
New Hybrid Phenalenone Dimer, Highly Conjugated Dihydroxylated C28 Steroid and Azaphilone from the Culture Extract of a Marine Sponge-Associated Fungus, Talaromyces pinophilus KUFA 1767
 , , , , ,
, , , , ,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
3. Experimental Sections
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Extraction and Isolation
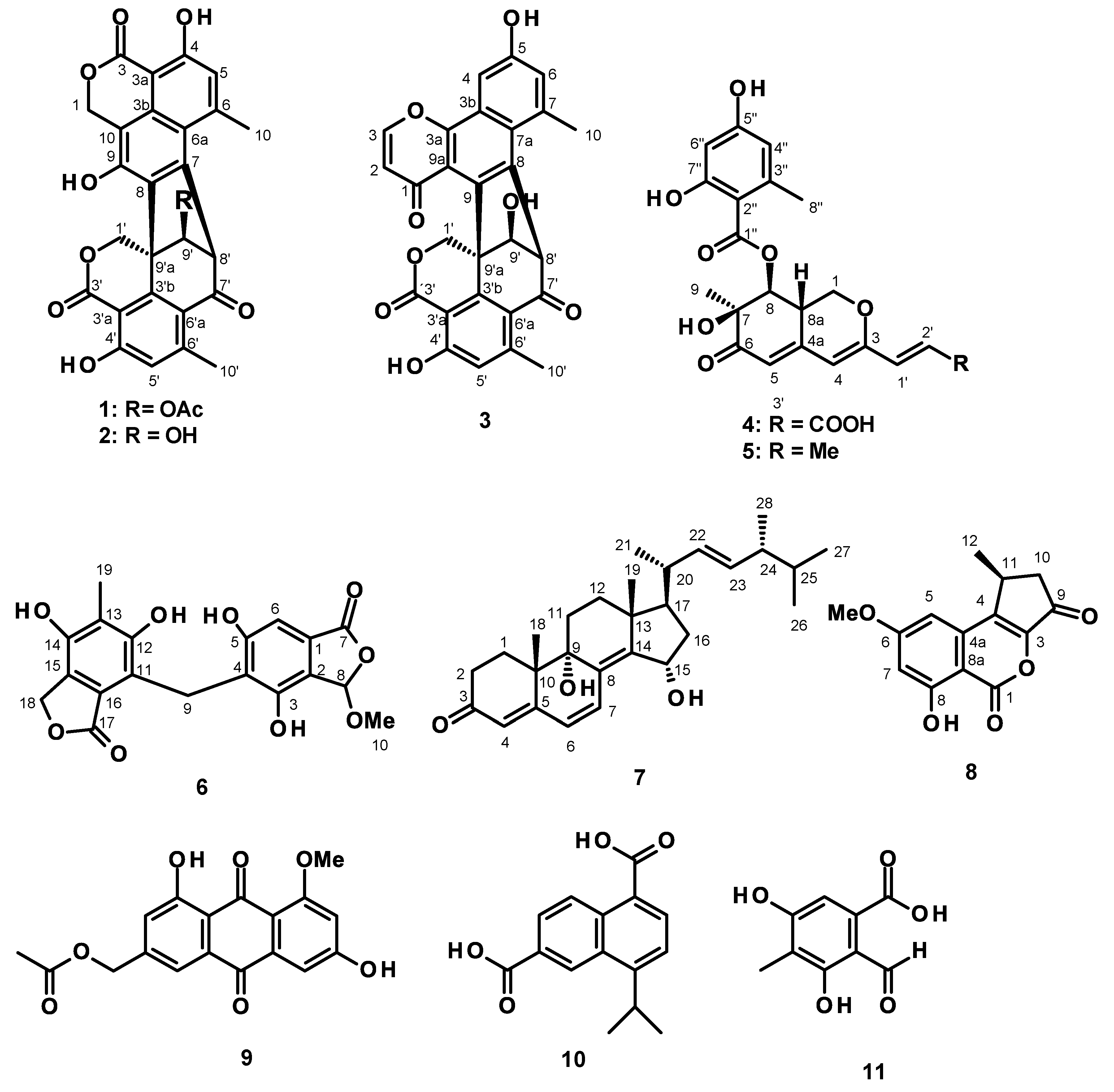
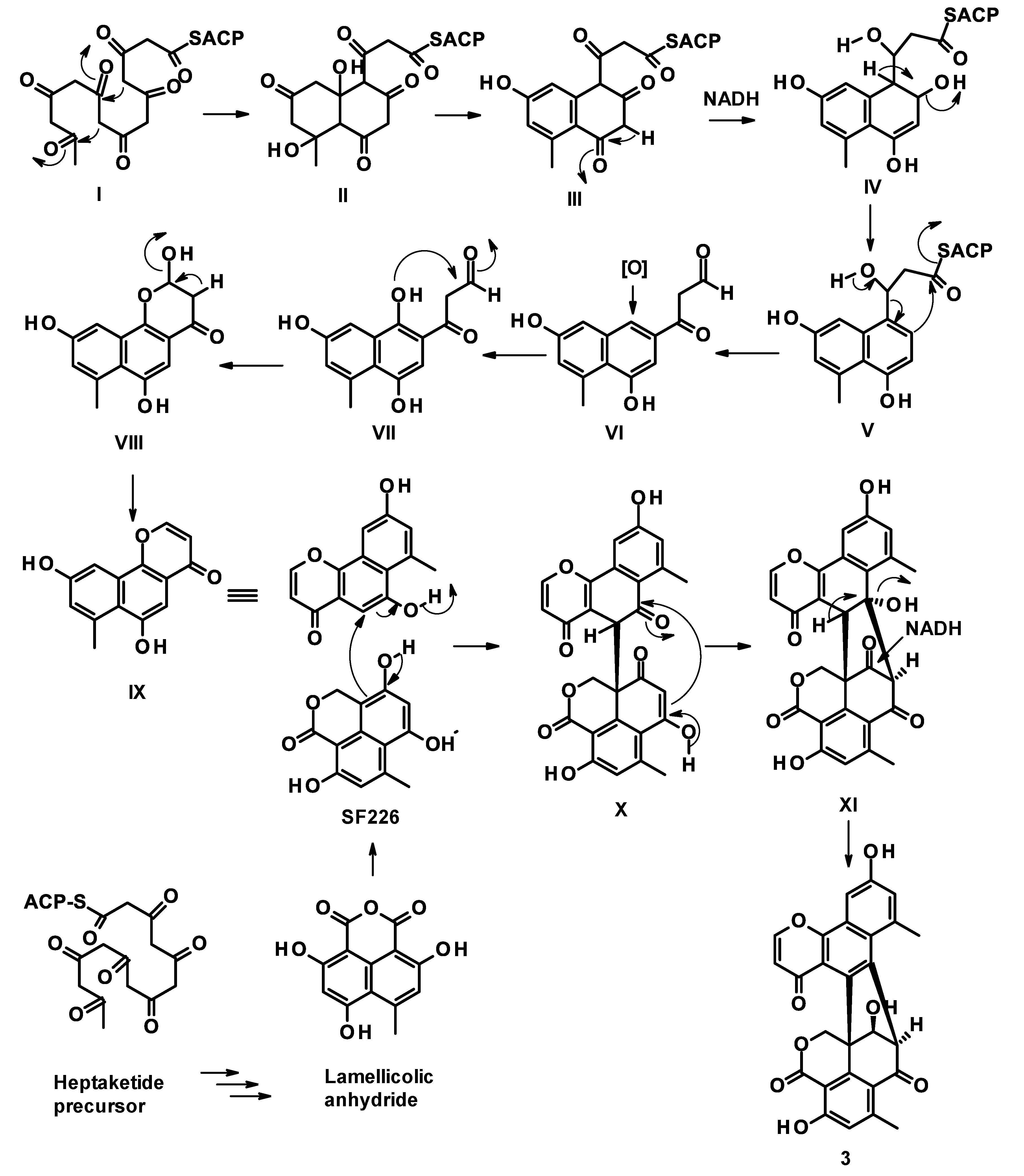
3.3.1. Talaropinophilone (3)
3.3.2. 7-Epi-pinazaphilone B (4)
3.3.3. Talaropinophilide (6)
3.3.4. 9 R,15S-Dihydroxy-ergosta-4,6,8 (14)-tetraen-3-one (7)
3.4. X-ray Crystal Structures
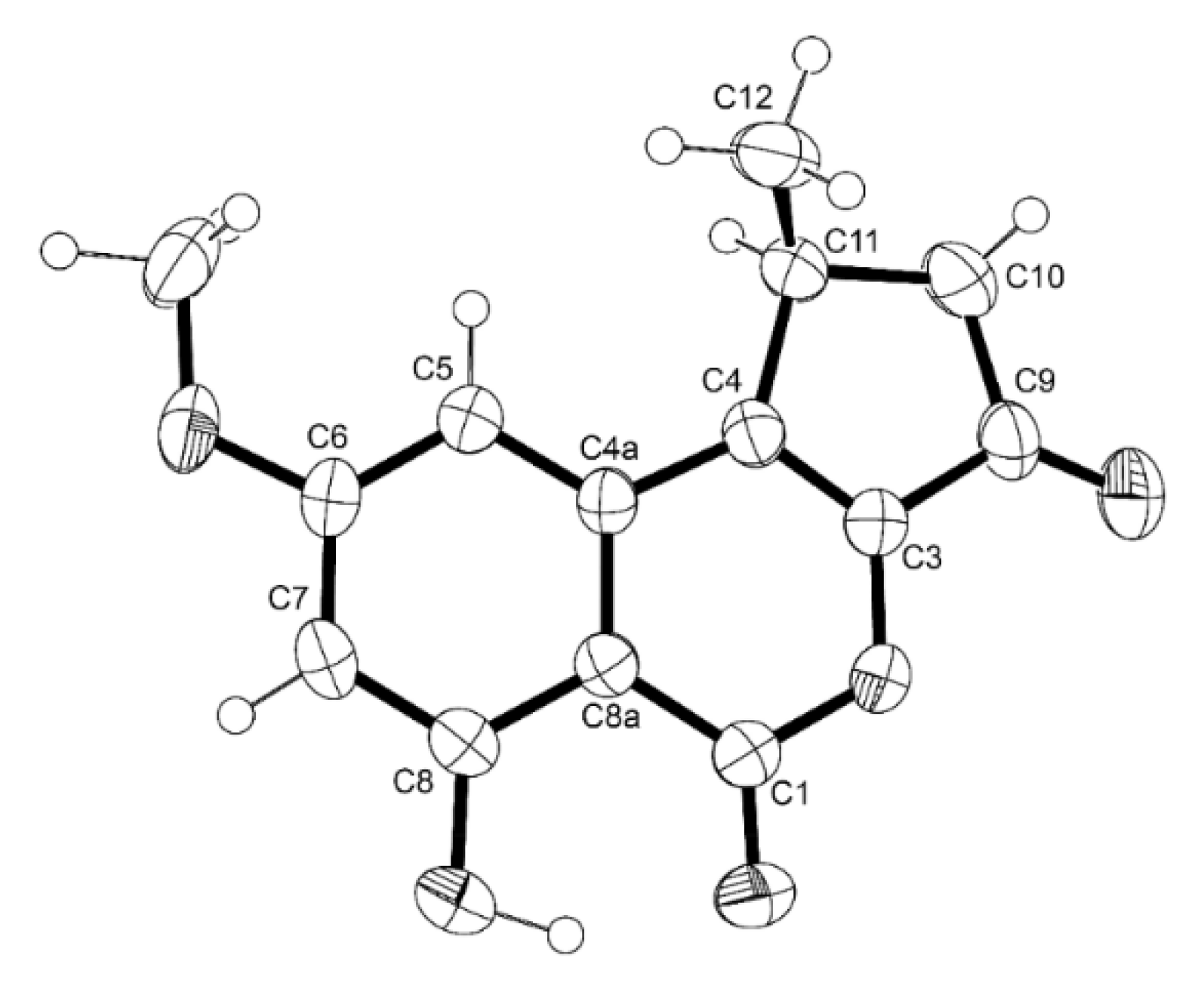
3.4.1. X-ray Crystal Structure of 7
3.4.2. X-ray Crystal Structure of 8
3.5. Electronic Circular Dichroism (ECD)
3.6. Antibacterial Activity Bioassays
3.6.1. Bacterial Strains and Testing Conditions
3.6.2. Antimicrobial Susceptibility Testing
3.6.3. Antibiotic Synergy Testing
3.6.4. Biofilm Formation Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dethoup, T.; Kaewsalong, N.; Songkumorn, P.; Jantasorn, A. Potential application of a marine-derived fungus, Talaromyces tratensis KUFA 0091 against rice diseases. Biol. Control 2018, 119, 1–6. [Google Scholar] [CrossRef]
- El-Naggar, N.E.A.; Haroun, S.A.; Oweis, E.A.; Sherief, A.A. Identification of newly isolated Talaromyces pinophilus and statistical optimization of β-glucosidase production under solid-state fermentation. Prep. Biochem. Biotechnol. 2015, 45, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Zhai, M.M.; Li, J.; Jiang, C.X.; Shi, Y.P.; Di, D.L.; Crews, P.; Wu, Q.X. The bioactive secondary metabolites from Talaromyces species. Nat. Prod. Bioprospect. 2016, 6, 1–24. [Google Scholar] [CrossRef]
- Lei, L.R.; Gong, L.Q.; Jin, M.Y.; Wang, R.; Liu, R.; Gao, J.; Liu, M.D.; Huang, L.; Wang, G.Z.; Wang, D.; et al. Research advances in the structures and biological activities of secondary metabolites from Talaromyces. Front. Microbiol. 2022, 13, 984801. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Uchida, R.; Ohte, S.; Miyachi, N.; Kobayashi, K.; Sato, N.; Nonaka, K.; Masuma, R.; Fukuda, T.; Yasuhara, T.; et al. New dinapinone derivatives, potent inhibitors of triacylglycerol synthesis in mammalian cells, produced by Talaromyces pinophilus FKI-3864. J. Antibiot. 2013, 66, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Vinale, F.; Nicoletti, R.; Lacatena, F.; Marra, R.; Sacco, A.; Lombardi, N.; d’Errico, G.; Digilio, M.C.; M Lorito, M.; Woo, S.L. Secondary metabolites from the endophytic fungus Talaromyces pinophilus. Nat. Prod. Res. 2017, 31, 1778–1785. [Google Scholar] [CrossRef]
- Salvatore, M.M.; DellaGreca, M.; Nicoletti, R.; Salvatore, F.; Vinale, F.; Naviglio, D.; Andolfi, A. Talarodiolide, a new 12-membered macrodiolide, and GC/MS investigation of culture filtrate and mycelial extracts of Talaromyces pinophilus. Molecules 2018, 23, 950. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.T.; Shi, X.; Xian, P.J.; Feng, Z.; Yang, J.; Yang, X.L. A new fusicoccane diterpene and a new polyene from the plant endophytic fungus Talaromyces pinophilus and their antimicrobial activities. Nat. Prod. Res. 2021, 35, 124–130. [Google Scholar] [CrossRef]
- Yang, S.W.; Chan, T.M.; Terracciano, J.; Patel, R.; Patel, M.; Gullo, V.; Chu, M.A. New hydrogenated azaphilone Sch 725680 from Aspergillus sp. J. Antibiot. 2006, 59, 720–723. [Google Scholar] [CrossRef]
- Dethoup, T.; Manoch, L.; Kijjoa, A.; Nascimento, M.S.J.; Puaparoj, P.; Silva, A.M.S.; Eaton, G.; Herz, W. Bacillisporins D and E, new oxyphenalenone dimers from Talaromyces bacillisporus. Planta Med. 2006, 72, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Q.; Xia, G.; Huang, H.; Li, H.; Ma, L.; Lu, Y.; He, L.; Xia, X.; Zhigang She, Z. Polyketides with α-glucosidase inhibitory activity from a mangrove endophytic fungus, Penicillium sp. HN29-3B1. J. Nat. Prod. 2015, 78, 1816–1822. [Google Scholar] [CrossRef]
- Naganuma, M.; Nishida, M.; Kuramochi, K.; Sugawara, F.; Yoshida, H.; Mizushina, Y. 1-Deoxyrubralactone, a novel specific inhibitor of families X and Y of eukaryotic DNA polymerases from a fungal strain derived from sea algae. Bioorg. Med. Chem. 2008, 16, 2939–2944. [Google Scholar] [CrossRef]
- May Zin, W.W.; Buttachon, S.; Dethoup, T.; Pereira, J.A.; Luís Gales, L.; Inácio, A.; Costa, P.M.; Lee, M.; Sekeroglu, N.; Silva, A.M.S.; et al. Antibacterial and antibiofilm activities of the metabolites isolated from the culture of the mangrove-derived endophytic fungus Eurotium chevalieri KUFA0006. Phytochemistry 2017, 141, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Ahammad, K.H.; Zaman, U.; Park, J.H.; DeVine, L.; Hu, Z.; Wu, X.; Kim, H.S.; Cao, S. Secondary metabolites from the leather coral-derived fungal strain Xylaria sp. FM1005 and their glycoprotein IIb/IIIa inhibitory activity. J. Nat. Prod. 2021, 84, 466–473. [Google Scholar] [CrossRef]
- Li, X.; Gao, J.; Li, M.; Cui, H.; Jiang, W.; Tu, Z.C.; Yuan, T. Aromatic cadinane sesquiterpenoids from the fruiting bodies of Phellinus pini block SARS-CoV-2 spike–ACE2 interaction. J. Nat. Prod. 2021, 84, 2385–2389. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Yoshinari, T.; Koshino, H.; Fujioka, S.; Okada, K.; Shimada, A. Rubralactone, rubralides A, B and C, and rubramin produced by Penicillium rubrum. Biosci. Biotechnol. Biochem. 2007, 71, 1896–1901. [Google Scholar] [CrossRef]
- Yamazaki, M.; Okuyama, E. Isolation and structures of oxaphenalenone dimers from Talaromyces bacillosporus. Chem. Pharm. Bull. 1980, 28, 3649–3655. [Google Scholar] [CrossRef]
- Cao, P.; Yang, J.; Miao, C.P.; Yan, Y.; Ma, Y.T.; Li, X.N.; Zhao, L.X.; Huang, S.X. New duclauxamide from Penicillium manginii YIM PH30375 and structure revision of the duclauxin family. Org. Lett. 2015, 17, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.W.; Chan, T.M.; Terracciano, J.; Loebenberg, D.; Patel, M.; Gullo, V.; Chu, M. Sch 1385568, a new azaphilone from Aspergillus sp. J. Antibiot. 2009, 62, 401–403. [Google Scholar] [CrossRef]
- Zhai, M.M.; Niu, H.T.; Li, J.; Xiao, H.; Shi, Y.P.; Di, D.L.; Crews, P.; Wu, Q.W. Talaromycolides A−C, novel phenyl-substituted phthalides isolated from the green Chinese onion-derived fungus Talaromyces pinophilus AF-02. J. Agric. Food Chem. 2015, 63, 9558–9564. [Google Scholar] [CrossRef]
- May Zin, W.W.; Buttachon, S.; Dethoup, T.; Fernandes, C.; Cravo, S.; Pinto, M.M.M.; Gales, L.; Pereira, J.A.; Silva, A.M.S.; Sekeroglu, N.; et al. New cyclotetrapeptides and a new diketopiperzine derivative from the marine sponge-associated fungus Neosartorya glabra KUFA 0702. Mar. Drugs 2016, 14, 136. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; pp. 315–322. [Google Scholar]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 72, 5463–5467. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Simões, R.R.; Aires-de-Sousa, M.; Conceição, T.; Antunes, F.; da Costa, P.M.; de Lencastre, H. High prevalence of EMRSA-15 in Portuguese public buses: A worrisome finding. PLoS ONE 2011, 6, e17630. [Google Scholar] [CrossRef] [PubMed]
- Bessa, L.J.; Barbosa-Vasconcelos, A.; Mendes, Â.; Vaz-Pires, P.; Da Costa, P.M. High prevalence of multidrug-resistant Escherichia coli and Enterococcus spp. in river water, upstream and downstream of a wastewater treatment plant. J. Water Health 2014, 12, 426–435. [Google Scholar] [CrossRef]
- CLSI Supplement M100; Performance Standards for Antimicrobial Susceptibility Testing. 28th ed. Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2018.
- CLSI Document M02-A11; Performance Standards for Antimicrobial Disk Susceptibility Tests, Approved Standard. 11th ed. Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2012.
- CLSI Standard M07; Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically. 11th ed. Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2018.
- CLSI Document M26-A; Methods for Determining Bactericidal Activity of Antimicrobial Agents. Approved Guideline. Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 1999.
- Kumla, D.; Dethoup, T.; Gales, L.; Pereira, J.A.; Freitas-Silva, J.; Costa, P.M.; Silva, A.M.S.; Pinto, M.M.M.; Kijjoa, A. Erubescensoic acid, a new polyketide and a xanthonopyrone SPF-3059-26 from the culture of the marine sponge-associated fungus Penicillium erubescens KUFA 0220 and antibacterial activity evaluation of some of its constituents. Molecules 2019, 24, 208. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.M.; Bessa, L.J.; Buttachon, S.; Costa, P.M.; Buaruang, J.; Dethoup, T.; Silva, A.M.S.; Kijjoa, A. Antibacterial and antibiofilm activities of tryptoquivalines and meroditerpenes isolated from the marine-derived fungi Neosartorya paulistensis, N. laciniosa, N. tsunodae, and the soil fungi N. fischeri and N. siamensis. Mar. Drugs 2014, 12, 822–839. [Google Scholar] [CrossRef]
- Odds, F.C. Synergy, antagonism, and what the chequer board puts between them. J. Antimicrob. Chemother. 2003, 52, 1. [Google Scholar] [CrossRef] [PubMed]
- Stepanović, S.; Vuković, D.; Hola, V.; Di Bonaventura, G.; Djukić, S.; Cirković, I.; Ruzicka, F. Quantification of biofilm in microtiter plates: Overview of testing conditions and practical recommendations for assessment of biofilm production by staphylococci. APMIS 2007, 115, 891–899. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC | ROESY |
|---|---|---|---|---|---|
| 1 | 67.2, CH2 | 5.64, d (15.1) 5.72, d (15.1) | C-3, 3b,9 C-3, 3b,9 | ||
| 3 | 169.9, CO | - | |||
| 3a | 97.9, C | - | |||
| 3b | 137.3, C | - | |||
| 4 | 161.9, C | - | |||
| 5 | 119.5, CH | 6.95, s | H-10 | C-3, 4, 6a, 10 | H3-10 |
| 6 | 146.4, C | - | |||
| 6a | 119.5 | - | |||
| 7 | 137.1, C | - | |||
| 8 | 135.2, C | - | |||
| 9 | 149.4, C | - | |||
| 9a | 110.1, C | - | |||
| 10 | 24.8, CH3 | 2.97, s | C-5, 6 | H-5 | |
| 1′α β | 70.5, CH2 | 4.98, d (12.4) 5.13 d (12.4) | C-3′, 3′b, 8, 9′, 9′b | ||
| 3′ | 168.3, CO | ||||
| 3′a | 104.2, C | - | |||
| 3′b | 148.2, C | - | |||
| 4′ | 163.6, C | - | |||
| 5′ | 120.1, CH | 6.82, d (0.6) | H-10′ | C-3′a, 4′, 6′a, 10′ | H3-10′ |
| 6′ | 152.8, C | - | |||
| 6′a | 117,2, C | - | |||
| 7′ | 193.2, CO | ||||
| 8′ | 65.0, CH | 4.82,s | C-6′a, 7, 7′, 8, 9′, 9′a | H3-10 | |
| 9′ | 85.7, CH | 4.76, s | C-7, 8. 8′ | H-1′β | |
| 9′a | 50.0, C | - | |||
| 10′ | 23.7, CH3 | 2.48, s | C-5′, 6′ 6′a | H-5′ | |
| OH-4′ | 11.85, brs | ||||
| OH-9′ | 6.25, brs |
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC | ROESY |
|---|---|---|---|---|---|
| 1 | 159.4, CO | C-3, 9a | |||
| 2 | 114.8, CH | 6.46, d (10.0) | H-3 | C-1,9a | H-3 |
| 3 | 141.2, CH | 8.60, d (10.0) | H-2 | C-1, 3a | H-2,4 |
| 3a | 151.1, C | - | |||
| 3b | 133.7, C | - | |||
| 4 | 103.9, CH | 7.54, d (2.0 | H-6 | C-5, 6, 7a | H-3 |
| 5 | 157.3, C | ||||
| 6 | 122.0, CH | 7.04, d (2.0)- | H-4, H3-10 | C-7a, 10a - | H3-10 |
| 7 | 137.9, C | ||||
| 7a | 122.4, C | - | - | ||
| 8 | 142.8, C | - | |||
| 9 | 130.3, C | - | |||
| 9a | 112.6, C | - | |||
| 10 | 24.9, CH3 | 3.01, s | H-4, 6 | C-6, 7, 7a | H-6, 8′ |
| 1′α β | 70.1, CH2 | 5.04, d (12.6) 5.17, d (12.6) | 1′β 1′α | C-3′, 3′b, 9, 9′a C-9 | |
| 3′ | 168.1, CO | - | |||
| 3′a | 104.5, C | - | |||
| 3′b | 148.1, C | - | |||
| 4′ | 161.0, C | ||||
| 5′ | 121.0, CH | 6.77, s | H3-10′ | C-6′a | H3-10′ |
| 6′ | 152.0, C | - | |||
| 6′a | 117.0, C | ||||
| 7′ | 192.4, CO | - | |||
| 8′ | 65.7CH | 4.97, s | C-7′, 8, 9, 9′, 9′a | H3-10 | |
| 9′ | 86.0, CH | 4.79 | C-8, C-9 | H-1′α | |
| 9′a | 50.0, C | - | |||
| 10′ | 23.7, CH3 | 2.46, s | H-5′ | C-5′, 6′, 6′a | H-6′ |
| OH-9′ | 6.33, br | ||||
| OH-4′ | 10.26, br |
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC | ROESY |
|---|---|---|---|---|---|
| 1α β | 68.2, CH2 | 3.85, dd (11.0, 13.5) 4.71, dd (11.0, 5.2) | H-1β, 8a H-1α, 8a | H-8 H-1α, 8a | |
| 3 | 156.9, C | - | |||
| 4 | 110.5, CH | 6.30, s | C-2′, 3, 5, 8a | H-5 | |
| 4a | 149.1, C | - | |||
| 5 | 119.7, CH | 5.88, d (2.0) | C-4, 7, 8a | H-4 | |
| 6 | 195.4, CO | - | |||
| 7 | 73.8, C | - | |||
| 8 | 75.2, CH | 5.12, d (9.8) | H-8a | C-1, 1″, 8a | H3-1α, 9 |
| 8a | 34.9, CH | 3.25, m | H-1α, 1β, 8 | ||
| 9 | 19.5, CH3 | 1.22, s | C-6, 7 | H-8 | |
| 1′ | 123.6, CH | 6.22, d (15.5) | C-3, 3′ | ||
| 2′ | 136.9, CH | 7.05, d (15.5) | H-1′ | C-3, 3′ | |
| 3′ | 167.3, CO | - | |||
| 1″ | 169.1, CO | - | |||
| 2″ | 109.9, C | - | |||
| 3″ | 161.0, C | - | |||
| 4″ | 100.7, CH | 6.20, d (2.0) | H-6″ | C-6″ | |
| 5″ | 159.5, C | - | |||
| 6″ | 109.9CH | 6.19,brs | H-4″ | C-4″, 8″ | H3-8″ |
| 7″ | 140.0, C | - | |||
| 8″ | 21.6, CH3 | 2.30, s | C-2″, 6″, 7″ | H-6″ | |
| OH-5″/7″ | - | 10.45, brs | |||
| OH-7 | - | 5.82, br |
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 1 | 126.0, C | - | ||
| 2 | 126.2, C | - | ||
| 3 | 152.3, C | - | ||
| 4 | 121.4, C | - | ||
| 5 | 158.8, C | - | ||
| 6 | 101.4, CH | 6.69, s | H-8, 9a, 9b | C-1, 4, 5, 7 |
| 7 | 169.0, CO | - | ||
| 8 | 102.8, CH | 6.29, s | H3-9, 10 | C-17, 10 |
| 9α β | 18.3, CH2 | 4.35, d (14.4) 4.47, d (14.4) | H-6, 18 H-6, 18 | C-3, 4, 5, 6, 11, 12 C-3, 4, 5, 6, 11, 12 |
| 10 | 56.6, OCH3 | 3.44, s | H-8 | C-8 |
| 11 | 121.6, C | - | ||
| 12 | 155.2, C | - | ||
| 13 | 118.9, C | - | ||
| 14 | 148.2, C | - | ||
| 15 | 125.2, C | - | ||
| 16 | 117.0, C | - | ||
| 17 | 171.7, CO | - | ||
| 18 | 67.0, CH2 | 5.14, s | H-9, 19 | C-11, 14, 15, 17 |
| 19 | 10.2, CH3 | 2.04, s | H-18 | C-12, 13, 14 |
| OH | 9.42, s |
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 1 | 27.5, CH2 | 1.66, m 1.81, m | ||
| 2 | 34.0, CH2 | 2.27, m 2.45, m | ||
| 3 | 198.6, CO | - | ||
| 4 | 124.9, CH | 5.70, s | C-2, 6, 10 | |
| 5 | 163.4, C | - | ||
| 6 | 124.8, CH | 6.08, d (9.6) | H-7 | C-4, 5, 8, 10 |
| 7 | 132.6, CH | 7.04, d (9.6) | H-6 | C-5, 8, 9, 14 |
| 8 | 131.3, C | - | ||
| 9 | 71.8, C | - | ||
| 10 | 42.0, C | - | ||
| 11 | 26.4, CH2 | 1.54, m 1.95, m | ||
| 12 | 31.7, CH2 | 1.66, m 1.81, m | ||
| 13 | 44.5, C | - | ||
| 14 | 158.2, C | - | ||
| 15 | 68.4, CH | 4.64, m | H-16 | |
| 16 | 40.3, CH2 | 1.69, m | H-15 | |
| 17 | 52.5, CH | 1.60, m | ||
| 18 | 21.0, CH3 | 1.04, s | C-1, 5, 9, 10 | |
| 19 | 19.3, CH3 | 0.85, s | C-13, 14, 17 | |
| 20 | 38.5, CH | 2.08, m | H-21, 22 | |
| 21 | 21.7, CH3 | 1.05, d (6.4) | H-20 | C-17, 22 |
| 22 | 135.5, CH | 5.25, m | H-20 | C-24 |
| 23 | 132.3, CH | 5.24, m | H-24 | C-20 |
| 24 | 42.6, CH | 1.87, m | H-23, 26 | |
| 25 | 33.0, CH | 1.47, m | H-28 | |
| 26 | 17.9, CH3 | 0.92, d (6.8) | H-24 | C-23, 24, 25 |
| 27 | 19.9, CH3 | 0.84, d (6.8) | C-24, 25, 28 | |
| 28 | 20.3, CH3 | 0.82, d (6.8) | H-25 | C-24, 25, 27 |
| OH-9 | - | 4.33, s | C-8, 9, 10 |
| Compound | E. faecalis ATCC 29212 | S. aureus ATCC 29213 | S. aureus 74/24 (MRSA) | |||
|---|---|---|---|---|---|---|
| MIC | MBC | MIC | MBC | MIC | MBC | |
| 1 | >64 | >64 | 4 | >64 | 4 | >64 |
| 2 | 32 | >64 | 8 | >64 | 16 | >64 |
| 4 | >64 | >64 | >64 | >64 | >64 | >64 |
| 5 | >64 | >64 | 64 | >64 | 16 | >64 |
| 6 | >64 | >64 | >64 | >64 | >64 | >64 |
| 7 | >64 | >64 | >64 | >64 | >64 | >64 |
| 8 | >64 | >32 | >32 | >32 | >32 | >32 |
| 10 | >64 | >64 | >64 | >64 | >64 | >64 |
| 11 | >32 | >32 | >32 | >32 | >32 | >32 |
| VAN | 4 | - | - | - | - | - |
| OXA | - | - | 0.2 | - | 64 | - |
| Compound | Concentration | Biofilm Biomass (% of Control) | |
|---|---|---|---|
| E. faecalis ATCC 29212 | S. aureus ATCC 29213 | ||
| 1 | 8 µg/mL | - | 0.08 ± 0.03 *** (2xMIC) |
| 4 µg/mL | - | 0.19 ± 0.17 *** (MIC) | |
| 2 | 64 µg/mL | 0.13 ± 0.02 *** (2xMIC) | - |
| 16 µg/mL | - | 0.13 ± 0.05 *** (2xMIC) | |
| 8 µg/mL | - | 0.29 ± 0.13 *** (MIC) | |
| DMSO | 1% (v/v) | 0.99 ± 0.02 *** | 1.00 ± 0.02 *** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, F.P.; Rodrigues, I.C.; Georgopolou, A.; Gales, L.; Pereira, J.A.; Costa, P.M.; Mistry, S.; Hafez Ghoran, S.; Silva, A.M.S.; Dethoup, T.; et al. New Hybrid Phenalenone Dimer, Highly Conjugated Dihydroxylated C28 Steroid and Azaphilone from the Culture Extract of a Marine Sponge-Associated Fungus, Talaromyces pinophilus KUFA 1767. Mar. Drugs 2023, 21, 194. https://doi.org/10.3390/md21030194
Machado FP, Rodrigues IC, Georgopolou A, Gales L, Pereira JA, Costa PM, Mistry S, Hafez Ghoran S, Silva AMS, Dethoup T, et al. New Hybrid Phenalenone Dimer, Highly Conjugated Dihydroxylated C28 Steroid and Azaphilone from the Culture Extract of a Marine Sponge-Associated Fungus, Talaromyces pinophilus KUFA 1767. Marine Drugs. 2023; 21(3):194. https://doi.org/10.3390/md21030194
Chicago/Turabian StyleMachado, Fátima P., Inês C. Rodrigues, Aikaterini Georgopolou, Luís Gales, José A. Pereira, Paulo M. Costa, Sharad Mistry, Salar Hafez Ghoran, Artur M. S. Silva, Tida Dethoup, and et al. 2023. "New Hybrid Phenalenone Dimer, Highly Conjugated Dihydroxylated C28 Steroid and Azaphilone from the Culture Extract of a Marine Sponge-Associated Fungus, Talaromyces pinophilus KUFA 1767" Marine Drugs 21, no. 3: 194. https://doi.org/10.3390/md21030194
APA StyleMachado, F. P., Rodrigues, I. C., Georgopolou, A., Gales, L., Pereira, J. A., Costa, P. M., Mistry, S., Hafez Ghoran, S., Silva, A. M. S., Dethoup, T., Sousa, E., & Kijjoa, A. (2023). New Hybrid Phenalenone Dimer, Highly Conjugated Dihydroxylated C28 Steroid and Azaphilone from the Culture Extract of a Marine Sponge-Associated Fungus, Talaromyces pinophilus KUFA 1767. Marine Drugs, 21(3), 194. https://doi.org/10.3390/md21030194