
Functional Genomics of a Collection of Gammaproteobacteria Isolated from Antarctica

 ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. General Genome Features and Creation of Custom Databases
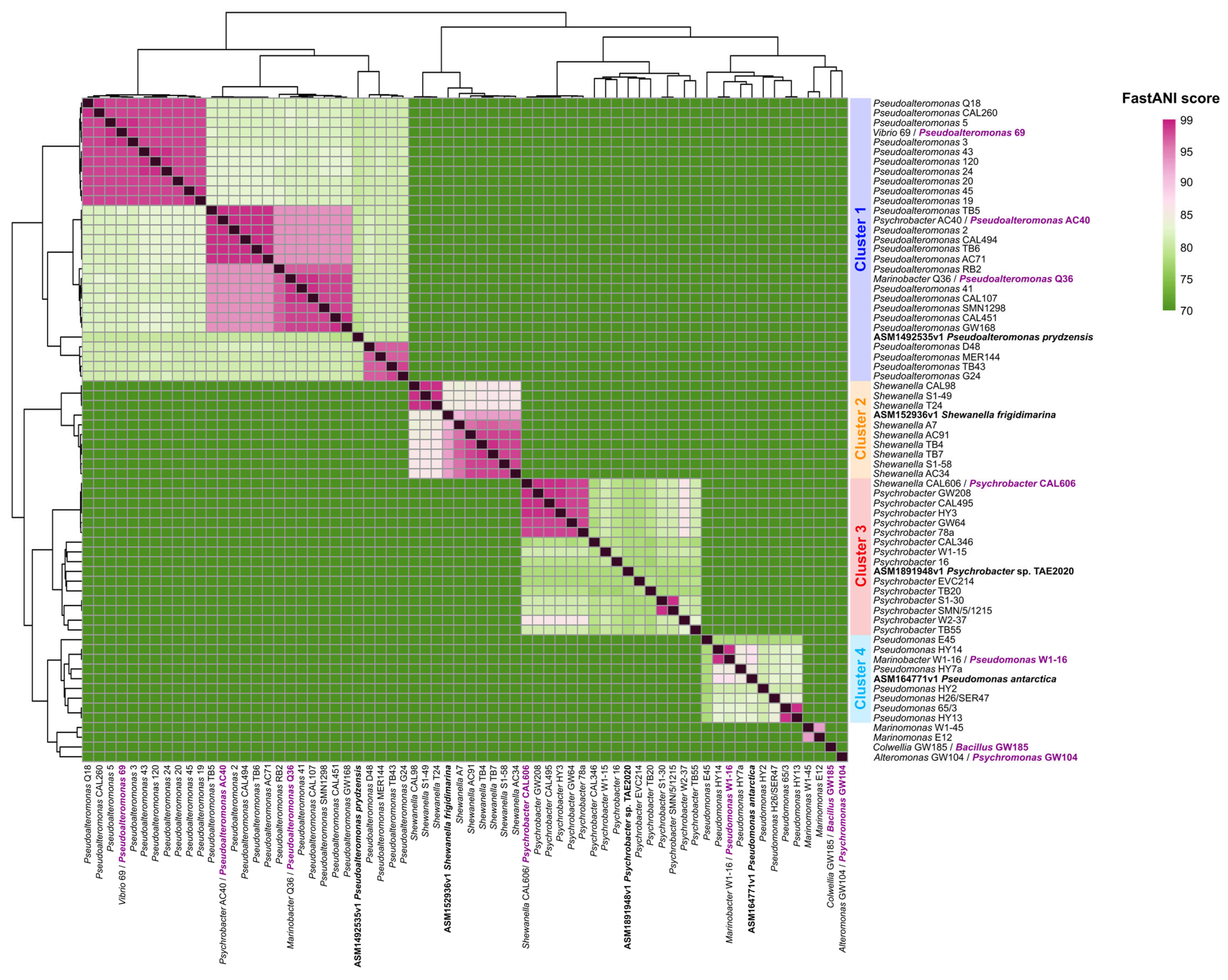
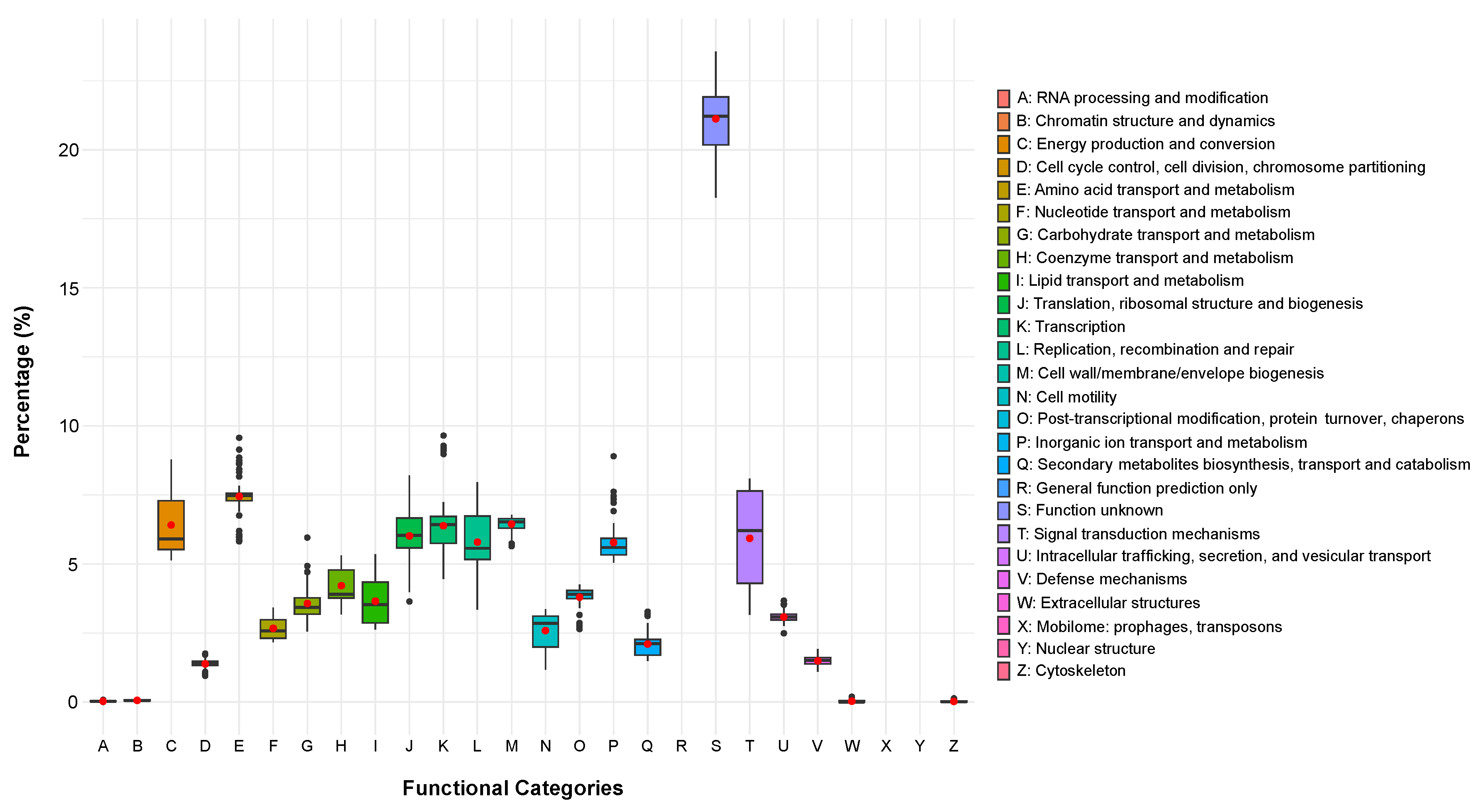
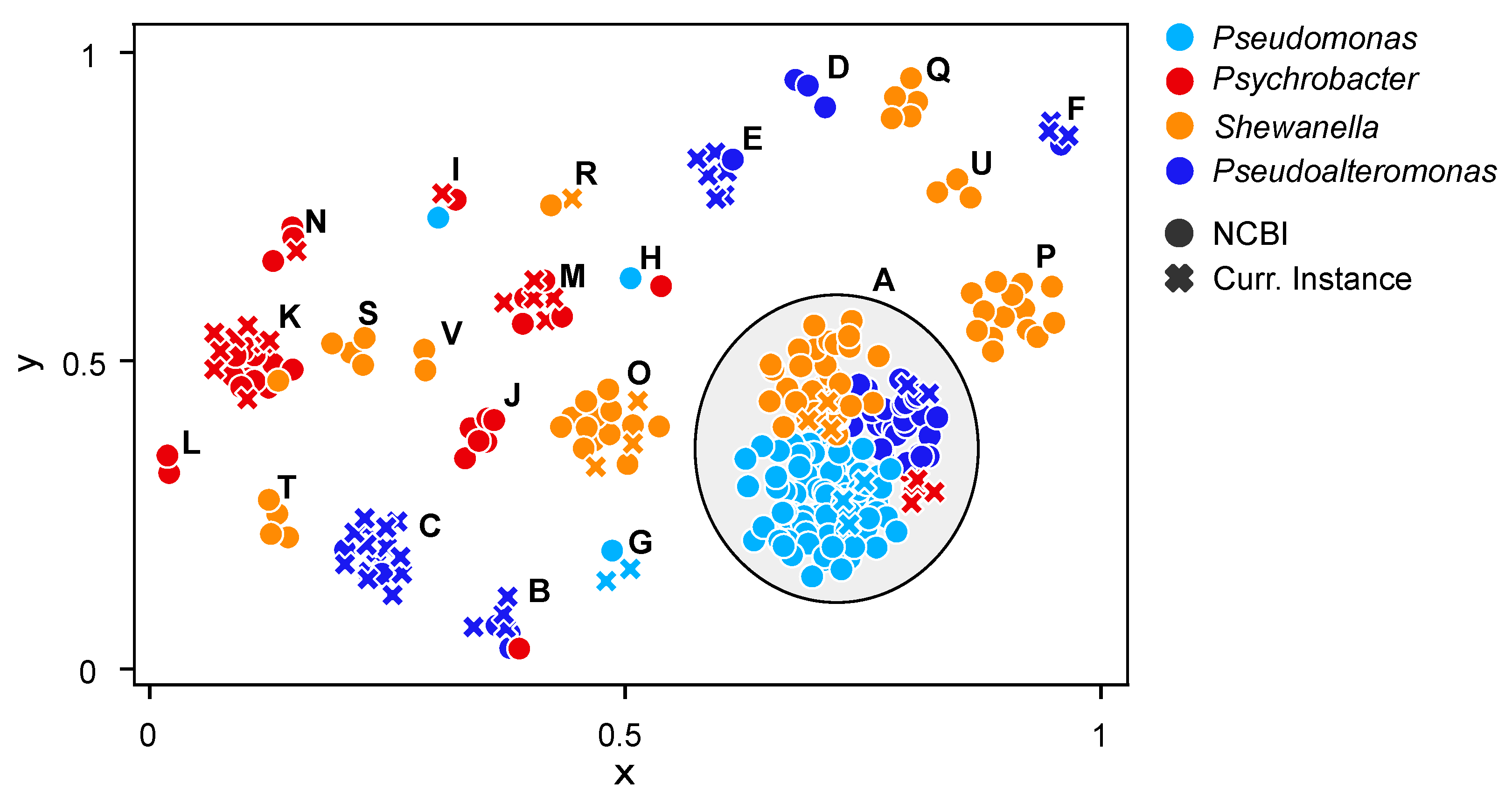
2.2. Phylogenomic Analyses and Functional Content of the Genomes
2.3. Genome Mining of Secondary Metabolites
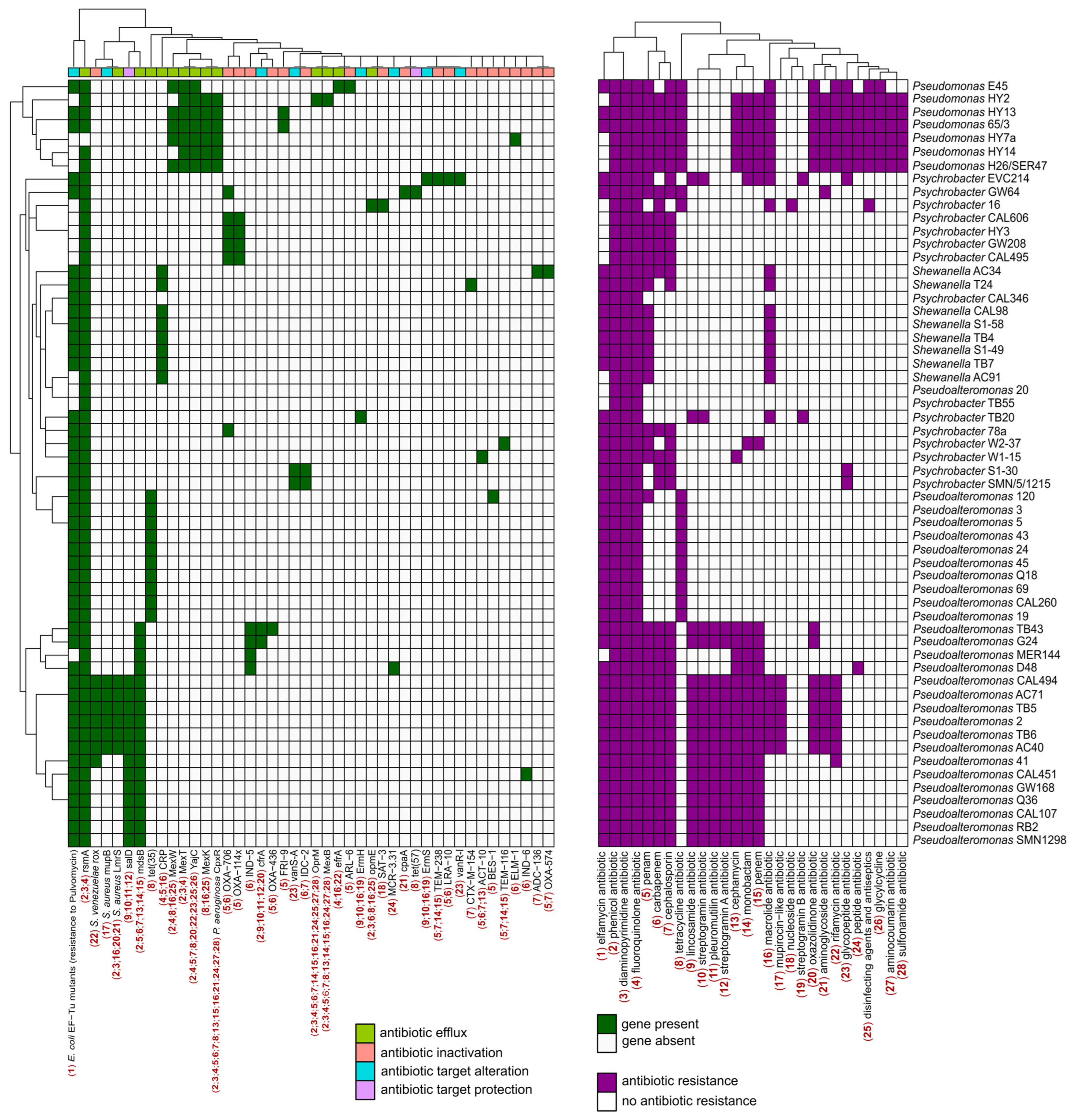
2.4. Characterization of Antibiotic Resistance Genes
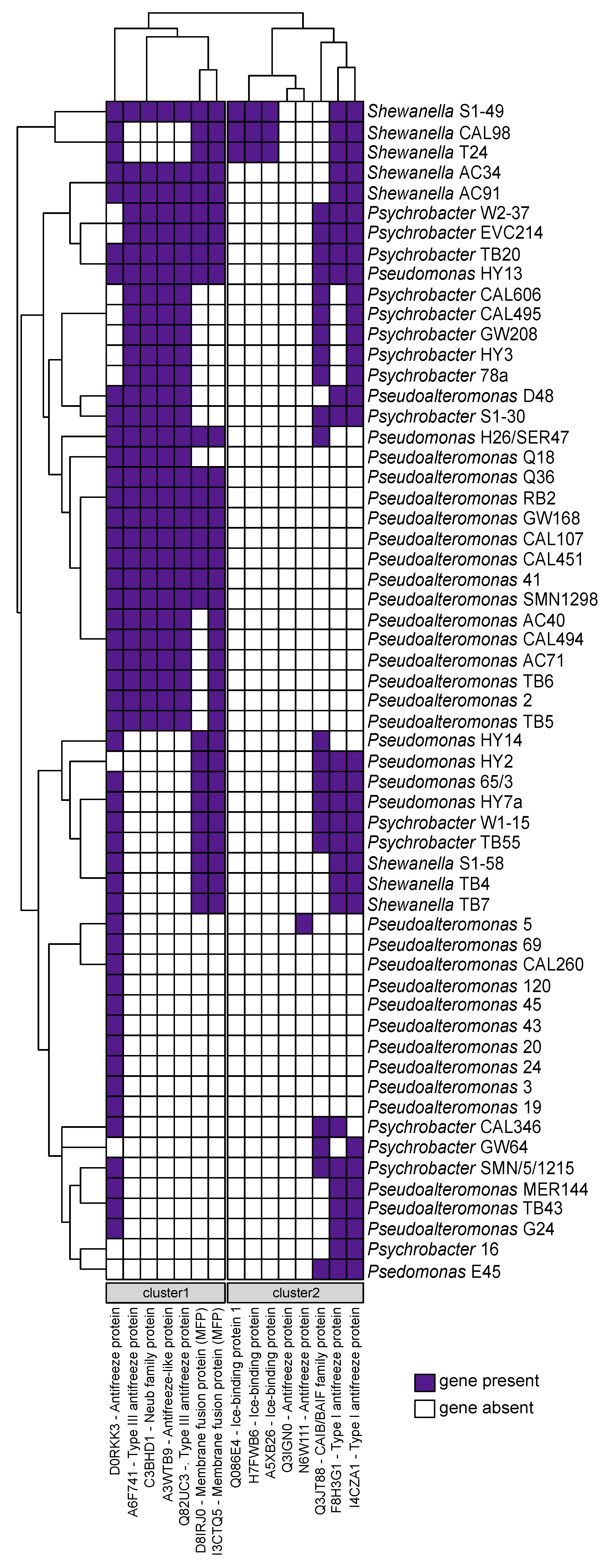
2.5. Cold-Adaptation Proteins (CAPs)
3. Materials and Methods
3.1. Bacterial Strains, Media, and Growth Conditions
3.2. Genome Extraction and Sequencing
3.3. Genus-Based Taxonomic Clustering and Database Construction
3.4. Genome Annotation and Genome Content Comparison
3.5. Detection of Secondary Metabolites
3.6. Phylogenomic Analyses
3.7. Antibiotic Resistance Genes
3.8. Cold-Shock Proteins
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Becker, P.; Bosschaerts, M.; Chaerle, P.; Daniel, H.-M.; Hellemans, A.; Olbrechts, A.; Rigouts, L.; Wilmotte, A.; Hendrickx, M. Public Microbial Resource Centers: Key Hubs for Findable, Accessible, Interoperable, and Reusable (FAIR) Microorganisms and Genetic Materials. Appl. Environ. Microbiol. 2019, 85, e01444-19. [Google Scholar] [CrossRef]
- Rappuoli, R.; Young, P.; Ron, E.; Pecetta, S.; Pizza, M. Save the Microbes to Save the Planet. A Call to Action of the International Union of the Microbiological Societies (IUMS). One Health Outlook 2023, 5, 5. [Google Scholar] [CrossRef]
- Locey, K.J.; Lennon, J.T. Scaling Laws Predict Global Microbial Diversity. Proc. Natl. Acad. Sci. USA 2016, 113, 5970–5975. [Google Scholar] [CrossRef]
- Smith, D.; Ryan, M.J. The Impact of Oecd Best Practice on the Validation of Cryopreservation Techniques for Microorganisms. Cryoletters 2008, 29, 63–72. [Google Scholar]
- Yadav, R.; Singh, P.K.; Shukla, P. Metabolic Engineering for Probiotics and Their Genome-Wide Expression Profiling. Curr. Protein Pept. Sci. 2018, 19, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Kumar, A.; Aparna, S.V.; Mallappa, R.H.; Grover, S.; Batish, V.K. Synthetic Biology: Applications in the Food Sector. Crit. Rev. Food Sci. Nutr. 2016, 56, 1777–1789. [Google Scholar] [CrossRef] [PubMed]
- Haiech, J.; Ranjeva, R.; Kilhoffer, M.-C. System biology and synthetic biology modify drug discovery and development. Med. Sci. 2012, 28, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Coelho, L.P.; Alves, R.; Del Río, Á.R.; Myers, P.N.; Cantalapiedra, C.P.; Giner-Lamia, J.; Schmidt, T.S.; Mende, D.R.; Orakov, A.; Letunic, I.; et al. Towards the Biogeography of Prokaryotic Genes. Nature 2022, 601, 252–256. [Google Scholar] [CrossRef]
- Laiolo, E.; Alam, I.; Uludag, M.; Jamil, T.; Agusti, S.; Gojobori, T.; Acinas, S.G.; Gasol, J.M.; Duarte, C.M. Metagenomic Probing toward an Atlas of the Taxonomic and Metabolic Foundations of the Global Ocean Genome. Front. Sci. 2024, 1, 1038696. [Google Scholar] [CrossRef]
- Martínez-Rosales, C.; Fullana, N.; Musto, H.; Castro-Sowinski, S. Antarctic DNA Moving Forward: Genomic Plasticity and Biotechnological Potential. FEMS Microbiol. Lett. 2012, 331, 1–9. [Google Scholar] [CrossRef]
- Kim, H.; Ducklow, H.W. A Decadal (2002–2014) Analysis for Dynamics of Heterotrophic Bacteria in an Antarctic Coastal Ecosystem: Variability and Physical and Biogeochemical Forcings. Front. Mar. Sci. 2016, 3, 214. [Google Scholar] [CrossRef]
- Doblin, M.A.; Van Sebille, E. Drift in Ocean Currents Impacts Intergenerational Microbial Exposure to Temperature. Proc. Natl. Acad. Sci. USA 2016, 113, 5700–5705. [Google Scholar] [CrossRef]
- Auger, M.; Morrow, R.; Kestenare, E.; Sallée, J.-B.; Cowley, R. Southern Ocean In-Situ Temperature Trends over 25 Years Emerge from Interannual Variability. Nat. Commun. 2021, 12, 514. [Google Scholar] [CrossRef]
- Mangano, S.; Michaud, L.; Caruso, C.; Brilli, M.; Bruni, V.; Fani, R.; Lo Giudice, A. Antagonistic Interactions between Psychrotrophic Cultivable Bacteria Isolated from Antarctic Sponges: A Preliminary Analysis. Res. Microbiol. 2009, 160, 27–37. [Google Scholar] [CrossRef]
- Savoca, S.; Lo Giudice, A.; Papale, M.; Mangano, S.; Caruso, C.; Spanò, N.; Michaud, L.; Rizzo, C. Antarctic Sponges from the Terra Nova Bay (Ross Sea) Host a Diversified Bacterial Community. Sci. Rep. 2019, 9, 16135. [Google Scholar] [CrossRef] [PubMed]
- Lo Giudice, A.; Brilli, M.; Bruni, V.; De Domenico, M.; Fani, R.; Michaud, L. Bacterium–Bacterium Inhibitory Interactions among Psychrotrophic Bacteria Isolated from Antarctic Seawater (Terra Nova Bay, Ross Sea): Antagonism among Psychrotrophic Antarctic Marine Bacteria. FEMS Microbiol. Ecol. 2007, 60, 383–396. [Google Scholar] [CrossRef]
- Michaud, L.; Di Marco, G.; Bruni, V.; Lo Giudice, A. Biodegradative Potential and Characterization of Psychrotolerant Polychlorinated Biphenyl-Degrading Marine Bacteria Isolated from a Coastal Station in the Terra Nova Bay (Ross Sea, Antarctica). Mar. Pollut. Bull. 2007, 54, 1754–1761. [Google Scholar] [CrossRef] [PubMed]
- Michaud, L.; Cello, F.; Brilli, M.; Fani, R.; Giudice, A.; Bruni, V. Biodiversity of Cultivable Psychrotrophic Marine Bacteria Isolated from Terra Nova Bay (Ross Sea, Antarctica). FEMS Microbiol. Lett. 2004, 230, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Lo Giudice, A.; Bruni, V.; Michaud, L. Characterization of Antarctic Psychrotrophic Bacteria with Antibacterial Activities against Terrestrial Microorganisms. J. Basic. Microbiol. 2007, 47, 496–505. [Google Scholar] [CrossRef]
- Romoli, R.; Papaleo, M.C.; De Pascale, D.; Tutino, M.L.; Michaud, L.; LoGiudice, A.; Fani, R.; Bartolucci, G. Characterization of the Volatile Profile of Antarctic Bacteria by Using Solid-phase Microextraction-gas Chromatography-mass Spectrometry. J. Mass. Spectrom. 2011, 46, 1051–1059. [Google Scholar] [CrossRef]
- Domenico, M.D.; Giudice, A.L.; Michaud, L.; Saitta, M.; Bruni, V. Diesel Oil and PCB-Degrading Psychrotrophic Bacteria Isolated from Antarctic Seawaters (Terra Nova Bay, Ross Sea). Polar Res. 2004, 23, 141–146. [Google Scholar] [CrossRef]
- Caruso, C.; Rizzo, C.; Mangano, S.; Poli, A.; Di Donato, P.; Nicolaus, B.; Di Marco, G.; Michaud, L.; Lo Giudice, A. Extracellular Polymeric Substances with Metal Adsorption Capacity Produced by Pseudoalteromonas Sp. MER144 from Antarctic Seawater. Environ. Sci. Pollut. Res. 2018, 25, 4667–4677. [Google Scholar] [CrossRef] [PubMed]
- Mangano, S.; Caruso, C.; Michaud, L.; Lo Giudice, A. First Evidence of Quorum Sensing Activity in Bacteria Associated with Antarctic Sponges. Polar Biol. 2018, 41, 1435–1445. [Google Scholar] [CrossRef]
- Lo Giudice, A.; Michaud, L.; De Pascale, D.; De Domenico, M.; Di Prisco, G.; Fani, R.; Bruni, V. Lipolytic Activity of Antarctic Cold-Adapted Marine Bacteria (Terra Nova Bay, Ross Sea). J. Appl. Microbiol. 2006, 101, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Lo Giudice, A.; Caruso, C.; Mangano, S.; Bruni, V.; De Domenico, M.; Michaud, L. Marine Bacterioplankton Diversity and Community Composition in an Antarctic Coastal Environment. Microb. Ecol. 2012, 63, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Mangano, S.; Michaud, L.; Caruso, C.; Lo Giudice, A. Metal and Antibiotic Resistance in Psychrotrophic Bacteria Associated with the Antarctic Sponge Hemigellius pilosus (Kirkpatrick, 1907). Polar Biol. 2014, 37, 227–235. [Google Scholar] [CrossRef]
- Lo Giudice, A.; Casella, P.; Caruso, C.; Mangano, S.; Bruni, V.; De Domenico, M.; Michaud, L. Occurrence and Characterization of Psychrotolerant Hydrocarbon-Oxidizing Bacteria from Surface Seawater along the Victoria Land Coast (Antarctica). Polar Biol. 2010, 33, 929–943. [Google Scholar] [CrossRef]
- Caruso, C.; Rizzo, C.; Mangano, S.; Poli, A.; Di Donato, P.; Finore, I.; Nicolaus, B.; Di Marco, G.; Michaud, L.; Lo Giudice, A. Production and Biotechnological Potential of Extracellular Polymeric Substances from Sponge-Associated Antarctic Bacteria. Appl. Environ. Microbiol. 2018, 84, e01624-17. [Google Scholar] [CrossRef]
- Lo Giudice, A.; Casella, P.; Bruni, V.; Michaud, L. Response of Bacterial Isolates from Antarctic Shallow Sediments towards Heavy Metals, Antibiotics and Polychlorinated Biphenyls. Ecotoxicology 2013, 22, 240–250. [Google Scholar] [CrossRef]
- Papaleo, M.C.; Fondi, M.; Maida, I.; Perrin, E.; Lo Giudice, A.; Michaud, L.; Mangano, S.; Bartolucci, G.; Romoli, R.; Fani, R. Sponge-Associated Microbial Antarctic Communities Exhibiting Antimicrobial Activity against Burkholderia Cepacia Complex Bacteria. Biotechnol. Adv. 2012, 30, 272–293. [Google Scholar] [CrossRef]
- Riccardi, C.; D’Angelo, C.; Calvanese, M.; Ricciardelli, A.; Tutino, M.L.; Parrilli, E.; Fondi, M. Genome Analysis of a New Biosurfactants Source: The Antarctic Bacterium Psychrobacter sp. TAE2020. Mar. Genom. 2022, 61, 100922. [Google Scholar] [CrossRef] [PubMed]
- Rumbaugh, K.P. Genomic Complexity and Plasticity Ensure Pseudomonas Success. FEMS Microbiol. Lett. 2014, 356, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Silby, M.W.; Winstanley, C.; Godfrey, S.A.C.; Levy, S.B.; Jackson, R.W. Pseudomonas Genomes: Diverse and Adaptable. FEMS Microbiol. Rev. 2011, 35, 652–680. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.J.; Jang, G.I.; Cho, B.C.; Lee, J.I.; Hwang, C.Y. Shewanella psychromarinicola Sp. Nov., a Psychrophilic Bacterium Isolated from Pelagic Sediment of the Ross Sea (Antarctica), and Reclassification of Shewanella arctica Kim et al. 2012 as a Later Heterotypic Synonym of Shewanella frigidimarina Bowman et al. 1997. Int. J. Syst. Evol. Microbiol. 2019, 69, 2415–2423. [Google Scholar] [CrossRef] [PubMed]
- Ninkuu, V.; Zhang, L.; Yan, J.; Fu, Z.; Yang, T.; Zeng, H. Biochemistry of Terpenes and Recent Advances in Plant Protection. Int. J. Mol. Sci. 2021, 22, 5710. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Solís, J.M.; Fernández, F.J. Endophytic Fungal Terpenoids: Natural Role and Bioactivities. Microorganisms 2022, 10, 339. [Google Scholar] [CrossRef] [PubMed]
- Avila, C. Terpenoids in Marine Heterobranch Molluscs. Mar. Drugs 2020, 18, 162. [Google Scholar] [CrossRef] [PubMed]
- Núñez-Pons, L.; Shilling, A.; Verde, C.; Baker, B.J.; Giordano, D. Marine Terpenoids from Polar Latitudes and Their Potential Applications in Biotechnology. Mar. Drugs 2020, 18, 401. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Kuzuyama, T.; Komatsu, M.; Shin-ya, K.; Omura, S.; Cane, D.E.; Ikeda, H. Terpene Synthases Are Widely Distributed in Bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 857–862. [Google Scholar] [CrossRef]
- Yamada, Y.; Cane, D.E.; Ikeda, H. Diversity and Analysis of Bacterial Terpene Synthases. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2012; Volume 515, pp. 123–162. ISBN 978-0-12-394290-6. [Google Scholar]
- Avalos, M.; Garbeva, P.; Vader, L.; Van Wezel, G.P.; Dickschat, J.S.; Ulanova, D. Biosynthesis, Evolution and Ecology of Microbial Terpenoids. Nat. Prod. Rep. 2022, 39, 249–272. [Google Scholar] [CrossRef]
- Yin, J.; Chen, J.-C.; Wu, Q.; Chen, G.-Q. Halophiles, Coming Stars for Industrial Biotechnology. Biotechnol. Adv. 2015, 33, 1433–1442. [Google Scholar] [CrossRef]
- Kang, J.Y.; Lee, B.; Kim, J.A.; Kim, M.-S.; Kim, C.H. Identification and Characterization of an Ectoine Biosynthesis Gene Cluster from Aestuariispira ectoiniformans sp. nov., Isolated from Seawater. Microbiol. Res. 2022, 254, 126898. [Google Scholar] [CrossRef]
- Gunde-Cimerman, N.; Plemenitaš, A.; Oren, A. Strategies of Adaptation of Microorganisms of the Three Domains of Life to High Salt Concentrations. FEMS Microbiol. Rev. 2018, 42, 353–375. [Google Scholar] [CrossRef] [PubMed]
- Mawji, E.; Gledhill, M.; Milton, J.A.; Tarran, G.A.; Ussher, S.; Thompson, A.; Wolff, G.A.; Worsfold, P.J.; Achterberg, E.P. Hydroxamate Siderophores: Occurrence and Importance in the Atlantic Ocean. Environ. Sci. Technol. 2008, 42, 8675–8680. [Google Scholar] [CrossRef] [PubMed]
- Uchgaonkar, P.; Padmadas, N.; Singh, S.; Dasgupta, D. Screening and Identification Of Siderophore Producing Marine Bacteria. Global. J. Biosci. Biotechnol. 2018, 7, 457–461. [Google Scholar]
- Lemaire, O.N.; Méjean, V.; Iobbi-Nivol, C. The Shewanella Genus: Ubiquitous Organisms Sustaining and Preserving Aquatic Ecosystems. FEMS Microbiol. Rev. 2020, 44, 155–170. [Google Scholar] [CrossRef]
- Hetrick, K.J.; Van Der Donk, W.A. Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Product Discovery in the Genomic Era. Curr. Opin. Chem. Biol. 2017, 38, 36–44. [Google Scholar] [CrossRef]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products: Overview and Recommendations for a Universal Nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef]
- Ongpipattanakul, C.; Desormeaux, E.K.; DiCaprio, A.; Van Der Donk, W.A.; Mitchell, D.A.; Nair, S.K. Mechanism of Action of Ribosomally Synthesized and Post-Translationally Modified Peptides. Chem. Rev. 2022, 122, 14722–14814. [Google Scholar] [CrossRef]
- Bartholomae, M.; Buivydas, A.; Viel, J.H.; Montalbán-López, M.; Kuipers, O.P. Major Gene-regulatory Mechanisms Operating in Ribosomally Synthesized and Post-translationally Modified Peptide (RiPP) Biosynthesis. Mol. Microbiol. 2017, 106, 186–206. [Google Scholar] [CrossRef]
- Cotter, P.D.; Ross, R.P.; Hill, C. Bacteriocins—A Viable Alternative to Antibiotics? Nat. Rev. Microbiol. 2013, 11, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Fondi, M.; Orlandini, V.; Perrin, E.; Maida, I.; Bosi, E.; Papaleo, M.C.; Michaud, L.; Lo Giudice, A.; De Pascale, D.; Tutino, M.L.; et al. Draft Genomes of Three Antarctic Psychrobacter Strains Producing Antimicrobial Compounds against Burkholderia cepacia Complex, Opportunistic Human Pathogens. Mar. Genom. 2014, 13, 37–38. [Google Scholar] [CrossRef] [PubMed]
- De Pascale, G.; Nazi, I.; Harrison, P.H.M.; Wright, G.D. β-Lactone Natural Products and Derivatives Inactivate Homoserine Transacetylase, a Target for Antimicrobial Agents. J. Antibiot. 2011, 64, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.; Özkaya, Ö.; Kümmerli, R. Bacterial Siderophores in Community and Host Interactions. Nat. Rev. Microbiol. 2020, 18, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mageed, W.M.; Lehri, B.; Jarmusch, S.A.; Miranda, K.; Al-Wahaibi, L.H.; Stewart, H.A.; Jamieson, A.J.; Jaspars, M.; Karlyshev, A.V. Whole Genome Sequencing of Four Bacterial Strains from South Shetland Trench Revealing Biosynthetic and Environmental Adaptation Gene Clusters. Mar. Genom. 2020, 54, 100782. [Google Scholar] [CrossRef] [PubMed]
- Olshvang, E.; Fritsch, S.; Scholtyssek, O.C.; Schalk, I.J.; Metzler-Nolte, N. Vectorization via Siderophores Increases Antibacterial Activity of K(RW) 3 Peptides against Pseudomonas aeruginosa. Chem. A Eur. J. 2023, 29, e202300364. [Google Scholar] [CrossRef] [PubMed]
- Kahlke, T.; Thorvaldsen, S. Molecular Characterization of Cold Adaptation of Membrane Proteins in the Vibrionaceae Core-Genome. PLoS ONE 2012, 7, e51761. [Google Scholar] [CrossRef] [PubMed]
- Ramón, A.; Esteves, A.; Villadóniga, C.; Chalar, C.; Castro-Sowinski, S. A General Overview of the Multifactorial Adaptation to Cold: Biochemical Mechanisms and Strategies. Braz. J. Microbiol. 2023, 54, 2259–2287. [Google Scholar] [CrossRef] [PubMed]
- Bale, N.J.; Rijpstra, W.I.C.; Sahonero-Canavesi, D.X.; Oshkin, I.Y.; Belova, S.E.; Dedysh, S.N.; Sinninghe Damsté, J.S. Fatty Acid and Hopanoid Adaption to Cold in the Methanotroph Methylovulum Psychrotolerans. Front. Microbiol. 2019, 10, 589. [Google Scholar] [CrossRef]
- Bianchi, A.C.; Olazábal, L.; Torre, A.; Loperena, L. Antarctic Microorganisms as Source of the Omega-3 Polyunsaturated Fatty Acids. World J. Microbiol. Biotechnol. 2014, 30, 1869–1878. [Google Scholar] [CrossRef]
- Nishida, T.; Orikasa, Y.; Watanabe, K.; Okuyama, H. The Cell Membrane-shielding Function of Eicosapentaenoic Acid for Escherichia coli against Exogenously Added Hydrogen Peroxide. FEBS Lett. 2006, 580, 6690–6694. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, H.; Orikasa, Y.; Nishida, T. Significance of Antioxidative Functions of Eicosapentaenoic and Docosahexaenoic Acids in Marine Microorganisms. Appl. Environ. Microbiol. 2008, 74, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, H. Hydrophilic and Hydrophobic Compounds Antithetically Affect the Growth of Eicosapentaenoic Acid-Synthesizing Escherichia Coli Recombinants. Open Microbiol. J. 2011, 5, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic Resistome Surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2019, 48, gkz935. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.C.; Lee, N.; Aw, T.G. Antibiotic Resistance in Minimally Human-Impacted Environments. Int. J. Environ. Res. Public Health 2020, 17, 3939. [Google Scholar] [CrossRef] [PubMed]
- Hwengwere, K.; Paramel Nair, H.; Hughes, K.A.; Peck, L.S.; Clark, M.S.; Walker, C.A. Antimicrobial Resistance in Antarctica: Is It Still a Pristine Environment? Microbiome 2022, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Cornforth, D.M.; Foster, K.R. Antibiotics and the Art of Bacterial War. Proc. Natl. Acad. Sci. USA 2015, 112, 10827–10828. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.L. Antibiotics and Antibiotic Resistance Genes in Natural Environments. Science 2008, 321, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.-A.T.; Chen, Q.-L.; He, J.-Z.; Hu, H.-W. Microbial Regulation of Natural Antibiotic Resistance: Understanding the Protist-Bacteria Interactions for Evolution of Soil Resistome. Sci. Total Environ. 2020, 705, 135882. [Google Scholar] [CrossRef]
- Bosi, E.; Fondi, M.; Orlandini, V.; Perrin, E.; Maida, I.; De Pascale, D.; Tutino, M.L.; Parrilli, E.; Lo Giudice, A.; Filloux, A.; et al. The Pangenome of (Antarctic) Pseudoalteromonas Bacteria: Evolutionary and Functional Insights. BMC Genom. 2017, 18, 93. [Google Scholar] [CrossRef]
- Celik, Y.; Graham, L.A.; Mok, Y.-F.; Bar, M.; Davies, P.L.; Braslavsky, I. Superheating of Ice Crystals in Antifreeze Protein Solutions. Proc. Natl. Acad. Sci. USA 2010, 107, 5423–5428. [Google Scholar] [CrossRef]
- Kristiansen, E.; Zachariassen, K.E. The Mechanism by Which Fish Antifreeze Proteins Cause Thermal Hysteresis. Cryobiology 2005, 51, 262–280. [Google Scholar] [CrossRef] [PubMed]
- Białkowska, A.; Majewska, E.; Olczak, A.; Twarda-Clapa, A. Ice Binding Proteins: Diverse Biological Roles and Applications in Different Types of Industry. Biomolecules 2020, 10, 274. [Google Scholar] [CrossRef] [PubMed]
- Shivaji, S.; Prakash, J.S.S. How Do Bacteria Sense and Respond to Low Temperature? Arch. Microbiol. 2010, 192, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, C.; Calvanese, M.; Ghini, V.; Alonso-Vásquez, T.; Perrin, E.; Turano, P.; Giurato, G.; Weisz, A.; Parrilli, E.; Tutino, M.L.; et al. Metabolic Robustness to Growth Temperature of a Cold-Adapted Marine Bacterium. mSystems 2023, 8, e01124-22. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Martinez, C.E.; Sgro, G.G.; Araujo, G.G.; Paiva, M.R.N.; Matsuyama, B.Y.; Guzzo, C.R.; Andrade, M.O.; Farah, C.S. Secrete or Perish: The Role of Secretion Systems in Xanthomonas Biology. Comput. Struct. Biotechnol. J. 2021, 19, 279–302. [Google Scholar] [CrossRef] [PubMed]
- Zgurskaya, H.I.; Yamada, Y.; Tikhonova, E.B.; Ge, Q.; Krishnamoorthy, G. Structural and Functional Diversity of Bacterial Membrane Fusion Proteins. Biochim. Biophys. Acta BBA Proteins Proteom. 2009, 1794, 794–807. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 7 May 2024).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A Web Server for Prokaryotic Species Circumscription Based on Pairwise Genome Comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA–DNA Hybridization Values and Their Relationship to Whole-Genome Sequence Similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. TYGS Is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast Genome and Metagenome Distance Estimation Using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving Cluster Detection and Comparison Capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A Computational Framework to Explore Large-Scale Biosynthetic Diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest-HPC: Fast selection of best-fit models of protein evolution. In Proceedings of the Euro-Par 2010 Parallel Processing Workshops: HeteroPar, HPCC, HiBB, CoreGrid, UCHPC, HPCF, PROPER, CCPI, VHPC, Ischia, Italy, 31 August–3 September 2010; Revised Selected Papers 16. Springer: Berlin/Heidelberg, Germany; pp. 177–184. [Google Scholar]
- Stamatakis, A. Using RAxML to Infer Phylogenies. CP Bioinformatics 2015, 51, 6–14. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T. ggtree: An r Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and Model-Centric Curation of the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Reen, F.; Gutiérrez-Barranquero, J.; Dobson, A.; Adams, C.; O’Gara, F. Emerging Concepts Promising New Horizons for Marine Biodiscovery and Synthetic Biology. Mar. Drugs 2015, 13, 2924–2954. [Google Scholar] [CrossRef] [PubMed]
- Reen, F.; Romano, S.; Dobson, A.; O’Gara, F. The Sound of Silence: Activating Silent Biosynthetic Gene Clusters in Marine Microorganisms. Mar. Drugs 2015, 13, 4754–4783. [Google Scholar] [CrossRef]
- Cowan, D.A.; Chown, S.L.; Convey, P.; Tuffin, M.; Hughes, K.; Pointing, S.; Vincent, W.F. Non-Indigenous Microorganisms in the Antarctic: Assessing the Risks. Trends Microbiol. 2011, 19, 540–548. [Google Scholar] [CrossRef]
- Hughes, K.A.; Fretwell, P.; Rae, J.; Holmes, K.; Fleming, A. Untouched Antarctica: Mapping a Finite and Diminishing Environmental Resource. Antarct. Sci. 2011, 23, 537–548. [Google Scholar] [CrossRef]
- Pertierra, L.R.; Hughes, K.A.; Vega, G.C.; Olalla-Tárraga, M.Á. High Resolution Spatial Mapping of Human Footprint across Antarctica and Its Implications for the Strategic Conservation of Avifauna. PLoS ONE 2017, 12, e0168280. [Google Scholar] [CrossRef]
- Ramírez-Rendon, D.; Passari, A.K.; Ruiz-Villafán, B.; Rodríguez-Sanoja, R.; Sánchez, S.; Demain, A.L. Impact of Novel Microbial Secondary Metabolites on the Pharma Industry. Appl. Microbiol. Biotechnol. 2022, 106, 1855–1878. [Google Scholar] [CrossRef]
- Andryukov, B.; Mikhailov, V.; Besednova, N. The Biotechnological Potential of Secondary Metabolites from Marine Bacteria. J. Mar. Sci. Eng. 2019, 7, 176. [Google Scholar] [CrossRef]
- Pham, J.V.; Yilma, M.A.; Feliz, A.; Majid, M.T.; Maffetone, N.; Walker, J.R.; Kim, E.; Cho, H.J.; Reynolds, J.M.; Song, M.C.; et al. A Review of the Microbial Production of Bioactive Natural Products and Biologics. Front. Microbiol. 2019, 10, 1404. [Google Scholar] [CrossRef]
- Srinivasan, R.; Kannappan, A.; Shi, C.; Lin, X. Marine Bacterial Secondary Metabolites: A Treasure House for Structurally Unique and Effective Antimicrobial Compounds. Mar. Drugs 2021, 19, 530. [Google Scholar] [CrossRef]
- Christner, B.C. Bioprospecting for Microbial Products That Affect Ice Crystal Formation and Growth. Appl. Microbiol. Biotechnol. 2010, 85, 481–489. [Google Scholar] [CrossRef]
- Jeon, S.M.; Naing, A.H.; Park, K.I.; Kim, C.K. The Effect of Antifreeze Protein on the Cryopreservation of Chrysanthemums. Plant Cell Tiss. Organ. Cult. 2015, 123, 665–671. [Google Scholar] [CrossRef]
- Zhou, H.; Infante Ferreira, C. Effect of Type-III Anti-Freeze Proteins (AFPs) on CO2 Hydrate Formation Rate. Chem. Eng. Sci. 2017, 167, 42–53. [Google Scholar] [CrossRef]
- Griffith, M.; Ewart, K.V. Antifreeze Proteins and Their Potential Use in Frozen Foods. Biotechnol. Adv. 1995, 13, 375–402. [Google Scholar] [CrossRef] [PubMed]
- Ustun, N.S.; Turhan, S. Antifreeze Proteins: Characteristics, Function, Mechanism of Action, Sources and Application to Foods: Antifreeze Proteins. J. Food Process. Preserv. 2015, 39, 3189–3197. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Average Length (Mbp) | Average Number of Contigs | Average % GC | Average Number of CDSs | |
|---|---|---|---|---|
| Psychrobacter | 4.29 | 1173 | 42.8 | 3394 |
| Shewanella | 4.96 | 457 | 41.7 | 4171 |
| Pseudomonas | 6.08 | 816 | 59.2 | 5364 |
| Pseudoalteromonas | 4.44 | 694 | 39.6 | 3856 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giovannini, M.; Vieri, W.; Bosi, E.; Riccardi, C.; Lo Giudice, A.; Fani, R.; Fondi, M.; Perrin, E. Functional Genomics of a Collection of Gammaproteobacteria Isolated from Antarctica. Mar. Drugs 2024, 22, 238. https://doi.org/10.3390/md22060238
Giovannini M, Vieri W, Bosi E, Riccardi C, Lo Giudice A, Fani R, Fondi M, Perrin E. Functional Genomics of a Collection of Gammaproteobacteria Isolated from Antarctica. Marine Drugs. 2024; 22(6):238. https://doi.org/10.3390/md22060238
Chicago/Turabian StyleGiovannini, Michele, Walter Vieri, Emanuele Bosi, Christopher Riccardi, Angelina Lo Giudice, Renato Fani, Marco Fondi, and Elena Perrin. 2024. "Functional Genomics of a Collection of Gammaproteobacteria Isolated from Antarctica" Marine Drugs 22, no. 6: 238. https://doi.org/10.3390/md22060238
APA StyleGiovannini, M., Vieri, W., Bosi, E., Riccardi, C., Lo Giudice, A., Fani, R., Fondi, M., & Perrin, E. (2024). Functional Genomics of a Collection of Gammaproteobacteria Isolated from Antarctica. Marine Drugs, 22(6), 238. https://doi.org/10.3390/md22060238