

Fucoxanthin from Laminaria japonica Targeting PANoptosis and Ferroptosis Pathways: Insights into Its Therapeutic Potential Against Ovarian Cancer


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Fucoxanthin Suppresses the Proliferation, Migration, and Invasion of Ovarian Cancer Cells
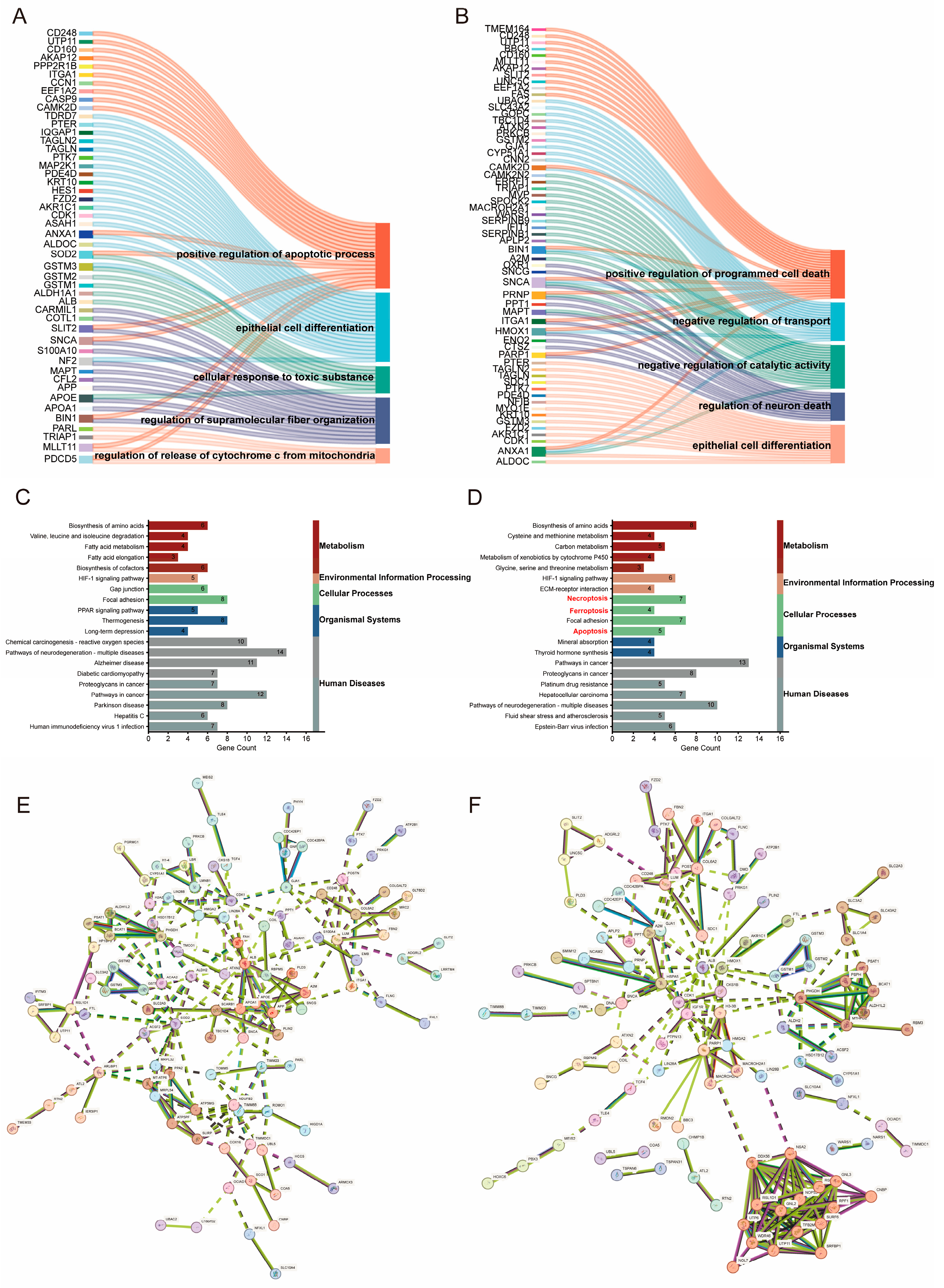
2.2. Proteomics-Based Study of the Mechanism of FX-Induced Programmed Cell Death in Ovarian Cancer Cells
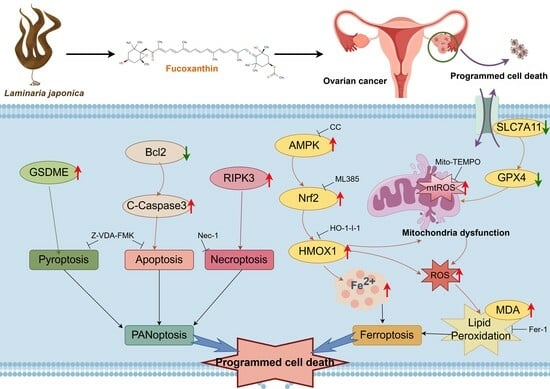
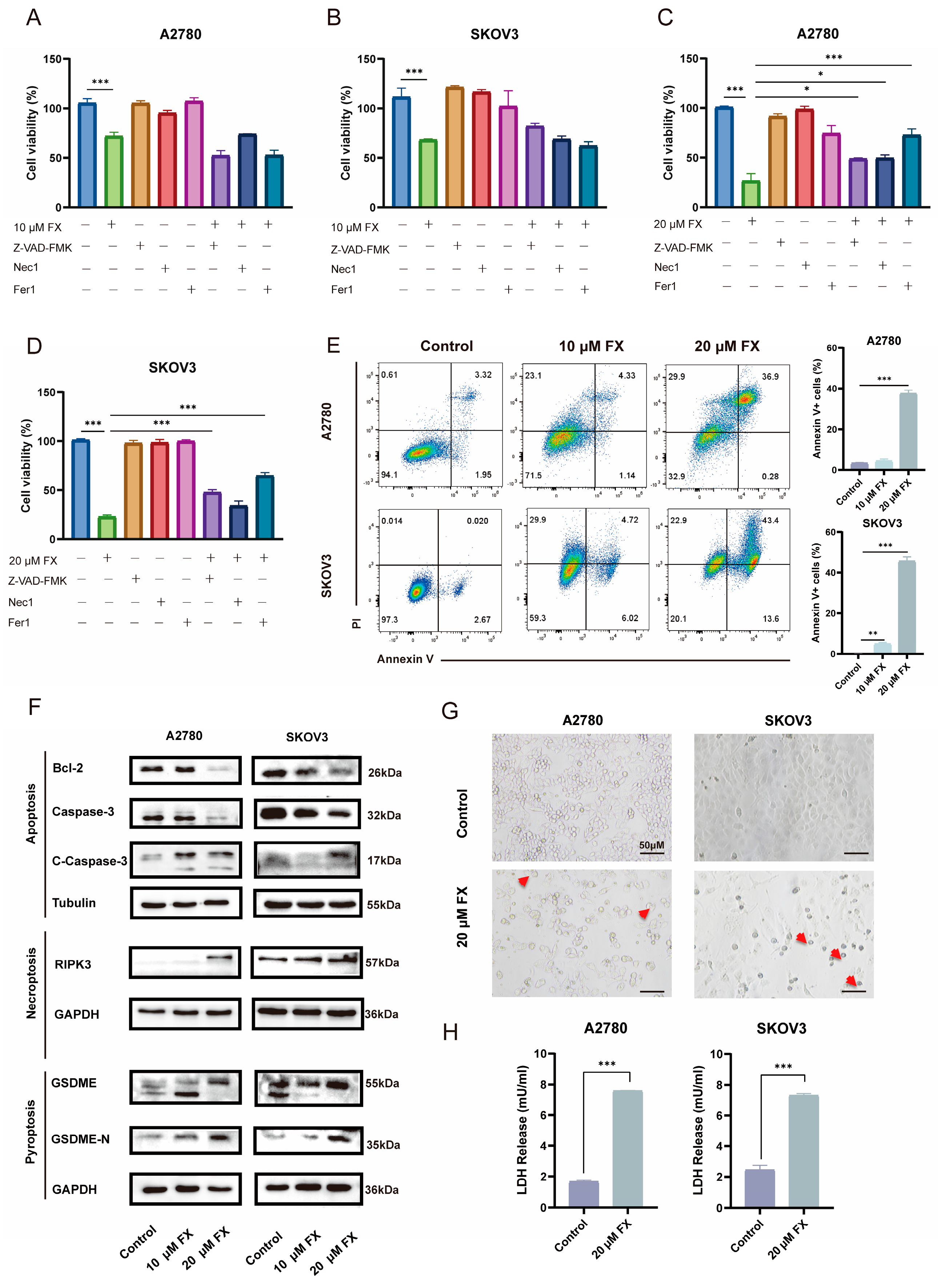
2.3. FX-Induced PANoptosis in Ovarian Cancer Cells
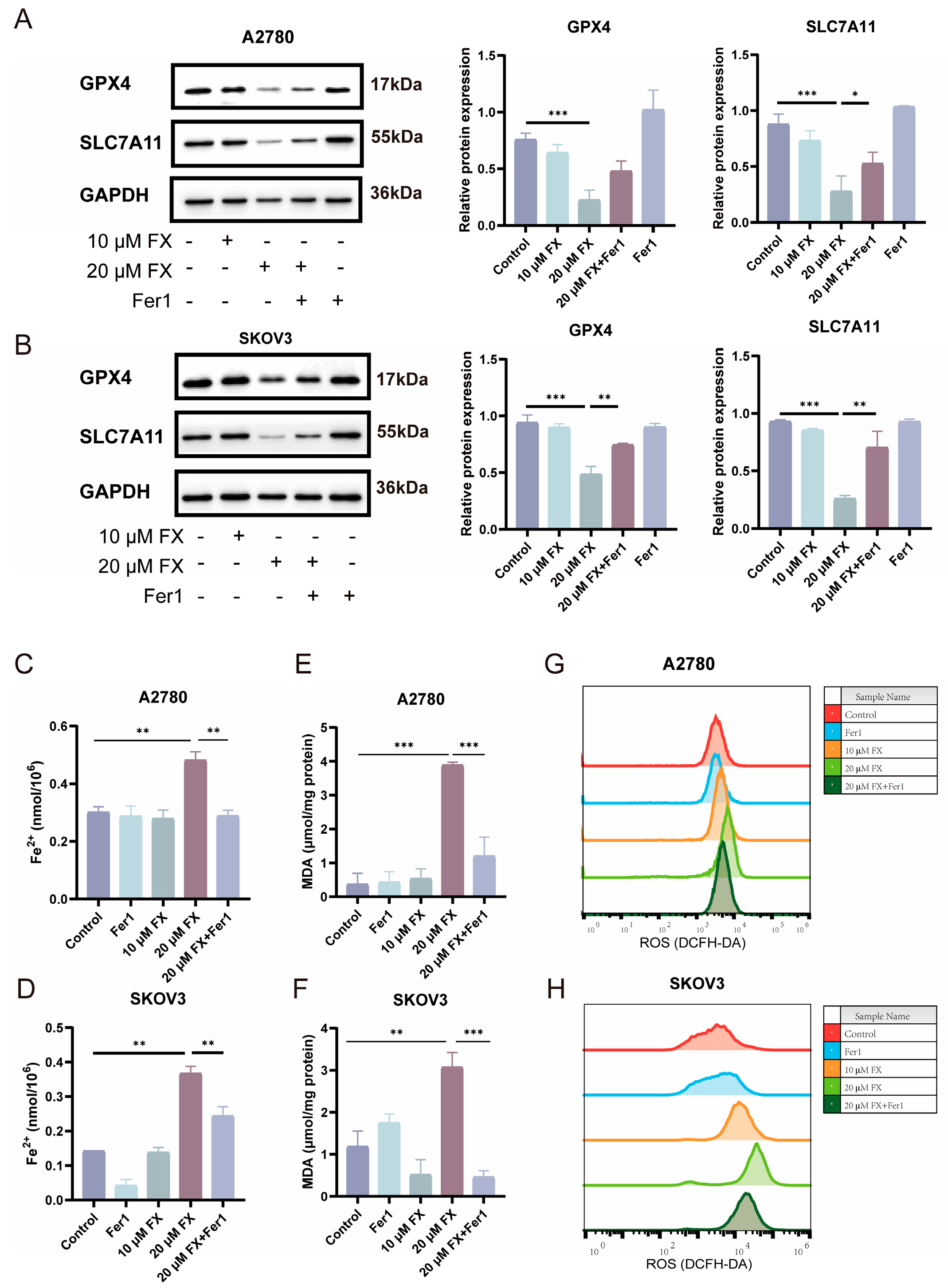
2.4. FX-Induced Ferroptosis in OC Cells
2.5. FX-Induced Ferroptosis in Ovarian Cancer Cells Is Associated with Mitochondrial Dysfunction
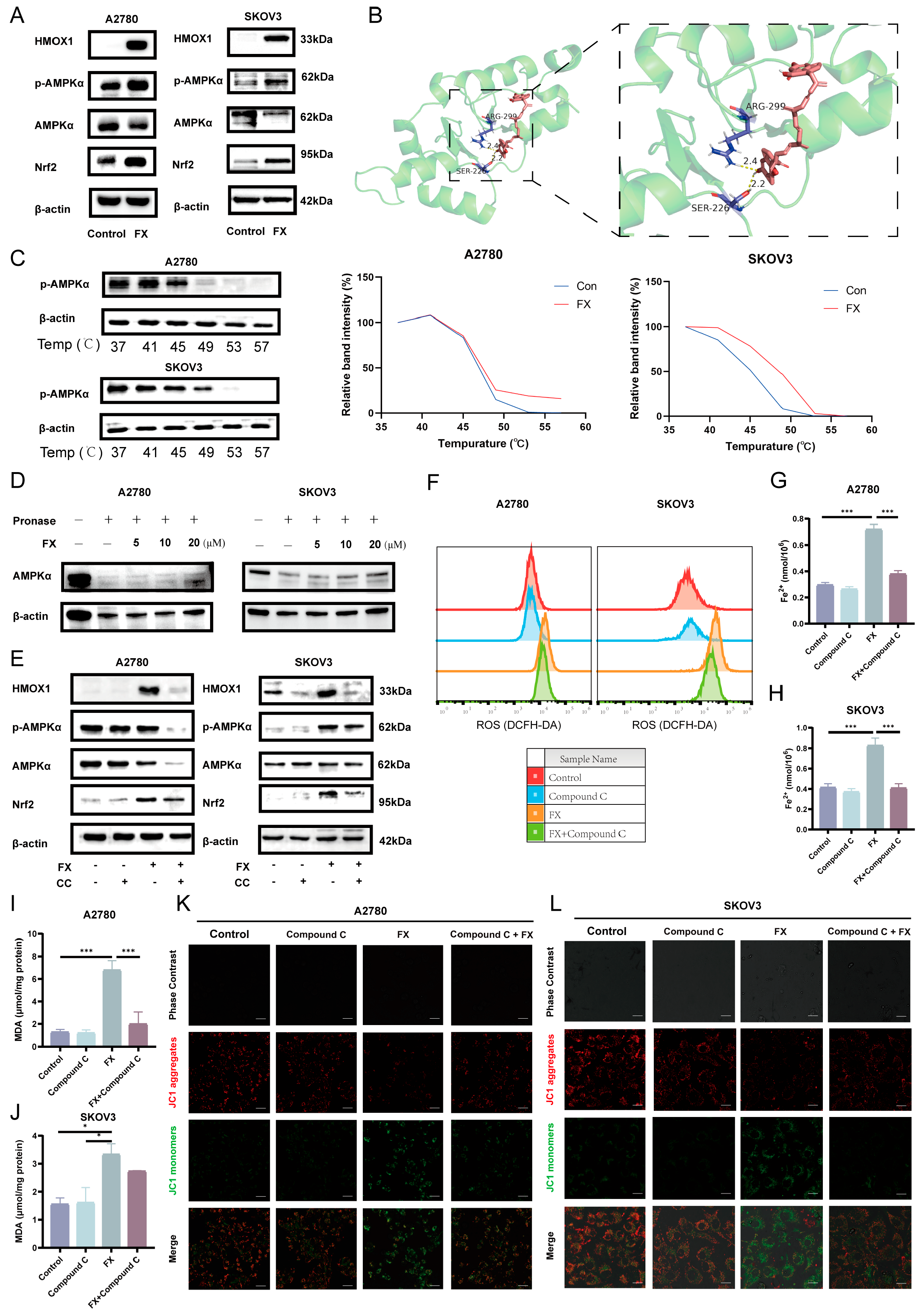
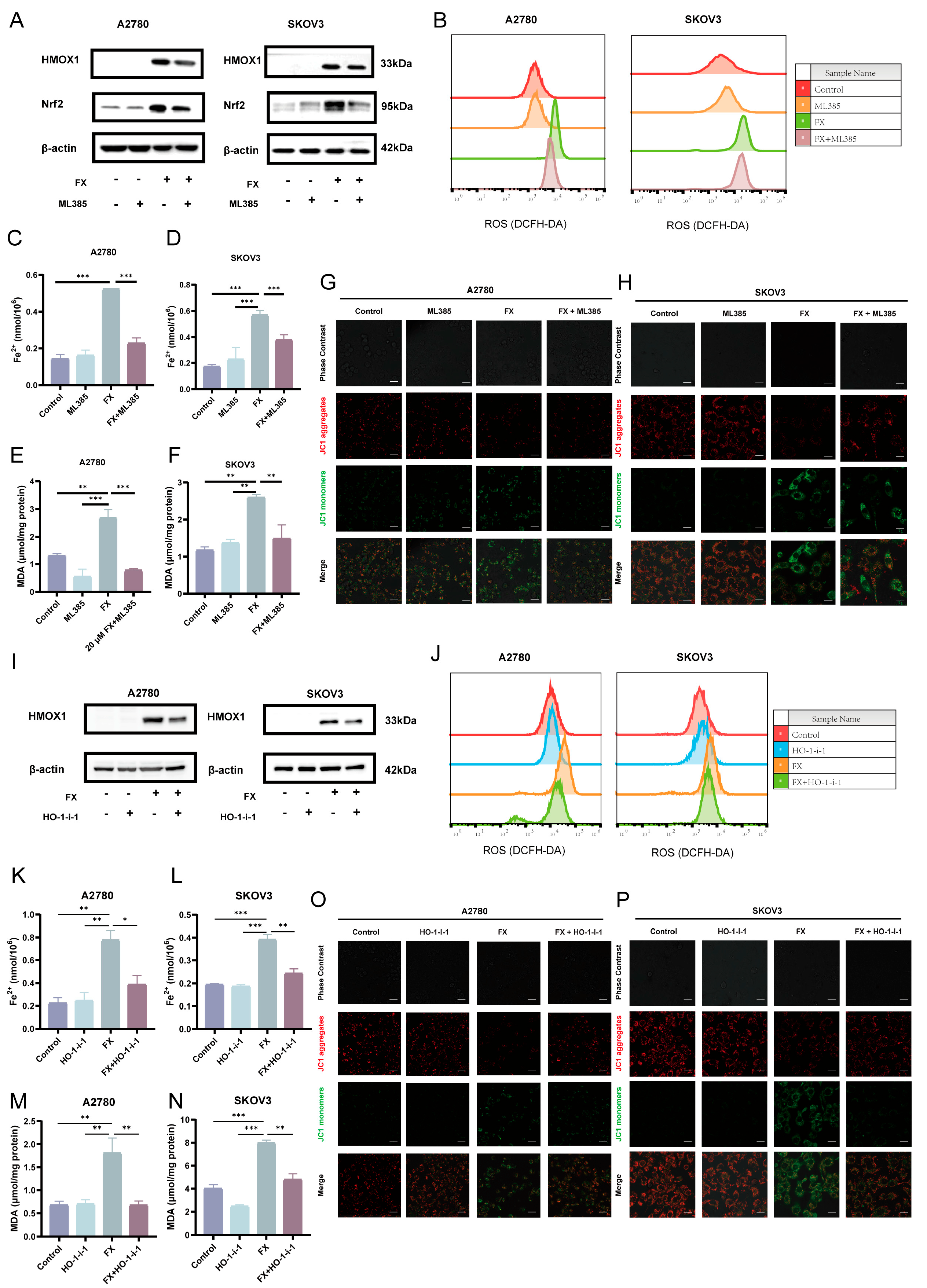
2.6. Fucoxanthin-Induced Ferroptosis via the AMPK/Nrf2/HMOX-1 Pathway in OC Cells
2.7. FX-Induced Ferroptosis in Ovarian Cancer and Suppressed Tumor Growth in a Mouse Model
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Reagents
4.3. Cell Viability Assay
4.4. Colony Formation Assay
4.5. Migration and Invasion Assays
4.6. Proteomics
4.7. Apoptosis Analysis
4.8. Western Blot Analysis
4.9. Measurement of Fe2+
4.10. Measurement of Malondialdehyde (MDA)
4.11. Measurement of ROS
4.12. Transmission Electron Microscopy (TEM)
4.13. Mitochondrial Membrane Potential
4.14. MitoSOX Red Staining
4.15. Real-Time Quantitative PCR Analysis
4.16. Auto Docking
4.17. Cellular Thermal Shift Assay (CETSA)
4.18. Drug Affinity Responsive Target Stability (DARTS)
4.19. In Vivo Tumor Xenograft Model
4.20. Immunohistochemistry (IHC) Assay
4.21. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CC | Compound C |
| DEPs | Differently expressed proteins |
| Fer-1 | Ferrostatin-1 |
| FX | Fucoxanthin |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| IC50 | The half inhibitory concentration |
| GPX4 | Glutathione peroxidase 4 |
| HO-1-i-1 | Heme oxygenase-1-IN-1 |
| LDH | Lactate dehydrogenase |
| PCD | Programmed cell death |
| PPI | Protein–protein interaction |
| SLC7A11 | Solute carrier family 7 member 11 |
| IHC | Immunohistochemistry |
| MDA | Malondialdehyde |
| MMP | Mitochondrial membrane potential |
| MTT | Methylthiazolyldiphenyl-tetrazolium bromide |
| Nec-1 | Necrostatin-1 |
| OC | Ovarian cancer |
| ROS | Reactive oxygen species |
| TEM | Transmission electron microscopy |
References
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer statistics, 2025. CA A Cancer J. Clin. 2025, 75, 10–45. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.L.; Eskander, R.N.; O’Malley, D.M. Advances in Ovarian Cancer Care and Unmet Treatment Needs for Patients with Platinum Resistance: A Narrative Review. JAMA Oncol. 2023, 9, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, L.; Guntupalli, S.R. Treatment of epithelial ovarian cancer. BMJ (Clin. Res. Ed.) 2020, 371, m3773. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA A Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef]
- Xiong, Y. The emerging role of PANoptosis in cancer treatment. Biomed. Pharmacother. = Biomed. Pharmacother. 2023, 168, 115696. [Google Scholar] [CrossRef]
- Ocansey, D.K.W.; Qian, F.; Cai, P.; Ocansey, S.; Amoah, S.; Qian, Y.; Mao, F. Current evidence and therapeutic implication of PANoptosis in cancer. Theranostics 2024, 14, 640–661. [Google Scholar] [CrossRef]
- Qi, Z.; Zhu, L.; Wang, K.; Wang, N. PANoptosis: Emerging mechanisms and disease implications. Life Sci. 2023, 333, 122158. [Google Scholar] [CrossRef]
- Chen, Y.; Deng, Z.; Chen, J.; Lin, J.; Zou, J.; Li, S.; Sun, Y. PANoptosis-Relevant Subgroups Predicts Prognosis and Characterizes the Tumour Microenvironment in Ovarian Cancer. J. Inflamm. Res. 2024, 17, 9773–9793. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Chen, Y.M.; Xu, W.; Liu, Y.; Zhang, J.H.; Yang, Y.Y.; Wang, Z.W.; Sun, D.J.; Li, H.; Liu, B.; Chen, L.X. Anomanolide C suppresses tumor progression and metastasis by ubiquitinating GPX4-driven autophagy-dependent ferroptosis in triple negative breast cancer. Int. J. Biol. Sci. 2023, 19, 2531–2550. [Google Scholar] [CrossRef]
- Xia, X.; Fan, X.; Zhao, M.; Zhu, P. The Relationship between Ferroptosis and Tumors: A Novel Landscape for Therapeutic Approach. Curr. Gene Ther. 2019, 19, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Basuli, D.; Tesfay, L.; Deng, Z.; Paul, B.; Yamamoto, Y.; Ning, G.; Xian, W.; McKeon, F.; Lynch, M.; Crum, C.P.; et al. Iron addiction: A novel therapeutic target in ovarian cancer. Oncogene 2017, 36, 4089–4099. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Huang, T.; Li, Y.; Zhang, H. Integrative Gut Microbiota and Metabolomic Analyses Reveal the PANoptosis- and Ferroptosis-Related Mechanisms of Chrysoeriol in Inhibiting Melanoma. J. Agric. Food Chem. 2024, 72, 25173–25185. [Google Scholar] [CrossRef]
- Saeed, A.; Su, J.; Ouyang, S. Marine-derived drugs: Recent advances in cancer therapy and immune signaling. Biomed. Pharmacother. = Biomed. Pharmacother. 2021, 134, 111091. [Google Scholar] [CrossRef]
- Florean, C.; Dicato, M.; Diederich, M. Immune-modulating and anti-inflammatory marine compounds against cancer. Semin. Cancer Biol. 2022, 80, 58–72. [Google Scholar] [CrossRef]
- Dong, H.; Dong, S.; Erik Hansen, P.; Stagos, D.; Lin, X.; Liu, M. Progress of Bromophenols in Marine Algae from 2011 to 2020: Structure, Bioactivities, and Applications. Mar. Drugs 2020, 18, 411. [Google Scholar] [CrossRef]
- Salido, M.; Soto, M.; Seoane, S. Seaweed: Nutritional and gastronomic perspective. A review. Algal Res. 2024, 77, 103357. [Google Scholar] [CrossRef]
- Karpiński, T.M.; Adamczak, A. Fucoxanthin-An Antibacterial Carotenoid. Antioxidants 2019, 8, 239. [Google Scholar] [CrossRef]
- Maeda, H.; Hosokawa, M.; Sashima, T.; Takahashi, N.; Kawada, T.; Miyashita, K. Fucoxanthin and its metabolite, fucoxanthinol, suppress adipocyte differentiation in 3T3-L1 cells. Int. J. Mol. Med. 2006, 18, 147–152. [Google Scholar] [CrossRef]
- Nishino, H. Cancer prevention by carotenoids. Mutat. Res. 1998, 402, 159–163. [Google Scholar] [CrossRef]
- Sachindra, N.M.; Sato, E.; Maeda, H.; Hosokawa, M.; Niwano, Y.; Kohno, M.; Miyashita, K. Radical scavenging and singlet oxygen quenching activity of marine carotenoid fucoxanthin and its metabolites. J. Agric. Food Chem. 2007, 55, 8516–8522. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, V.; Mamatha, B.S. Fucoxanthin, a Functional Food Ingredient: Challenges in Bioavailability. Curr. Nutr. Rep. 2023, 12, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Rengarajan, T.; Rajendran, P.; Nandakumar, N.; Balasubramanian, M.P.; Nishigaki, I. Cancer preventive efficacy of marine carotenoid fucoxanthin: Cell cycle arrest and apoptosis. Nutrients 2013, 5, 4978–4989. [Google Scholar] [CrossRef] [PubMed]
- Rwigemera, A.; Mamelona, J.; Martin, L.J. Comparative effects between fucoxanthinol and its precursor fucoxanthin on viability and apoptosis of breast cancer cell lines MCF-7 and MDA-MB-231. Anticancer Res. 2015, 35, 207–219. [Google Scholar]
- Lu, Y.; Wang, Y.; Yao, T.; Dong, X.; Liu, Y.; Nakamura, Y.; Qi, H. Mechanism of inhibition of melanoma by fucoxanthin simulated in vitro digestion products in cell models constructed using human malignant melanoma cells (A375) and keratinocytes (HaCaT). Food Chem. 2025, 462, 141003. [Google Scholar] [CrossRef]
- Wang, W.; Fu, C.; Lin, M.; Lu, Y.; Lian, S.; Xie, X.; Zhou, G.; Li, W.; Zhang, Y.; Jia, L.; et al. Fucoxanthin prevents breast cancer metastasis by interrupting circulating tumor cells adhesion and transendothelial migration. Front. Pharmacol. 2022, 13, 960375. [Google Scholar] [CrossRef]
- Gangadhar, K.N.; Rodrigues, M.J.; Pereira, H.; Gaspar, H.; Malcata, F.X.; Barreira, L.; Varela, J. Anti-Hepatocellular Carcinoma (HepG2) Activities of Monoterpene Hydroxy Lactones Isolated from the Marine Microalga Tisochrysis Lutea. Mar. Drugs 2020, 18, 567. [Google Scholar] [CrossRef]
- Ming, J.X.; Wang, Z.C.; Huang, Y.; Ohishi, H.; Wu, R.J.; Shao, Y.; Wang, H.; Qin, M.Y.; Wu, Z.L.; Li, Y.Y.; et al. Fucoxanthin extracted from Laminaria Japonica inhibits metastasis and enhances the sensitivity of lung cancer to Gefitinib. J. Ethnopharmacol. 2021, 265, 113302. [Google Scholar] [CrossRef]
- Mei, C.; Zhou, S.; Zhu, L.; Ming, J.; Zeng, F.; Xu, R. Antitumor Effects of Laminaria Extract Fucoxanthin on Lung Cancer. Mar. Drugs 2017, 15, 39. [Google Scholar] [CrossRef]
- Sani, F.V.; Kamalian, S.; Abdi, H.; Ghofrani, S.; Boustan, A.; Mosaffa, F. Fucoxanthin Inhibits the Proliferation of ABCC2-Over Expressing Cisplatin-Resistance Ovarian Cancer Cells via Inducing Apoptosis. Pharm. Sci. 2023, 29, 320–327. [Google Scholar] [CrossRef]
- Gevorgiz, R.G.; Gureev, M.A.; Zheleznova, S.N.; Gureeva, E.V.; Nechoroshev, M.V. Production of Diadinoxanthin in an Intensive Culture of the Diatomaceous Alga Cylindrotheca closterium (Ehrenb.) Reimann et Lewin. and Its Proapoptotic Activity. Appl. Biochem. Microbiol. 2022, 58, 261–268. [Google Scholar] [CrossRef]
- Mosaffa, F.; Sani, F.V.; Ghofrani, S.; Kamalian, S.; Gharaee, M.E. Evaluating the Anti-Migratory Effect of Fucoxanthin in Human Cisplatin-Resistant Ovarian Cancer Cells. J. Adv. Med. Biomed. Res. 2024, 32, 135–143. [Google Scholar] [CrossRef]
- Eid, S.Y.; Althubiti, M.A.; Abdallah, M.E.; Wink, M.; El-Readi, M.Z. The carotenoid fucoxanthin can sensitize multidrug resistant cancer cells to doxorubicin via induction of apoptosis, inhibition of multidrug resistance proteins and metabolic enzymes. Phytomedicine 2020, 77, 153280. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, Y.; Li, J. Fucoxanthin suppresses the malignant progression of ovarian cancer by inactivating STAT3/c-Myc signaling. Am. J. Transl. Res. 2023, 15, 2528–2540. [Google Scholar]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef]
- Li, Q.; Shi, N.; Cai, C.; Zhang, M.; He, J.; Tan, Y.; Fu, W. The Role of Mitochondria in Pyroptosis. Front. Cell Dev. Biol. 2020, 8, 630771. [Google Scholar] [CrossRef]
- Weindel, C.G.; Martinez, E.L.; Zhao, X.; Mabry, C.J.; Bell, S.L.; Vail, K.J.; Coleman, A.K.; VanPortfliet, J.J.; Zhao, B.; Wagner, A.R.; et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell 2022, 185, 3214–3231.e23. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Wei, S.; Nguyen, T.H.; Jo, Y.; Zhang, Y.; Park, W.; Gariani, K.; Oh, C.M.; Kim, H.H.; Ha, K.T.; et al. Mitochondria-associated programmed cell death as a therapeutic target for age-related disease. Exp. Mol. Med. 2023, 55, 1595–1619. [Google Scholar] [CrossRef]
- Wei, S.; Qiu, T.; Yao, X.; Wang, N.; Jiang, L.; Jia, X.; Tao, Y.; Wang, Z.; Pei, P.; Zhang, J.; et al. Arsenic induces pancreatic dysfunction and ferroptosis via mitochondrial ROS-autophagy-lysosomal pathway. J. Hazard. Mater. 2020, 384, 121390. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef]
- Li, Y.; Cen, Y.; Fang, Y.; Tang, S.; Li, S.; Ren, Y.; Zhang, H.; Lu, W.; Xu, J. Breaking the Iron Homeostasis: A “Trojan Horse” Self-Assembled Nanodrug Sensitizes Homologous Recombination Proficient Ovarian Cancer Cells to PARP Inhibition. ACS Nano 2022, 16, 12786–12800. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, P.; Li, Z.; Xu, C.; Wang, H.; Jia, B.; Gong, A.; Xu, M. Vitamin C Sensitizes Pancreatic Cancer Cells to Erastin-Induced Ferroptosis by Activating the AMPK/Nrf2/HMOX1 Pathway. Oxidative Med. Cell. Longev. 2022, 2022, 5361241. [Google Scholar] [CrossRef]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef]
- Matzinger, M.; Fischhuber, K.; Pölöske, D.; Mechtler, K.; Heiss, E.H. AMPK leads to phosphorylation of the transcription factor Nrf2, tuning transactivation of selected target genes. Redox Biol. 2020, 29, 101393. [Google Scholar] [CrossRef]
- Wang, X.; Chen, X.; Zhou, W.; Men, H.; Bao, T.; Sun, Y.; Wang, Q.; Tan, Y.; Keller, B.B.; Tong, Q.; et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm. Sin. B 2022, 12, 708–722. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, Y.; Lei, H.; Cai, Y.; Shen, J.; Zhu, P.; He, Q.; Zhao, M. The Nrf-2/HO-1 Signaling Axis: A Ray of Hope in Cardiovascular Diseases. Cardiol. Res. Pract. 2020, 2020, 5695723. [Google Scholar] [CrossRef]
- Ji, X.; Chen, Z.; Lin, W.; Wu, Q.; Wu, Y.; Hong, Y.; Tong, H.; Wang, C.; Zhang, Y. Esculin induces endoplasmic reticulum stress and drives apoptosis and ferroptosis in colorectal cancer via PERK regulating eIF2α/CHOP and Nrf2/HO-1 cascades. J. Ethnopharmacol. 2024, 328, 118139. [Google Scholar] [CrossRef]
- Zhou, Q.; Meng, Y.; Li, D.; Yao, L.; Le, J.; Liu, Y.; Sun, Y.; Zeng, F.; Chen, X.; Deng, G. Ferroptosis in cancer: From molecular mechanisms to therapeutic strategies. Signal Transduct. Target. Ther. 2024, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.M.; Sessa, C.; Krasner, C.N.; Vermorken, J.B.; Colombo, N.; Kaye, S.; Gore, M.; Zintl, P.; Gómez, J.; Parekh, T.; et al. Trabectedin as single agent in relapsed advanced ovarian cancer: Results from a retrospective pooled analysis of three phase II trials. Med. Oncol. 2013, 30, 435. [Google Scholar] [CrossRef]
- Morgan, R.J., Jr.; Leong, L.; Chow, W.; Gandara, D.; Frankel, P.; Garcia, A.; Lenz, H.J.; Doroshow, J.H. Phase II trial of bryostatin-1 in combination with cisplatin in patients with recurrent or persistent epithelial ovarian cancer: A California cancer consortium study. Investig. New Drugs 2012, 30, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Miyake, R.; Yamanaka, S.; Matsubara, S.; Mabuchi, S. Preclinical Activity of Plitidepsin Against Clear Cell Carcinoma of the Ovary. Anticancer Res. 2021, 41, 4277–4285. [Google Scholar] [CrossRef]
- El-Seedi, H.R.; Refaey, M.S.; Elias, N.; El-Mallah, M.F.; Albaqami, F.M.K.; Dergaa, I.; Du, M.; Salem, M.F.; Tahir, H.E.; Dagliaa, M.; et al. Marine natural products as a source of novel anticancer drugs: An updated review (2019–2023). Nat. Prod. Bioprospecting 2025, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.A.; Mendonca, P.; Elhag, R.; Soliman, K.F.A. Anticancer Effects of Fucoxanthin through Cell Cycle Arrest, Apoptosis Induction, Angiogenesis Inhibition, and Autophagy Modulation. Int. J. Mol. Sci. 2022, 23, 16091. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.C.; Kolb, E.A.; Sampson, V.B. Development of Chemotherapy with Cell-Cycle Inhibitors for Adult and Pediatric Cancer Therapy. Cancer Res. 2018, 78, 320–325. [Google Scholar] [CrossRef]
- Ahmed, S.A.; Mendonca, P.; Messeha, S.S.; Soliman, K.F.A. Anticancer Effects of Fucoxanthin through Cell Cycle Arrest, Apoptosis Induction, and Angiogenesis Inhibition in Triple-Negative Breast Cancer Cells. Molecules 2023, 28, 6536. [Google Scholar] [CrossRef]
- Almeida, T.P.; Ferreira, J.; Vettorazzi, A.; Azqueta, A.; Rocha, E.; Ramos, A.A. Cytotoxic activity of fucoxanthin, alone and in combination with the cancer drugs imatinib and doxorubicin, in CML cell lines. Environ. Toxicol. Pharmacol. 2018, 59, 24–33. [Google Scholar] [CrossRef]
- Zhu, Y.; Cheng, J.; Min, Z.; Yin, T.; Zhang, R.; Zhang, W.; Hu, L.; Cui, Z.; Gao, C.; Xu, S.; et al. Effects of fucoxanthin on autophagy and apoptosis in SGC-7901cells and the mechanism. J. Cell. Biochem. 2018, 119, 7274–7284. [Google Scholar] [CrossRef]
- Hou, G.; Chen, Y.; Lei, H.; Lu, S.; Cheng, L. Nanomaterials-Induced PANoptosis: A Promising Anti-Tumor Strategy. Angew. Chem. (Int. Ed. Engl.) 2025, 64, e202419649. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.; Jang, Y.; Karki, R.; Han, J.H. Implications of inflammatory cell death-PANoptosis in health and disease. Arch. Pharmacal Res. 2024, 47, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, Y.; Guo, Y.; Shi, X.; Chen, X.; Feng, W.; Wu, L.L.; Zhang, J.; Yu, S.; Wang, Y.; et al. An Overview: The Diversified Role of Mitochondria in Cancer Metabolism. Int. J. Biol. Sci. 2023, 19, 897–915. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, C.; Wang, X.; Sun, Y.; Zhang, J.; Chen, J.; Shi, Y. An Epigenetic Role of Mitochondria in Cancer. Cells 2022, 11, 2518. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, Y. Mitochondria as a target in cancer treatment. MedComm 2020, 1, 129–139. [Google Scholar] [CrossRef]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc− Activity. Curr. Biol. CB 2018, 28, 2388–2399.e5. [Google Scholar] [CrossRef]
- Zimmermann, K.; Baldinger, J.; Mayerhofer, B.; Atanasov, A.G.; Dirsch, V.M.; Heiss, E.H. Activated AMPK boosts the Nrf2/HO-1 signaling axis—A role for the unfolded protein response. Free Radic. Biol. Med. 2015, 88 Pt B, 417–426. [Google Scholar] [CrossRef]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Gao, W.; Wang, Z.; Jian, H.; Peng, L.; Yu, X.; Xue, P.; Peng, W.; Li, K.; Zeng, P. Polyphyllin I induced ferroptosis to suppress the progression of hepatocellular carcinoma through activation of the mitochondrial dysfunction via Nrf2/HO-1/GPX4 axis. Phytomedicine Int. J. Phytother. Phytopharm. 2024, 122, 155135. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Mao, Y.; Liu, H.; Huang, Y.; Xu, R. Fucoxanthin from Laminaria japonica Targeting PANoptosis and Ferroptosis Pathways: Insights into Its Therapeutic Potential Against Ovarian Cancer. Mar. Drugs 2025, 23, 123. https://doi.org/10.3390/md23030123
Wang Y, Mao Y, Liu H, Huang Y, Xu R. Fucoxanthin from Laminaria japonica Targeting PANoptosis and Ferroptosis Pathways: Insights into Its Therapeutic Potential Against Ovarian Cancer. Marine Drugs. 2025; 23(3):123. https://doi.org/10.3390/md23030123
Chicago/Turabian StyleWang, Yaze, Yiru Mao, Hui Liu, Yi Huang, and Rong Xu. 2025. "Fucoxanthin from Laminaria japonica Targeting PANoptosis and Ferroptosis Pathways: Insights into Its Therapeutic Potential Against Ovarian Cancer" Marine Drugs 23, no. 3: 123. https://doi.org/10.3390/md23030123
APA StyleWang, Y., Mao, Y., Liu, H., Huang, Y., & Xu, R. (2025). Fucoxanthin from Laminaria japonica Targeting PANoptosis and Ferroptosis Pathways: Insights into Its Therapeutic Potential Against Ovarian Cancer. Marine Drugs, 23(3), 123. https://doi.org/10.3390/md23030123