
New Polyketides from a Marine Sponge-Derived Fungus, Neopestalotiopsis sp., with Anti-Renal Fibrosis Activity

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
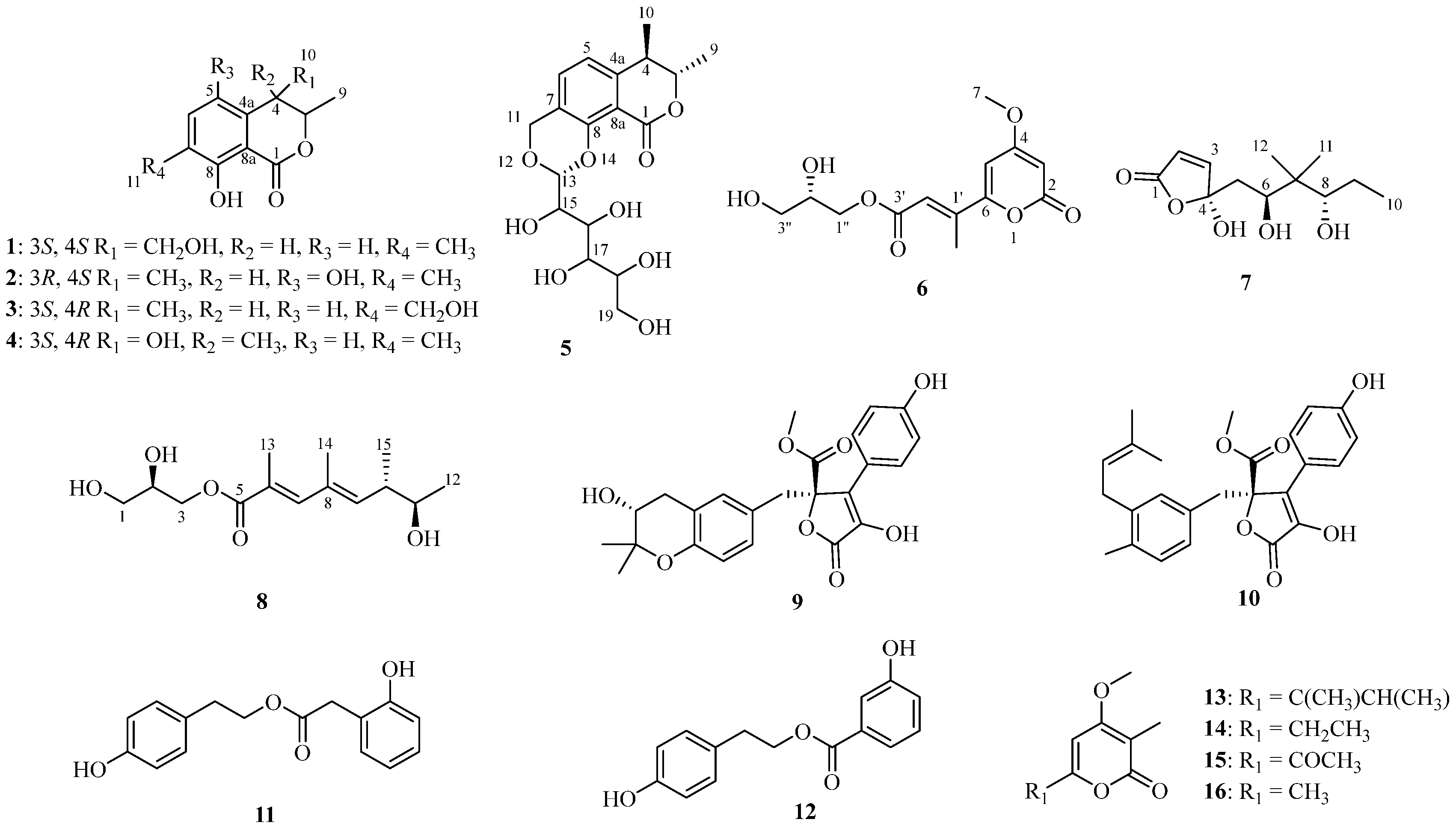
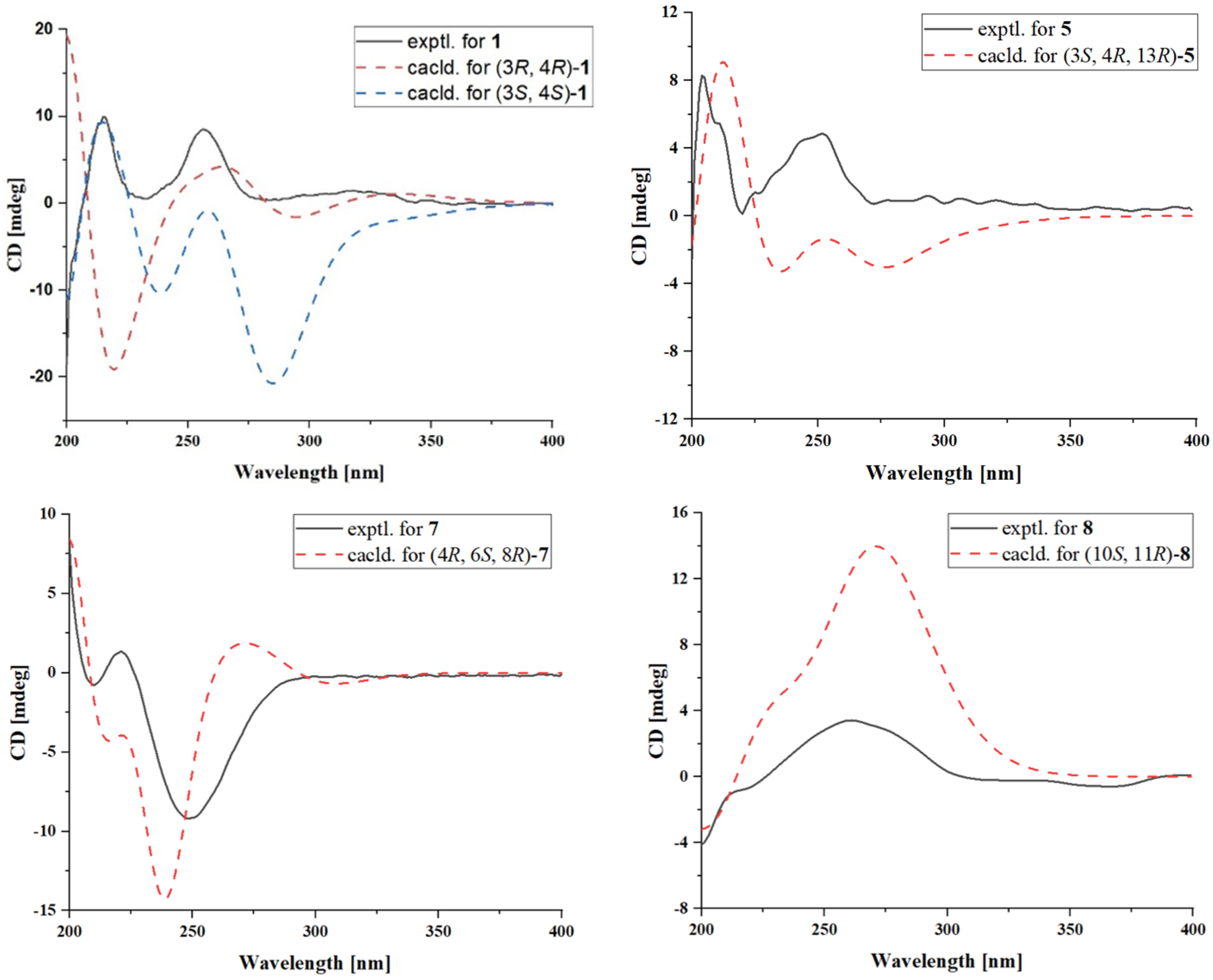
2.1. Structural Determination
2.2. Bioactivity Assay
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation and Extraction
3.4. Isolation and Purification
3.5. Spectroscopic Data of Compounds
3.6. X-Ray Crystallographic Analysis
3.7. ECD Computational and DP4+ Probability Analysis Methods
3.8. Antioxidant Activity Assay
3.9. Antimicrobial Activity Assay
3.10. TGF-β1-Stimulated HK-2 Cell Model
3.11. Western Blot (WB) Analysis
3.12. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carney, E.F. The impact of chronic kidney disease on global health. Nat. Rev. Nephrol. 2020, 16, 251. [Google Scholar] [CrossRef] [PubMed]
- Cockwell, P.; Fisher, L.-A. The global burden of chronic kidney disease. Lancet 2020, 395, 662–664. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef]
- Xiang, Y.; Yuan, Z.; Deng, Q.; Xie, L.; Yu, D.; Shi, J. Potential therapeutic medicines for renal fibrosis: Small-molecule compounds and natural products. Bioorg. Chem. 2024, 143, 106999. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Horowitz, J.C.; Thannickal, V.J. Mechanisms for the Resolution of Organ Fibrosis. Physiology 2019, 34, 43–55. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Lamas, S.; Ortiz, A. Antifibrotic Agents for the Management of CKD: A Review. Am. J. Kidney Dis. 2022, 80, 251–263. [Google Scholar] [CrossRef]
- Kim, E.S.; Keating, G.M. Pirfenidone: A review of its use in idiopathic pulmonary fibrosis. Drugs 2015, 75, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Corte, T.J.; Fischer, A.; Kreuter, M.; Lederer, D.J.; Molina-Molina, M.; Axmann, J.; Kirchgaessler, K.-U.; Samara, K.; Gilberg, F.; et al. Pirfenidone in patients with unclassifiable progressive fibrosing inte rstitial lung disease: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2020, 8, 147–157. [Google Scholar] [CrossRef]
- Lin, X.; Wu, Q.; Yu, Y.; Liang, Z.; Liu, Y.; Zhou, L.; Tang, L.; Zhou, X. Penicilliumin B, a novel sesquiterpene methylcyclopentenedione from a deep sea-derived Penicillium strain with renoprotective activities. Sci. Rep. 2017, 7, 10757. [Google Scholar] [CrossRef]
- Zhong, M.; Li, Y.; Deng, L.; Fang, J.; Yu, X. Insight into the adaptation mechanisms of high hydrostatic pressure in physiology and metabolism of hadal fungi from the deepest ocean sediment. mSystems 2024, 9, e01085-23. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, M.; Gérard, L.; Milhau, C.; Vinale, F.; Dufossé, L.; Fouillaud, M. Salinity and Temperature Influence Growth and Pigment Production in the Marine-Derived Fungal Strain Talaromyces albobiverticillius 30548. Microorganisms 2019, 7, 10. [Google Scholar] [CrossRef]
- Giordano, D. Bioactive Molecules from Extreme Environments. Mar. Drugs 2020, 18, 640. [Google Scholar] [CrossRef]
- Jones, E.B.G.; Ramakrishna, S.; Vikineswary, S.; Das, D.; Bahkali, A.H.; Guo, S.-Y.; Pang, K.-L. How Do Fungi Survive in the Sea and Respond to Climate Change? J. Fungi 2022, 8, 291. [Google Scholar] [CrossRef]
- Li, P.; Lu, H.; Zhang, Y.; Zhang, X.; Liu, L.; Wang, M.; Liu, L. The natural products discovered in marine sponge-associated microorgan isms: Structures, activities, and mining strategy. Front. Mar. Sci. 2023, 10, 1191858. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2020, 37, 175–223. [Google Scholar] [CrossRef]
- Koshino, H.; Yoshihara, T.; Okuno, M.; Sakamura, S.; Tajimi, A.; Shimanuki, T. Gamahonolides A, B, and Gamahorin, Novel Antifungal Compounds from Str omata of Epichloe typhina on Phleum pratense. Biosci. Biotechnol. Biochem. 1992, 56, 1096–1099. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, W.; Yang, Y.; Liu, T.; Yang, H.; Xin, Z. New phenolic compounds from Coreopsis tinctoria Nutt. and their antiox idant and angiotensin i-converting enzyme inhibitory activities. J. Agric. Food Chem. 2015, 63, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Rönsberg, D.; Debbab, A.; Mándi, A.; Wray, V.; Dai, H.; Kurtán, T.; Proksch, P.; Aly, A.H. Secondary metabolites from the endophytic fungus Pestalotiopsis virgat ula isolated from the mangrove plant Sonneratia caseolaris. Tetrahedron Lett. 2013, 54, 3256–3259. [Google Scholar] [CrossRef]
- Xu, Z.; Wu, X.; Li, G.; Feng, Z.; Xu, J. Pestalotiopisorin B, a new isocoumarin derivative from the mangrove en dophytic fungus Pestalotiopsis sp. HHL101. Nat. Prod. Res. 2020, 34, 1002–1007. [Google Scholar] [CrossRef]
- Xu, J.; Aly, A.H.; Wray, V.; Proksch, P. Polyketide derivatives of endophytic fungus Pestalotiopsis sp. isolate d from the Chinese mangrove plant Rhizophora mucronata. Tetrahedron Lett. 2011, 52, 21–25. [Google Scholar] [CrossRef]
- Bi, Y.-M.; Bi, X.-B.; Fang, A.; Zhao, Q.-R. Metabolites from the fungus Cephalosporium sp. AL031. Arch. Pharm. Res. 2007, 30, 267–269. [Google Scholar] [CrossRef]
- Hirota, A.; Nemoto, A.; Tsuchiya, Y.; Hojo, H.; Abe, N. Isolation of a 2-Pyrone Compound as an Antioxidant from a Fungus and Its New Reaction Product with 1,1-Diphenyl-2-picrylhydrazyl Radical. Biosci. Biotechnol. Biochem. 1999, 63, 418–420. [Google Scholar] [CrossRef]
- Nagia, M.M.; El-Metwally, M.M.; Shaaban, M.; El-Zalabani, S.M.; Hanna, A.G. Four butyrolactones and diverse bioactive secondary metabolites from t errestrial Aspergillus flavipes MM2: Isolation and structure determination. Org. Med. Chem. Lett. 2012, 2, 9. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Zhang, Y.; Yao, Y.-B.; Lei, X.-L.; Qian, Z.-J. Butyrolactone-I from Coral-Derived Fungus Aspergillus terreus Attenuates Neuro-Inflammatory Response via Suppression of NF-κB Pathway in BV-2 Cells. Mar. Drugs 2018, 16, 202. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.; Du, J.; Pettit, R.; Knight, J.C.; Doubek, D.L. Antineoplastic Agents. 575. The Fungus Aspergillus phoenicis. Heterocycles 2009, 79, 909. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, Q.; Zhu, J.; Xia, G.; Zang, H. Synthesis, α-Glucosidase inhibition and molecular docking studies of tyrosol derivatives. Nat. Prod. Res. 2021, 35, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Tao, H.; Lin, X.; Wang, J.; Liao, S.; Dong, J.; Zhou, X.; Liu, Y. Prenylated indole alkaloids and chromone derivatives from the fungus Penicillium sp. SCSIO041218. Tetrahedron 2018, 74, 77–82. [Google Scholar] [CrossRef]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An Improved Probability for the Stereochemical Assignment of Isomeric Compounds using Quantum Chemical Calculations of NMR Shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef]
- Huang, Z.; Nong, X.; Ren, Z.; Wang, J.; Zhang, X.; Qi, S. Anti-HSV-1, antioxidant and antifouling phenolic compounds from the de ep-sea-derived fungus Aspergillus versicolor SCSIO 41502. Bioorg. Med. Chem. Lett. 2017, 27, 787–791. [Google Scholar] [CrossRef]
- Huang, Z.-H.; Nong, X.-H.; Liang, X.; Qi, S.-H. New tetramic acid derivatives from the deep-sea-derived fungus Cladosp orium sp. SCSIO z0025. Tetrahedron 2018, 74, 2620–2626. [Google Scholar] [CrossRef]
- Gan, L.; Jiang, Q.; Huang, D.; Wu, X.; Zhu, X.; Wang, L.; Xie, W.; Huang, J.; Fan, R.; Jing, Y.; et al. A natural small molecule alleviates liver fibrosis by targeting apolip oprotein L2. Nat. Chem. Biol. 2025, 21, 80–90. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pos. | 1 | 2 | 5 | |||
|---|---|---|---|---|---|---|
| δC, Type | δH, (J in Hz) | δC, Type | δH, (J in Hz) | δC, Type | δH, (J in Hz) | |
| 1 | 170.5, C | 170.7, C | 170.9, C | |||
| 3 | 77.6, CH | 5.05, qd, (6.7, 2.0) | 83, CH | 4.73, q, (6.7) | 82.6, CH | 4.57, m |
| 4 | 46.7, CH | 2.89, ddd, (7.6, 5.6, 1.8) | 32.7, CH | 3.19, q, (6.8) | 38.5, CH | 2.96, p, (7.0) |
| 4a | 137.9, C | 126.5, C | 145.3, C | |||
| 5 | 120.1, CH | 6.79, d, (7.5) | 147, C | 117.9, CH | 6.90, d, (7.7) | |
| 6 | 138.4, CH | 7.39, d, (7.5) | 126.6, CH | 6.95, s | 137.6, CH | 7.74, d, (7.7) |
| 7 | 126.6, C | 126, C | 125.9, C | |||
| 8 | 161.3, C | 154.7, C | 160.9, C | |||
| 8a | 108.2, C | 107, C | 108.2, C | |||
| 9 | 19.9, CH3 | 1.37, d, (6.7) | 20.3, CH3 | 1.30, d, (6.7) | 19.9, CH3 | 1.43, d, (6.5) |
| 10 | 64.7, CH2 | 3.66, m | 19.9, CH3 | 1.27, d, (7.1) | 18.2, CH3 | 1.36, d, (6.9) |
| 11 | 15.5, CH3 | 2.22, s | 15.5, CH3 | 2.17, s | 64.7, CH2 | 4.85, d, (12.5)4.61, d, (12.5) |
| 13 | 100.0, CH | 4.95, d, (3.8) | ||||
| 15 | 73.7, CH | 3.44, dd, (9.7, 3.8) | ||||
| 16 | 75.1, CH | 3.70, m | ||||
| 17 | 71.7, CH | 3.34, m | ||||
| 18 | 73.8, CH | 3.66, td, (4.9, 2.1) | ||||
| 19 | 62.5, CH2 | 3.77, dd, (11.2, 2.1)3.70, m | ||||
| Pos. | 6 | |
|---|---|---|
| δC, Type | δH, (J in Hz) | |
| 2 | 165.6, C | |
| 3 | 91.3, CH | 5.74, d, (1.9) |
| 4 | 172.9, C | |
| 5 | 104, CH | 6.55, d, (2.1) |
| 6 | 160.1, C | |
| 7 | 57.3, CH3 | 3.9, s |
| 1′ | 144, C | |
| 2′ | 120.8, CH | 6.65, d, (1.6) |
| 3′ | 167.9, C | |
| 4′ | 13.6, CH3 | 2.37, d, (1.1) |
| 1″ | 64.4, CH2 | 3.59, dd, (11.2, 4.9) |
| 2″ | 73.9, CH | 3.65, m |
| 3″ | 64.4, CH2 | 3.51, dd, (11.2, 6.0) |
| Pos. | 7 | 8 | ||
|---|---|---|---|---|
| δC, Type | δH, (J in Hz) | δC, Type | δH, (J in Hz) | |
| 1 | 170.5, C | 64.1, CH2 | 3.61, m | |
| 2 | 122.4, CH | 6.29, d, (5.6) | 71.3, CH | 3.90, m |
| 3 | 155.5, CH | 7.55, d, (5.6) | 66.9, CH2 | 4.25, dd, (11.4, 4.8)4.16, dd, (11.4, 6.1) |
| 4 | 107.5, C | |||
| 5 | 36.4, CH2 | 1.91, m1.65, dd, (13.5, 4.9) | 170.5, C | |
| 6 | 70.2, CH | 3.54, dd, (11.8, 4.7) | 126.4, C | |
| 7 | 38.1, C | 144.8, CH | 7.21, s | |
| 8 | 82.8, CH | 3.36, m | 133.1, C | |
| 9 | 21.3, CH2 | 1.55, m1.26, m | 140.3, CH | 5.51, d, (10.0) |
| 10 | 11.4, CH3 | 0.83, t, (7.3) | 41.9, CH | 2.5, m |
| 11 | 11.7, CH3 | 0.77, s | 72.5, CH | 3.58, m |
| 12 | 22.2, CH3 | 0.90, s | 21.4, CH3 | 1.17, d, (6.3) |
| 13 | 14.3, CH3 | 2.04, s | ||
| 14 | 16.8, CH3 | 1.91, s | ||
| 15 | 16.8, CH3 | 1.07, d, (6.7) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; She, J.; Cai, M.; Chen, X.; Qiu, R.; Luo, X.; Liu, Y.; Zhou, X.; Tang, L. New Polyketides from a Marine Sponge-Derived Fungus, Neopestalotiopsis sp., with Anti-Renal Fibrosis Activity. Mar. Drugs 2025, 23, 148. https://doi.org/10.3390/md23040148
Li X, She J, Cai M, Chen X, Qiu R, Luo X, Liu Y, Zhou X, Tang L. New Polyketides from a Marine Sponge-Derived Fungus, Neopestalotiopsis sp., with Anti-Renal Fibrosis Activity. Marine Drugs. 2025; 23(4):148. https://doi.org/10.3390/md23040148
Chicago/Turabian StyleLi, Xinlong, Jianglian She, Meiqun Cai, Xinqi Chen, Rongxiang Qiu, Xiaowei Luo, Yonghong Liu, Xuefeng Zhou, and Lan Tang. 2025. "New Polyketides from a Marine Sponge-Derived Fungus, Neopestalotiopsis sp., with Anti-Renal Fibrosis Activity" Marine Drugs 23, no. 4: 148. https://doi.org/10.3390/md23040148
APA StyleLi, X., She, J., Cai, M., Chen, X., Qiu, R., Luo, X., Liu, Y., Zhou, X., & Tang, L. (2025). New Polyketides from a Marine Sponge-Derived Fungus, Neopestalotiopsis sp., with Anti-Renal Fibrosis Activity. Marine Drugs, 23(4), 148. https://doi.org/10.3390/md23040148