

Comparative Evaluation of the Antibacterial and Antitumor Activities of Marine Alkaloid 3,10-Dibromofascaplysin


 , , , and
, , , and
Abstract
1. Introduction
2. Results and Discussion
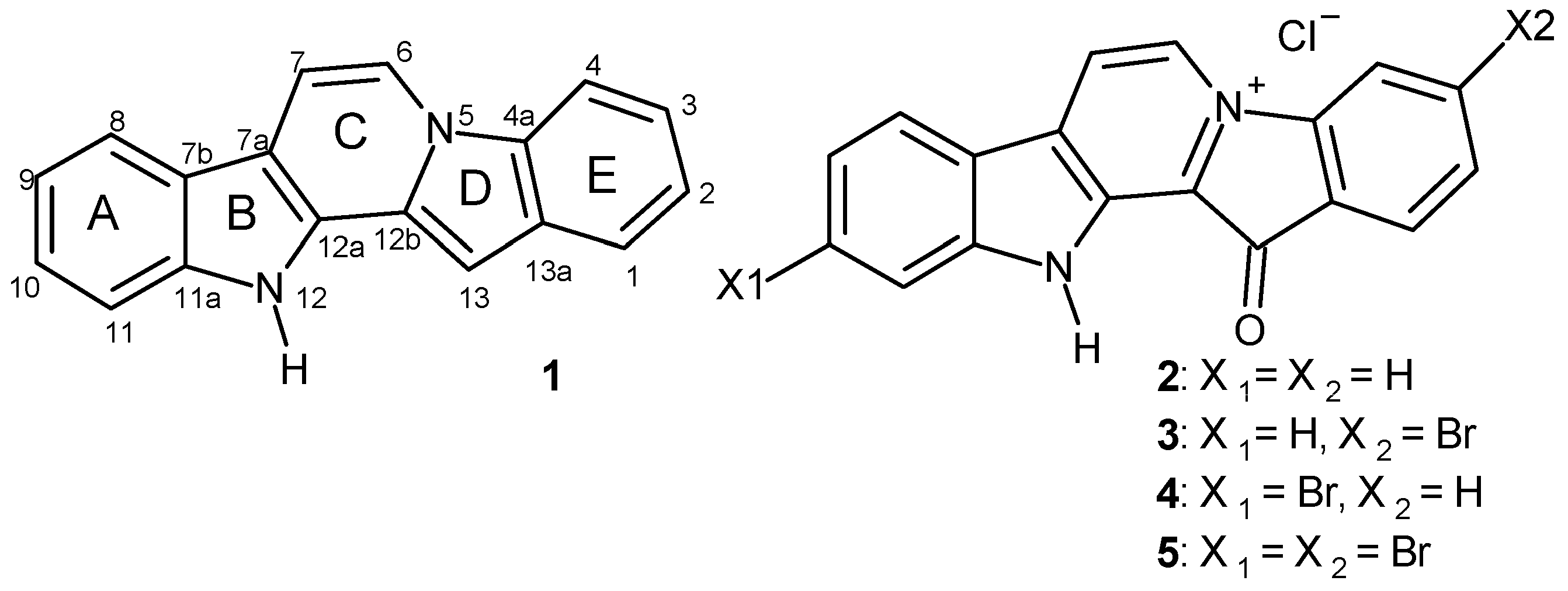
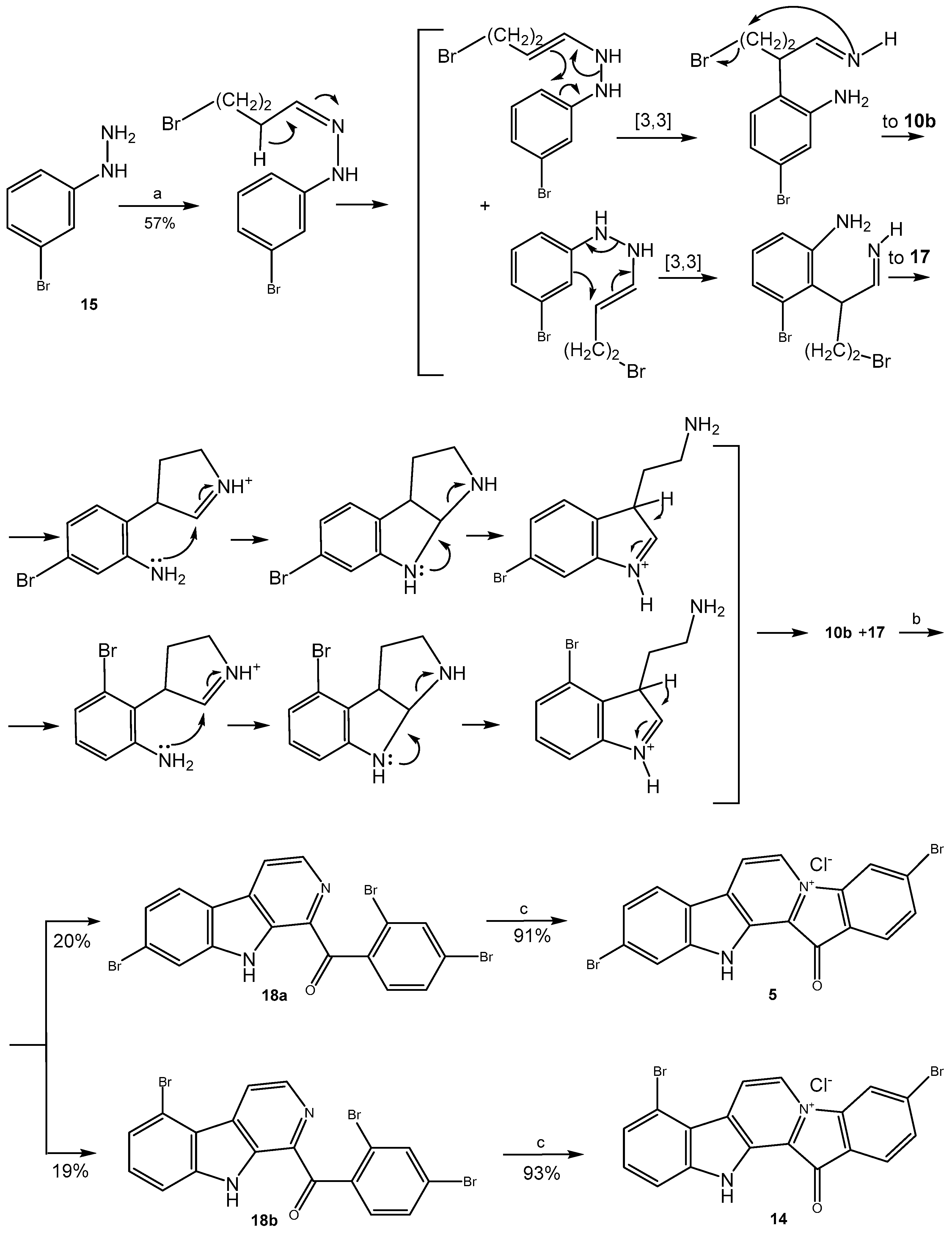
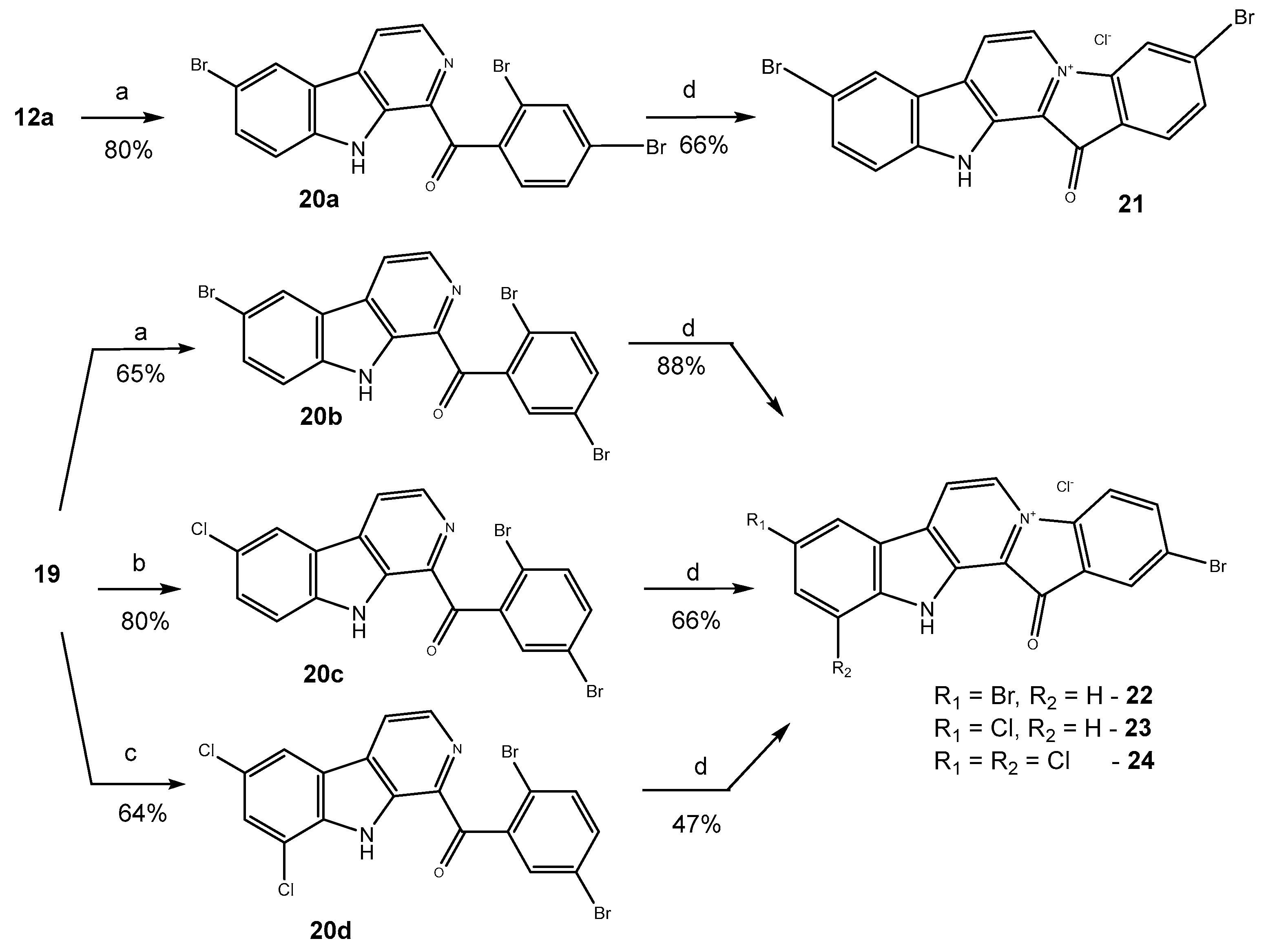
2.1. Preparation of Fascaplysins 3–5, Its Isomers, and Some Analogs
2.2. Antibacterial and Antiproliferative Activities of 3-Bromo-, 10-Bromo-, and 3,10-Dibromofascaplysins, Their Isomers, and Analogs In Vitro
2.3. Study of the Therapeutic Potential of 3,10-Dibromofascaplysin In Vivo
2.3.1. Antibacterial Efficacy of 3,10-Dibromofascaplysin
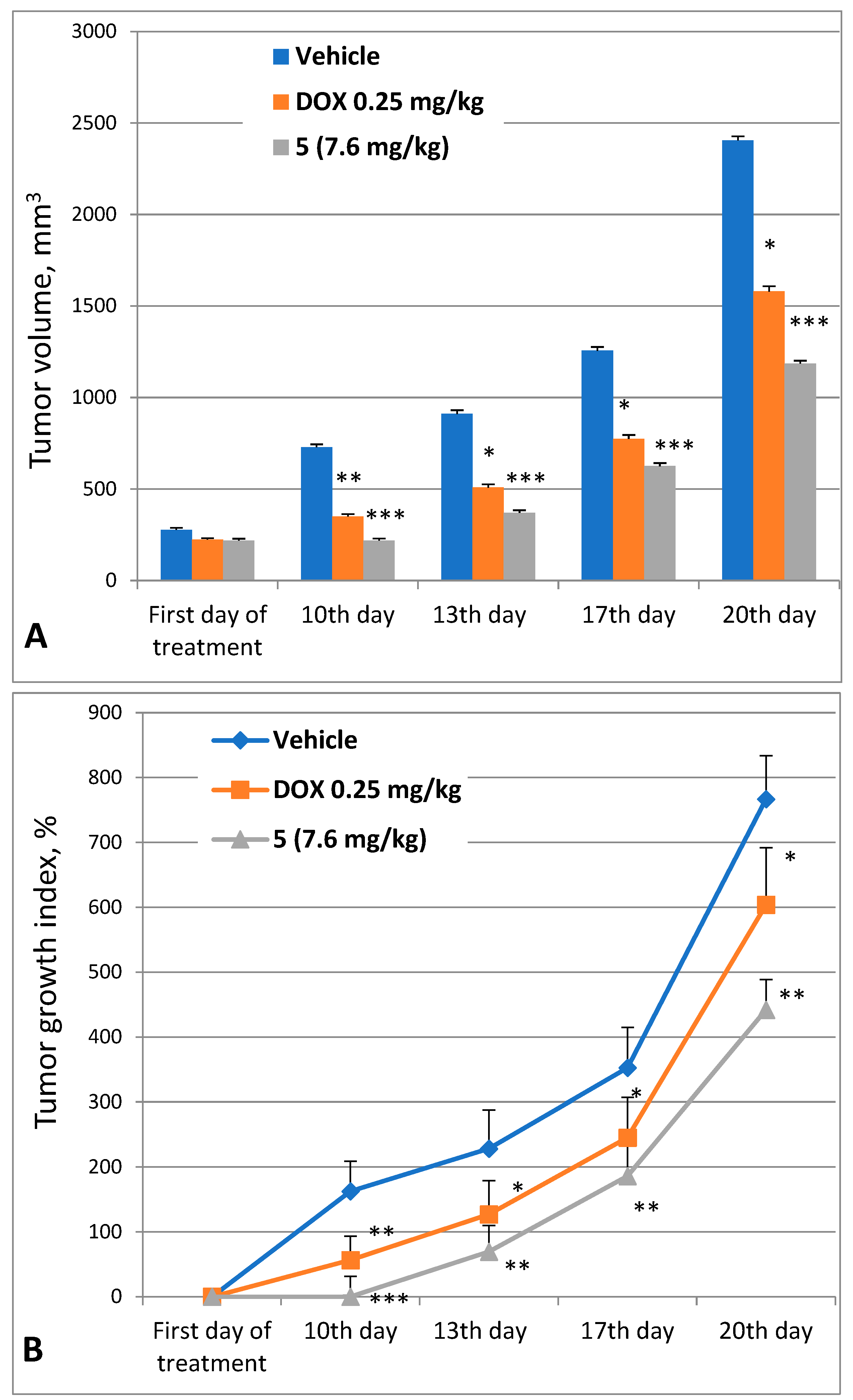
2.3.2. Antitumor Efficacy of 3,10-Dibromofascaplysin (5)
3. Materials and Methods
3.1. Chemistry
3.1.1. Preparation of Mixture of Tryptamines 10b and 17
3.1.2. Preparation of Substituted 1-Benzoyl-β-Carbolines 12a-b, 18a-b, 19
3.1.3. Preparation of Substituted 1-Benzoyl-β-Carbolines 20a–20b
3.1.4. Synthesis of Compound 20c
3.1.5. Preparation of Compound 20d
3.1.6. Preparation of Fascaplysin Derivatives
3.2. Biological Assay
3.2.1. MIC Values Determination
3.2.2. In Vivo Efficiency Study of Antibacterial Activity
3.2.3. In Vivo Study of Antitumor Activity
3.2.4. A Murine Model of Ascite Ehrlich Adenocarcinoma
3.2.5. A Murine Model of Solid Ehrlich Adenocarcinoma
3.3. Statistical Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Roll, D.M.; Ireland, C.M.; Lu, H.S.M.; Clardy, J. Fascaplysin, an unusual antimicrobial pigment from the marine sponge Fascaplysinopsis sp. J. Org. Chem. 1988, 53, 3276–3278. [Google Scholar] [CrossRef]
- Wang, C.; Wang, S.; Li, H.; Hou, Y.; Cao, H.; Hua, H.; Li, D. Marine-Derived Lead Fascaplysin: Pharmacological Activity, Total Synthesis, and Structural Modification. Mar. Drugs 2023, 21, 226. [Google Scholar] [CrossRef]
- Soni, R.; Muller, L.; Furet, P.; Schoepfer, J.; Stephan, C.; Zumstein-Mecker, S.; Fretz, H.; Chaudhuri, B. Inhibition of cyclin-dependent kinase 4 (Cdk4) by fascaplysin, a marine natural product. Biochem. Biophys. Res. Commun. 2000, 275, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Hörmann, A.; Chaudhuri, B.; Fretz, H. DNA binding properties of the marine sponge pigment fascaplysin. Bioorgan. Med. Chem. 2001, 9, 917–921. [Google Scholar] [CrossRef]
- Aubry, C.; Jenkins, P.R.; Mahale, S.; Chaudhuri, B.; Maréchal, J.-D.; Sutcliffe, M.J. New fascaplysin-based CDK4-specific inhibitors: Design, synthesis and biological activity. Chem. Commun. 2004, 35, 1696–1697. [Google Scholar] [CrossRef] [PubMed]
- Aubry, C.; Wilson, A.J.; Jenkins, P.R.; Mahale, S.; Chaudhuri, B.; Maréchal, J.-D.; Sutcliffe, M.J. Design, synthesis and biological activity of new CDK4-specific inhibitors, based on fascaplysin. Org. Biomol. Chem. 2006, 4, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Mahale, S.; Aubry, C.; Wilson, A.J.; Jenkins, P.R.; Maréchal, J.-D.; Sutcliffe, M.J.; Chaudhuri, B. CA224, a non-planar analogue of fascaplysin, inhibits Cdk4 but not Cdk2 and arrests cells at G0/G1 inhibiting pRB phosphorylation. Bioorgan. Med. Chem. Lett. 2006, 16, 4272–4278. [Google Scholar] [CrossRef]
- García, M.D.; Wilson, A.J.; Emmerson, D.P.G.; Jenkins, P.R.; Mahale, S.; Chaudhuri, B. Synthesis, crystal structure and biological activity of β-carboline based selective CDK4-cyclin D1 inhibitors. Org. Biomol. Chem. 2006, 4, 4478–4484. [Google Scholar] [CrossRef]
- Jenkins, P.R.; Wilson, J.; Emmerson, D.; Garcia, M.D.; Smith, M.R.; Gray, S.J.; Britton, R.G.; Mahale, S.; Chaudhuri, B. Design, synthesis and biological evaluation of new tryptamine and tetrahydro-β-carboline-based selective inhibitors of CDK4. Bioorgan. Med. Chem. 2008, 16, 7728–7739. [Google Scholar] [CrossRef]
- Aubry, C.; Wilson, A.J.; Emmerson, D.; Murphy, E.; Chan, Y.Y.; Dickens, M.P.; García, M.D.; Jenkins, P.R.; Mahale, S.; Chaudhuri, B. Fascaplysin-inspired diindolyls as selective inhibitors of CDK4/cyclin D1. Bioorgan. Med. Chem. 2009, 17, 6073–6084. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.L.; Lu, X.L.; Lin, J.; Chen, H.M.; Yan, X.J.; Wang, F.; Xu, W.F. Direct effects of fascaplysin on human umbilical vein endothelial cells attributing the anti-angiogenesis activity. Biomed. Pharmacother. 2010, 64, 527–533. [Google Scholar] [CrossRef]
- Meng, N.; Mu, X.; Lv, X.; Wang, L.; Li, N.; Gong, Y. Autophagy represses fascaplysin-induced apoptosis and angiogenesis inhibition via ROS and p8 in vascular endothelia cells. Biomed. Pharmacother. 2019, 114, 108866. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.-I.; Lee, Y.-M.; Nam, T.-J.; Ko, Y.-S.; Mah, S.; Kim, J.; Kim, Y.; Reddy, R.H.; Kim, Y.J.; Hong, S.; et al. Fascaplysin exerts anti-cancer effects through the downregulation of survivin and HIF-1 and Inhibition of VEGFR2 and TRKA. Int. J. Mol. Sci. 2017, 18, 2074. [Google Scholar] [CrossRef]
- Kumar, S.; Guru, S.K.; Pathania, A.S.; Manda, S.; Kumar, A.; Bharate, S.B.; Vishwakarma, R.A.; Malik, F.; Bhushan, S. Fascaplysin induces caspase mediated crosstalk between apoptosis and autophagy through the inhibition of PI3K/AKT/mTOR signaling cascade in human leukemia HL-60 cells. J. Cell. Biochem. 2015, 116, 985–997. [Google Scholar] [CrossRef]
- Popov, A.M.; Stonik, V.A. Physiological activity of fascaplisine--an unusual pigment from tropical sea sponges. Antibiot. Chemother. 1991, 36, 12–14. [Google Scholar]
- Subramanian, B.; Nakeff, A.; Tenney, K.; Crews, P.; Gunatilaka, L.; Valeriote, F. A new paradigm for the development of anticancer agents from natural products. J. Exp. Ther. Oncol. 2006, 5, 195–204. [Google Scholar] [PubMed]
- Yan, X.; Chen, H.; Lu, X.; Wang, F.; Xu, W.; Jin, H.; Zhu, P. Fascaplysin exert anti-tumor effects through apoptotic and anti-angiogenesis pathways in sarcoma mice model. Eur. J. Pharm. Sci. 2011, 43, 251–259. [Google Scholar] [CrossRef]
- Sharma, S.; Guru, S.K.; Manda, S.; Kumar, A.; Mintoo, M.J.; Prasad, V.D.; Sharma, P.R.; Mondhe, D.M.; Bharate, S.B.; Bhushan, S. A marine sponge alkaloid derivative 4-chloro fascaplysin inhibits tumor growth and VEGF mediated angiogenesis by disrupting PI3K/Akt/mTOR signaling cascade. Chem. Interact. 2017, 275, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Zhidkov, M.E.; Sidorova, M.A.; Smirnova, P.A.; Tryapkin, O.A.; Kachanov, A.V.; Kantemirov, A.V.; Dezhenkova, L.G.; Grammatikova, N.E.; Isakova, E.B.; Shchekotikhin, A.E.; et al. Comparative evaluation of the antibacterial and antitumor activities of 9-phenylfascaplysin and its analogs. Mar. Drugs 2024, 22, 53. [Google Scholar] [CrossRef] [PubMed]
- Mc Carlie, S.; Boucher, C.E.; Bragg, R.R. Molecular basis of bacterial disinfectant resistance. Drug Resist. Updates 2020, 48, 100672. [Google Scholar] [CrossRef]
- Kong, Q.; Yang, Y. Recent advances in antibacterial agents. Bioorg. Med. Chem. Lett. 2021, 35, 127799. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qiu, H.; Yang, N.; Xie, H.; Liang, W.; Lin, J.; Zhu, H.; Zhou, Y.; Wang, N.; Tan, X.; et al. Fascaplysin derivatives binding to DNA via unique cationic five-ring coplanar backbone showed potent antimicrobial/antibiofilm activity against MRSA in vitro and in vivo. Eur. J. Med. Chem. 2022, 230, 114099. [Google Scholar] [CrossRef]
- Qiu, H.; Zhao, X.; Jiang, Y.; Liang, W.; Wang, W.; Jiang, X.; Jiang, M.; Wang, X.; Cui, W.; Li, Y.; et al. Design and synthesis of fascaplysin derivatives as inhibitors of FtsZ with potent antibacterial activity and mechanistic study. Eur. J. Med. Chem. 2023, 254, 115348. [Google Scholar] [CrossRef] [PubMed]
- Lock, R.L.; Harry, E.J. Cell-division inhibitors: New insights for future antibiotics. Nat. Rev. Drug Discov. 2008, 7, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Silber, N.; de Opitz, C.L.M.; Mayer, C.; Sass, P. Cell division protein FtsZ: From structure and mechanism to antibiotic target. Future Microbiol. 2020, 15, 801–831. [Google Scholar] [CrossRef]
- Haydon, D.J.; Stokes, N.R.; Ure, R.; Galbraith, G.; Bennett, J.M.; Brown, D.R.; Baker, P.J.; Barynin, V.V.; Rice, D.W.; Sedelnikova, S.E.; et al. An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science 2008, 321, 1673–1675. [Google Scholar] [CrossRef]
- Bisson, A.W.; Hsu, Y.P.; Squyres, G.R.; Kuru, E.; Wu, F.B.; Jukes, C.; Sun, Y.J.; Dekker, C.; Holden, S.; Van Nieuwenhze, M.S.; et al. Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science 2017, 355, 739–743. [Google Scholar] [CrossRef]
- Privalsky, T.M.; Soohoo, A.M.; Wang, J.H.; Walsh, C.T.; Wright, G.D.; Gordon, E.M.; Gray, N.S.; Khosla, C. Prospects for antibacterial discovery and development. J. Am. Chem. Soc. 2021, 143, 21127–21142. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.B.; Qiu, H.D.; Zhang, K.; Zhang, P.P.; Liang, W.D.; Yang, M.X.; Mou, C.Y.; Lin, M.M.; He, M.; Xiao, X.; et al. Fascaplysin derivatives are potent multitarget agents against alzheimer’s disease: In vitro and in vivo evidence. ACS Chem. Neurosci. 2019, 10, 4741–4756. [Google Scholar] [CrossRef]
- Sun, Q.M.; Liu, F.F.; Sang, J.C.; Lin, M.M.; Ma, J.L.; Xiao, X.; Yan, S.C.; Naman, C.B.; Wang, N.; He, S.; et al. 9-Methylfascaplysin is a more potent Aβ aggregation inhibitor than the marine-derived alkaloid, fascaplysin, and produces nanomolar neuroprotective effects in SH-SY5Y cells. Mar. Drugs 2019, 17, 121. [Google Scholar] [CrossRef]
- Zhao, X.; Cao, X.; Qiu, H.; Liang, W.; Jiang, Y.; Wang, Q.; Wang, W.; Li, C.; Li, Y.; Han, B.; et al. Rational molecular design converting fascaplysin derivatives to potent broad-spectrum inhibitors against bacterial pathogens via targeting FtsZ. Eur. J. Med. Chem. 2024, 270, 116347. [Google Scholar] [CrossRef] [PubMed]
- Kuzmich, A.S.; Fedorov, S.N.; Shastina, V.V.; Shubina, L.K.; Radchenko, O.S.; Balaneva, N.N.; Zhidkov, M.E.; Park, J.-I.; Kwak, J.Y.; Stonik, V.A. The anticancer activity of 3- and 10-bromofascaplysins is mediated by caspase-8, -9, -3-dependent apoptosis. Bioorgan. Med. Chem. 2010, 18, 3834–3840. [Google Scholar] [CrossRef] [PubMed]
- Lyakhova, I.A.; Bryukhovetsky, I.S.; Kudryavtsev, I.V.; Khotimchenko, Y.S.; Zhidkov, M.E.; Kantemirov, A.V. Antitumor activity of fascaplysin derivatives on glioblastoma model in vitro. Bull. Exp. Biol. Med. 2018, 164, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Zhidkov, M.E.; Smirnova, P.A.; Tryapkin, O.A.; Kantemirov, A.V.; Khudyakova, Y.V.; Malyarenko, O.S.; Ermakova, S.P.; Grigorchuk, V.P.; Kaune, M.; Von Amsberg, G.; et al. Total syntheses and preliminary biological evaluation of brominated fascaplysin and reticulatine alkaloids and their analogues. Mar. Drugs 2019, 17, 496. [Google Scholar] [CrossRef] [PubMed]
- Spirin, P.; Shyrokova, E.; Lebedev, T.; Vagapova, E.; Smirnova, P.; Kantemirov, A.; Dyshlovoy, S.A.; von Amsberg, G.; Zhidkov, M.; Prassolov, V. Cytotoxic marine alkaloid 3,10-dibromofascaplysin induces apoptosis and synergizes with cytarabine resulting in leukemia cell death. Mar. Drugs 2021, 19, 489. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Kaune, M.; Hauschild, J.; Kriegs, M.; Hoffer, K.; Busenbender, T.; Smirnova, P.A.; Zhidkov, M.E.; Poverennaya, E.V.; Oh-Hohenhorst, S.J.; et al. Efficacy and mechanism of action of marine alkaloid 3,10-dibromofascaplysin in drug-resistant prostate cancer cells. Mar. Drugs 2020, 18, 609. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W.; Pelcman, B. Total syntheses of the marine sponge pigments fascaplysin and homofascaplysin B and C. J. Org. Chem. 1992, 57, 3636–3642. [Google Scholar] [CrossRef]
- Rocca, P.; Marsais, F.; Godard, A.; Quéguiner, G. A short synthesis of the antimicrobial marine sponge pigment fascaplysin. Tetrahedron Lett. 1993, 34, 7917–7918. [Google Scholar] [CrossRef]
- Molina, P.; Fresneda, P.M.; García-Zafra, S.; Almendros, P. Iminophosphorane-mediated syntheses of the fascaplysin alkaloid of marine origin and nitramarine. Tetrahedron Lett. 1994, 35, 8851–8854. [Google Scholar] [CrossRef]
- Radchenko, O.S.; Novikov, V.L.; Elyakov, G.B. A simple and practical approach to the synthesis of the marine sponge pigment fascaplysin and related compounds. Tetrahedron Lett. 1997, 38, 5339–5342. [Google Scholar] [CrossRef]
- Waldmann, H.; Eberhardt, L.; Wittstein, K.; Kumar, K. Silver catalyzed cascade synthesis of alkaloid ring systems: Concise total synthesis of fascaplysin, homofascaplysin C and analogues. Chem. Commun. 2010, 46, 4622–4624. [Google Scholar] [CrossRef] [PubMed]
- Zhidkov, M.E.; Baranova, O.V.; Kravchenko, N.S.; Dubovitskii, S.V. A new method for the synthesis of the marine alkaloid fascaplysin. Tetrahedron Lett. 2010, 51, 6498–6499. [Google Scholar] [CrossRef]
- Tryapkin, O.A.; Kantemirov, A.V.; Dyshlovoy, S.A.; Prassolov, V.S.; Spirin, P.V.; von Amsberg, G.; Sidorova, M.A.; Zhidkov, M.E. A new mild method for synthesis of marine alkaloid fascaplysin and its therapeutically promising derivatives. Mar. Drugs 2023, 21, 424. [Google Scholar] [CrossRef] [PubMed]
- Zhidkov, M.E.; Kaminskii, V.A. A new method for the synthesis of the marine alkaloid fascaplysin based on the microwave-assisted Minisci reaction. Tetrahedron Lett. 2013, 54, 3530–3532. [Google Scholar] [CrossRef]
- Zhu, Y.-P.; Liu, M.-C.; Cai, Q.; Jia, F.-C.; Wu, A.-X. A cascade coupling strategy for one-pot total synthesis of β-carboline and isoquinoline-containing natural products and derivatives. Chem. A Eur. J. 2013, 19, 10132–10137. [Google Scholar] [CrossRef] [PubMed]
- Zhidkov, M.E.; Kantemirov, A.V.; Koisevnikov, A.V.; Andin, A.N.; Kuzmich, A.S. Syntheses of the marine alkaloids 6-oxofascaplysin, fascaplysin and their derivatives. Tetrahedron Lett. 2018, 59, 708–711. [Google Scholar] [CrossRef]
- Palani, V.; Perea, M.A.; Gardner, K.E.; Sarpong, R. A pyrone remodeling strategy to access diverse heterocycles: Application to the synthesis of fascaplysin natural products. Chem. Sci. 2021, 12, 1528–1534. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Baranova, O.V.; Balaneva, N.N.; Fedorov, S.N.; Radchenko, O.S.; Dubovitskii, S.V. The first syntheses of 3-bromofascaplysin, 10-bromofascaplysin and 3,10-dibromofascaplysin—Marine alkaloids from Fascaplysinopsis reticulata and Didemnum sp. by application of a simple and effective approach to the pyrido[1,2-a:3,4-b′]diindole system. Tetrahedron Lett. 2007, 48, 7998–8000. [Google Scholar] [CrossRef]
- Schumacher, R.W.; Davidson, B.S. Synthesis of didemnolines A-D, N9-substituted β-carboline alkaloids from the marine ascidian Didemnum sp. Tetrahedron 1999, 55, 935–942. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Kaune, M.; Kantemirov, A.V.; Smirnova, P.A.; Spirin, P.V.; Sidorova, M.A.; Stadnik, S.A.; Shyrokova, E.Y.; Kaluzhny, D.N.; Tryapkin, O.A.; et al. Study of structure–activity relationships of the marine alkaloid fascaplysin and its derivatives as potent anticancer agents. Mar. Drugs 2022, 20, 185. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standarts Institute M100 Performance Standards for Antimicrobial Susceptibility Testing. 2020. Available online: https://clsi.org/standards/products/microbiology/documents/m100/ (accessed on 23 March 2021).
- Council of Europe European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Purposes. Strasbourg: 1986, 18.III.1986, Council of Europe, ETS No.123. Available online: https://rm.coe.int/168007a67b (accessed on 28 August 2018).
- Directive 2010/63/EU on the Protection of Animals Used for Scientific Purposes EN. Official Journal of the European Union, L 276/33-276/79 (20.10.2010). Available online: https://eur-lex.europa.eu/eli/dir/2010/63/oj/eng/pdf (accessed on 29 January 2025).
- National State Standard GOST 33044-2014 the Russian Federation Standard “The Principles of Good Laboratory Practice” (Approved and Put into Effect by the Order of the Federal Agency for Technical Regulation and Metrology of October 20, 2014), No 1700. Available online: https://docs.cntd.ru/document/1200115791 (accessed on 1 August 2015). (In Russian).
- Behrens, B. Zur Auswertung der Digitalisblätter im Forschversuch. Arch. Exp. Pathol. Pharmakol. 1929, 140, 237. [Google Scholar] [CrossRef]
- National State Standard GOST 33216-2014 the Russian Federation Standard “Guidelines for Accommodation and Care of Animals. Species-Specific Provisions for Laboratory Rodents and Rabbits” (Approved and Put into Effect by the Order of the Federal Agency for Technical Regulation and Metrology of December 22, 2014), No 73P. Available online: https://docs.cntd.ru/document/1200127506 (accessed on 1 August 2015). (In Russian).
- Elsherbiny, N.M.; Younis, N.N.; Shaheen, M.A.; Elseweidy, M.M. The synergistic effect between vanillin and doxorubicin in Ehrlich ascites carcinoma solid tumour and MCF-7 human breast cancer cell line. Pathol. Res. Pract. 2016, 212, 767–777. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MIC, µg/mL | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S. aureus ATCC 29213 | B. cereus ATCC 10702 | E. faecalis ATCC 29212 | E. faecium 132 | E. faecium 130 (VRE) | E. faecalis 583 (VRE) | S. aureus 88 (MRSA) | S. aureus PE3R (MRSA) | S. epidermidis 2001 MR | S. aureus 21555 | M. smegmatis ATCC 607 | E. coli ATCC 25922 | |
| Van | 0.5 | 1.0 | 2.0 | 0.5 | >32.0 | 32.0 | 0.5 | 1.0 | 1.0 | 2.0 | - | - |
| Rif | 0.018 | 0.25 | - | - | - | - | - | - | - | - | 0.03 | 8.0 |
| 2 | 1.0 | 0.125 | 8.0 | 1.0 | 1.0 | ≥8.0 | 1.0 | 0.5 | 0.0075 | 0.03 | 0.03 | 8.0 |
| 3 | 0.06 | 0.03 | 0.25 | 2.0 | 1.0 | 0.5 | 0.06 | 0.06 | 0.03 | 0.06 | 0.5 | 8.0 |
| 4 | 0.25 | 0.13 | 1.0 | 8.0 | 8.0 | 0.5 | 0.25 | 0.25 | 0.03 | 0.25 | 0.5 | 16.0 |
| 5 | 0.06 | 0.06 | 0.25 | 4.0 | 0.25 | 0.125 | 0.06 | 0.06 | 0.015 | 0.03 | 1.0 | >8.0 |
| 13 | 0.03 | 0.015 | 0.03 | 0.5 | 0.06 | 0.03 | 0.03 | 0.015 | 0.0075 | 0.015 | 0.5 | >16.0 |
| 14 | 0.03 | 0.015 | 0.015 | 0.5 | 0.015 | 0.0075 | 0.015 | 0.0075 | 0.0075 | 0.00375 | 0.25 | 4.0 |
| 21 | 0.03 | 0.015 | 0.015 | 0.25 | 0.03 | 0.015 | 0.015 | 0.015 | 0.0075 | 0.015 | 0.25 | 4.0 |
| 22 | 0.015 | 0.015 | 0.015 | 0.25 | 0.03 | 0.015 | 0.015 | 0.015 | 0.00375 | 0.015 | 0.25 | 4.0 |
| 23 | 0.015 | 0.015 | 0.015 | 0.25 | 0.03 | 0.015 | 0.03 | 0.015 | 0.0018 | 0.015 | 0.25 | >16.0 |
| 24 | 0.03 | 0.03 | 0.03 | 0.25 | 0.015 | 0.06 | 0.015 | 0.015 | 0.0075 | 0.015 | 0.5 | >16.0 |
| 9a | 0.03 | 0.03 | 0.25 | 4.0 | 2.0 | 0.25 | 0.015 | 0.03 | 0.00375 | 0.03 | 0.25 | 8.0 |
| Group | Daily Dose | MST, Days | ILS, % | Survival, % |
|---|---|---|---|---|
| Control | - | 16.1 ± 5.0 | - | 0 |
| Compound 5 | 7.56 mg/kg | 49.3 ± 14.63 * | 206 | 40 |
| Dox | 0.25 mg/kg | 62.0 ± 0.0 ** | 284.8 | 60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhidkov, M.E.; Smirnova, P.A.; Grammatikova, N.E.; Isakova, E.B.; Shchekotikhin, A.E.; Styshova, O.N.; Klimovich, A.A.; Popov, A.M. Comparative Evaluation of the Antibacterial and Antitumor Activities of Marine Alkaloid 3,10-Dibromofascaplysin. Mar. Drugs 2025, 23, 68. https://doi.org/10.3390/md23020068
Zhidkov ME, Smirnova PA, Grammatikova NE, Isakova EB, Shchekotikhin AE, Styshova ON, Klimovich AA, Popov AM. Comparative Evaluation of the Antibacterial and Antitumor Activities of Marine Alkaloid 3,10-Dibromofascaplysin. Marine Drugs. 2025; 23(2):68. https://doi.org/10.3390/md23020068
Chicago/Turabian StyleZhidkov, Maxim E., Polina A. Smirnova, Natalia E. Grammatikova, Elena B. Isakova, Andrey E. Shchekotikhin, Olga N. Styshova, Anna A. Klimovich, and Aleksandr M. Popov. 2025. "Comparative Evaluation of the Antibacterial and Antitumor Activities of Marine Alkaloid 3,10-Dibromofascaplysin" Marine Drugs 23, no. 2: 68. https://doi.org/10.3390/md23020068
APA StyleZhidkov, M. E., Smirnova, P. A., Grammatikova, N. E., Isakova, E. B., Shchekotikhin, A. E., Styshova, O. N., Klimovich, A. A., & Popov, A. M. (2025). Comparative Evaluation of the Antibacterial and Antitumor Activities of Marine Alkaloid 3,10-Dibromofascaplysin. Marine Drugs, 23(2), 68. https://doi.org/10.3390/md23020068