Zhao, Y.; Yang, F.; Zheng, L.; Ma, Y.; Liu, Q.; Cai, L.; Gong, W.; Wang, B. Preparation and structure-activity relationship of highly active black garlic polysaccharides. Int. J. Biol. Macromol. 2022, 220, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Ai, C.; Guo, L.; Fu, Y.; Cao, C.; Song, S. Characteristic oligosaccharides released from acid hydrolysis for the structural analysis of chondroitin sulfate. Carbohydr. Res. 2017, 449, 114–119. [Google Scholar] [CrossRef]
- Mindell, J.A. Lysosomal acidification mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Wang, R.; Wang, J.; Hassan, A.; Lee, C.H.; Xie, X.S.; Li, X. Molecular basis of V-ATPase inhibition by bafilomycin A1. Nat. Commun. 2021, 12, 1782. [Google Scholar] [CrossRef]
- Rajak, S. Dynamics of cellular plasticity in non-alcoholic steatohepatitis (NASH). Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167102. [Google Scholar] [CrossRef]
- Ma, Y.; Lee, G.; Heo, S.; Roh, Y. Oxidative stress is a key modulator in the development of nonalcoholic fatty liver disease. Antioxidants 2022, 11, 91. [Google Scholar] [CrossRef]
- Ramos, M.J.; Bandiera, L.; Menolascina, F.; Fallowfield, J.A. In vitro models for non-alcoholic fatty liver disease: Emerging platforms and their applications. Iscience 2022, 25, 103549. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.Y.; Yu, L.; Zhao, X.L.; Wang, L.T.; Zhao, C.J.; Huang, H.; Zhu, H.L.; Efferth, T.; Gu, C.B.; Fu, Y.J. Seed oil of Rosa roxburghii Tratt against non-alcoholic fatty liver disease in vivo and in vitro through PPARalpha/PGC-1alpha-mediated mitochondrial oxidative metabolism. Phytomedicine 2022, 98, 153919. [Google Scholar] [CrossRef] [PubMed]
- Waseem, T.; Ahmed, M.; Rajput, T.A.; Babar, M.M. Molecular implications of glycosaminoglycans in diabetes pharmacotherapy. Int. J. Biol. Macromol. 2023, 247, 125821. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Nicholls, S.J.; Langsted, A.; Ray, K.K.; Tybjoerg-Hansen, A. Advances in lipid-lowering therapy through gene-silencing technologies. Nat. Rev. Cardiol. 2018, 15, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Bartel, K.; Pein, H.; Popper, B.; Schmitt, S.; Janaki-Raman, S.; Schulze, A.; Lengauer, F.; Koeberle, A.; Werz, O.; Zischka, H.; et al. Connecting lysosomes and mitochondria—A novel role for lipid metabolism in cancer cell death. Cell Commun. Signal. 2019, 17, 87. [Google Scholar] [CrossRef]
- Folts, C.J.; Scott-Hewitt, N.; Proschel, C.; Mayer-Proschel, M.; Noble, M. Lysosomal re-acidification prevents lysosphingolipid-induced lysosomal impairment and cellular toxicity. PLoS Biol. 2016, 14, e1002583. [Google Scholar] [CrossRef]
- Schulze, R.J.; Krueger, E.W.; Weller, S.G.; Johnson, K.M.; Casey, C.A.; Schott, M.B.; Mcniven, M.A. Direct lysosome-based autophagy of lipid droplets in hepatocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 32443–32452. [Google Scholar] [CrossRef]
- Zeng, J.; Acin-Perez, R.; Assali, E.A.; Martin, A.; Brownstein, A.J.; Petcherski, A.; Fernandez-Del-Rio, L.; Xiao, R.; Lo, C.H.; Shum, M.; et al. Restoration of lysosomal acidification rescues autophagy and metabolic dysfunction in non-alcoholic fatty liver disease. Nat. Commun. 2023, 14, 2573. [Google Scholar] [CrossRef]
- Ying, C.; Jin, C.; Zeng, S.; Chao, M.; Hu, X. Alkalization of cellular pH leads to cancer cell death by disrupting autophagy and mitochondrial function. Oncogene 2022, 41, 3886–3897. [Google Scholar] [CrossRef]
- Lakpa, K.L.; Khan, N.; Afghah, Z.; Chen, X.; Geiger, J.D. Lysosomal stress response (LSR): Physiological importance and pathological relevance. J. Neuroimmune Pharm. 2021, 16, 219–237. [Google Scholar] [CrossRef]
- Chen, J.; Mei, M.S.; Xu, Y.; Xiong, S.; Zhao, Y.; Liu, R.; Shi, S.; Wang, H.; Wang, S. The impact of the methyl esters of homogalacturonan on cellular uptake dependent hypoglycemic activity in IR-HepG2 cells. Carbohydr. Polym. 2022, 293, 119741. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Pan, X.; Wang, H.; Wu, Z.; Sullivan, M.A.; Liu, Y.; Liu, J.; Wang, K.; Zhang, Y. Mechanism of lentinan intes-tinal absorption: Clathrin-mediated endocytosis and macropinocytosis. J. Agric. Food Chem. 2021, 69, 7344–7352. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Tanaka, Y.; Mori, S.; Mitsuzumi, K.; Fujii, S.; Sasakura, H.; Morioka, Y.; Sugioka, K.; Takeuchi, K.; Nishi-waki, K. Endogenous chondroitin extends lifespan by inhibiting VHA-7-mediated tubular lysosome formation. Sci. Rep. 2024, 14, 29651. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Ballabio, A. Lysosome: Regulator of lipid degradation pathways. Trends Cell Biol. 2014, 24, 743–750. [Google Scholar] [CrossRef]
- Reggiori, F.; Gabius, H.J.; Aureli, M.; Romer, W.; Sonnino, S.; Eskelinen, E.L. Glycans in autophagy, endocytosis and lysosomal functions. Glycoconj. J. 2021, 38, 625–647. [Google Scholar] [CrossRef]
- Takeichi, Y.; Miyazawa, T.; Sakamoto, S.; Hanada, Y.; Wang, L.; Gotoh, K.; Uchida, K.; Katsuhara, S.; Sakamoto, R.; Ishihara, T.; et al. Non-alcoholic fatty liver disease in mice with hepatocyte-specific deletion of mitochondrial fission factor. Diabetologia 2021, 64, 2092–2107. [Google Scholar] [CrossRef]
- Angelova, P.R.; Esteras, N.; Abramov, A.Y. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: Finding ways for prevention. Med. Res. Rev. 2021, 41, 770–784. [Google Scholar] [CrossRef]
- Gustafsson, A.B.; Dorn, G.W. Evolving and expanding the roles of mitophagy as a homeostatic and pathogenic process. Physiol. Rev. 2019, 99, 853–892. [Google Scholar] [CrossRef]
- Masclaux-Daubresse, C.; Chen, Q.; Have, M. Regulation of nutrient recycling via autophagy. Curr. Opin. Plant Biol. 2017, 39, 8–17. [Google Scholar] [CrossRef]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376. [Google Scholar] [CrossRef]
- Shihan, M.H.; Novo, S.G.; Le Marchand, S.J.; Wang, Y.; Duncan, M.K. A simple method for quantitating confocal fluorescent images. Biochem. Biophys. Rep. 2021, 25, 100916. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Shao, H.; Shao, X.; Yu, L.; Gao, Y.; Ren, Y.; Liu, F.; Meng, C.; Ling, P.; Chen, Q. In situ visualization of the cellular uptake and sub—Cellular distribution of mussel oligosaccharides. J. Pharm. Anal. 2024, 14, 100932. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Wang, H.; Zhang, X.; Ling, P.; Xiao, Y.; Chen, Q.; Chen, X. Ion monitoring at nanoscale sites of interorganelle membrane contact in living cells. Small Struct. 2024, 5, 2300350. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, T.; Lv, H.; Shao, H.; Zhang, X.; Wang, A.; Zhang, W.; Liu, F.; Ling, P. Chondroitin Sulfate as a Lysosomal Enhancer Attenuates Lipid-Driven Inflammation via Lipophagy and Mitophagy. Mar. Drugs 2025, 23, 228. https://doi.org/10.3390/md23060228
Sun T, Lv H, Shao H, Zhang X, Wang A, Zhang W, Liu F, Ling P. Chondroitin Sulfate as a Lysosomal Enhancer Attenuates Lipid-Driven Inflammation via Lipophagy and Mitophagy. Marine Drugs. 2025; 23(6):228. https://doi.org/10.3390/md23060228
Chicago/Turabian StyleSun, Ting, Huimin Lv, Huarong Shao, Xiuhua Zhang, Anqi Wang, Wei Zhang, Fei Liu, and Peixue Ling. 2025. "Chondroitin Sulfate as a Lysosomal Enhancer Attenuates Lipid-Driven Inflammation via Lipophagy and Mitophagy" Marine Drugs 23, no. 6: 228. https://doi.org/10.3390/md23060228
APA StyleSun, T., Lv, H., Shao, H., Zhang, X., Wang, A., Zhang, W., Liu, F., & Ling, P. (2025). Chondroitin Sulfate as a Lysosomal Enhancer Attenuates Lipid-Driven Inflammation via Lipophagy and Mitophagy. Marine Drugs, 23(6), 228. https://doi.org/10.3390/md23060228