
Marine-Derived Natural Product HDYL-GQQ-495 Targets P62 to Inhibit Autophagy

 ,
,  and
and
Abstract
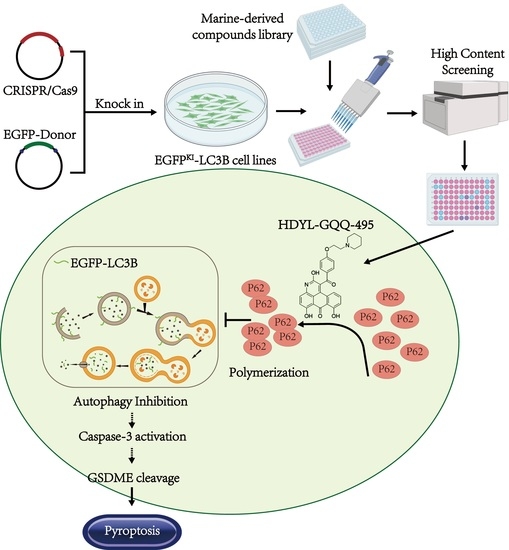
1. Introduction
2. Results
2.1. EGFP-LC3B Knock-In Cell Line Construction for Screening of Marine-Derived Compounds
2.2. HDYL-GQQ-495 Is a Novel Autophagy Inhibitor
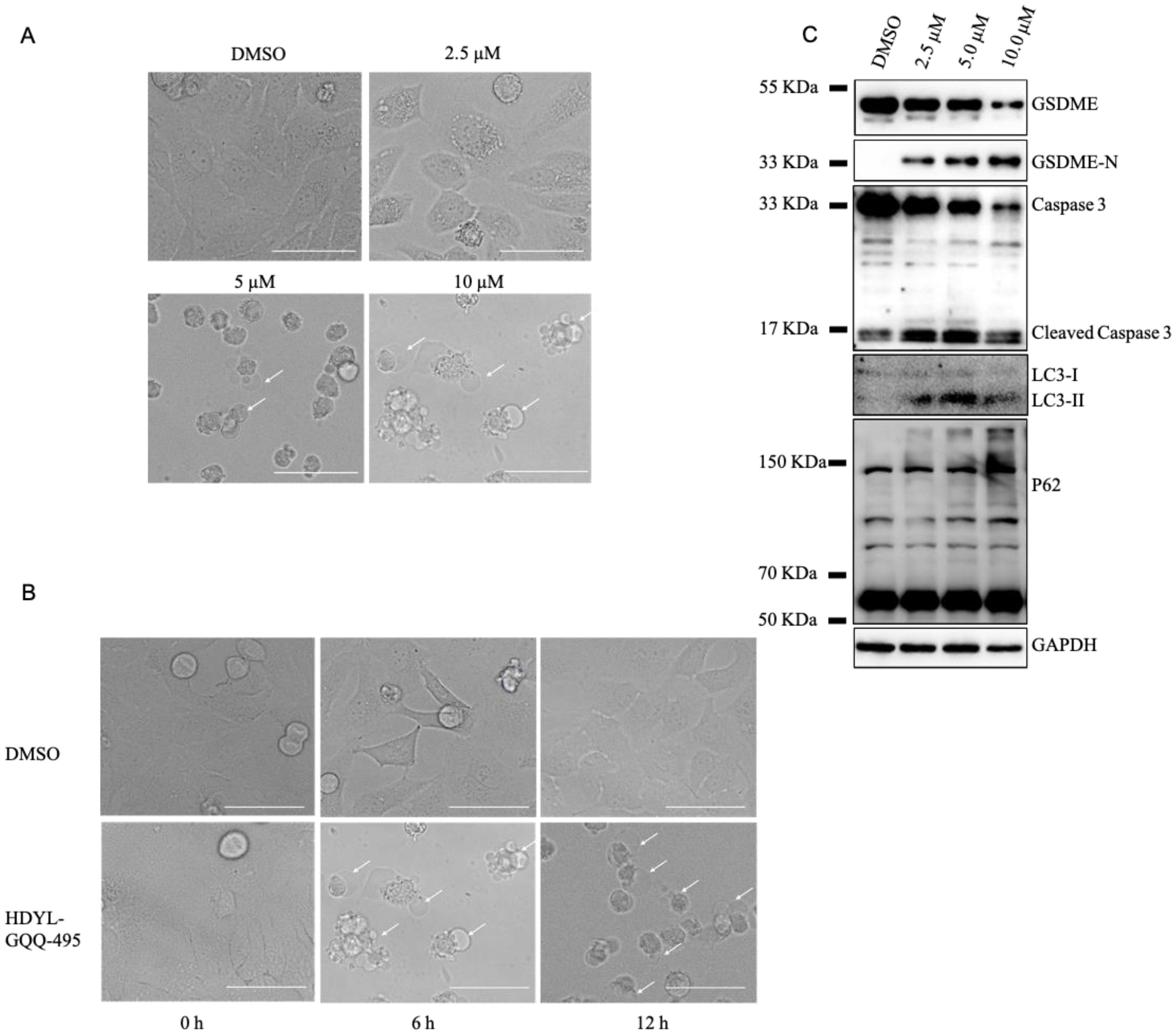
2.3. HDYL-GQQ-495 Inhibits Autophagy and Promotes Pyroptosis
2.4. HDYL-GQQ-495 Targets P62 to Inhibit Autophagy and Promote Pyroptosis
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture
4.3. CRISPR/Cas9 and EGFP-Donor Construction
4.4. Transfection and Single Clone Isolation
4.5. High Throughput Screening
4.6. Immunofluorescence
4.7. Microscale Thermophoresis Assay
4.8. Synthesis HDYL-GQQ-495
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2013, 24, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Fang, Y.; Yang, Y.; Qin, Y.; Wu, P.; Wang, T.; Lai, H.; Meng, L.; Wang, D.; Zheng, Z.; et al. Elaiophylin, a novel autophagy inhibitor, exerts antitumor activity as a single agent in ovarian cancer cells. Autophagy 2015, 11, 1849–1863. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, J.; Pan, H.; Hu, P.; Hao, Y.; Cai, W.; Zhu, H.; Yu, A.D.; Xie, X.; Ma, D.; et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc. Natl. Acad. Sci. USA 2007, 104, 19023–19028. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the Autophagosome Maturation Process by a Novel Reporter Protein, Tandem Fluorescent-Tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef]
- Pampaloni, F.; Mayer, B.; Vel-Job, K.K.; Ansari, N.; Hötte, K.; Kögel, D.; Stelzer, E.H. A Novel Cellular Spheroid-Based Autophagy Screen Applying Live Fluorescence Microscopy Identifies Nonactin as a Strong Inducer of Autophagosomal Turnover. SLAS Discov. 2017, 22, 558–570. [Google Scholar] [CrossRef]
- Kaizuka, T.; Morishita, H.; Hama, Y.; Tsukamoto, S.; Matsui, T.; Toyota, Y.; Kodama, A.; Ishihara, T.; Mizushima, T.; Mizushima, N. An Autophagic Flux Probe that Releases an Internal Control. Mol. Cell 2016, 64, 835–849. [Google Scholar] [CrossRef]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Du, L.; Zhu, T.; Fang, Y.; Liu, H.; Gu, Q.; Zhu, W. Aspergiolide A, a novel anthraquinone derivative with naphtho[1,2,3-de]chromene-2,7-dione skeleton isolated from a marine-derived fungus Aspergillus glaucus. Tetrahedron 2007, 63, 1085–1088. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kumanomidou, T.; Sou, Y.-S.; Mizushima, T.; Ezaki, J.; Ueno, T.; Kominami, E.; Yamane, T.; Tanaka, K.; Komatsu, M. Structural Basis for Sorting Mechanism of p62 in Selective Autophagy. J. Biol. Chem. 2008, 283, 22847–22857. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Donohue, E.; Balgi, A.D.; Komatsu, M.; Roberge, M. Induction of Covalently Crosslinked p62 Oligomers with Reduced Binding to Polyubiquitinated Proteins by the Autophagy Inhibitor Verteporfin. PLoS ONE 2014, 9, e114964. [Google Scholar] [CrossRef] [PubMed]
- Donohue, E.; Tovey, A.; Vogl, A.W.; Arns, S.; Sternberg, E.; Young, R.; Roberge, M. Inhibition of Autophagosome Formation by the Benzoporphyrin Derivative Verteporfin. J. Biol. Chem. 2011, 286, 7290–7300. [Google Scholar] [CrossRef]
- Liu, J.; Wang, C.; Li, J.; Yu, Y.; Liu, Y.; Liu, H.; Peng, Q.; Guan, X. Autophagy blockage promotes the pyroptosis of ox-LDL-treated macrophages by modulating the p62/Nrf2/ARE axis. J. Physiol. Biochem. 2021, 77, 419–429. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Broz, P.; Pelegrín, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Yang, X.; Chen, G.; Yu, K.N.; Yang, M.; Peng, S.; Ma, J.; Qin, F.; Cao, W.; Cui, S.; Nie, L.; et al. Cold atmospheric plasma induces GSDME-dependent pyroptotic signaling pathway via ROS generation in tumor cells. Cell Death Dis. 2020, 11, 295. [Google Scholar] [CrossRef]
- Zhao, P.; Wang, M.; Chen, M.; Chen, Z.; Peng, X.; Zhou, F.; Song, J.; Qu, J. Programming cell pyroptosis with biomimetic nanoparticles for solid tumor immunotherapy. Biomaterials 2020, 254, 120142. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fang, Y.; Chen, X.; Wang, Z.; Liang, X.; Zhang, T.; Liu, M.; Zhou, N.; Lv, J.; Tang, K.; et al. Gasdermin E–mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol. 2020, 5, eaax7969. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, S.; Qi, J.; Chen, Z.; Wu, Y.; Guo, J.; Wang, K.; Sun, X.; Zheng, J. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019, 10, 193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; González-Polo, R.-A.; Casares, N.; Perfettini, J.-L.; Dessen, P.; Larochette, N.; Métivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of Macroautophagy Triggers Apoptosis. Mol. Cell. Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mul, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nature 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Tanida, I. Autophagosome Formation and Molecular Mechanism of Autophagy. Antioxidants Redox Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef]
- Ratovitski, E.A. Tumor Protein (TP)-p53 Members as Regulators of Autophagy in Tumor Cells upon Marine Drug Exposure. Mar. Drugs 2016, 14, 154. [Google Scholar] [CrossRef]
- Yu, H.; Wu, C.-L.; Wang, X.; Ban, Q.; Quan, C.; Liu, M.; Dong, H.; Li, J.; Kim, G.-Y.; Choi, Y.H.; et al. SP600125 enhances C-2-induced cell death by the switch from autophagy to apoptosis in bladder cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 448. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Menchinskaya, E.S.; Venz, S.; Rast, S.; Amann, K.; Hauschild, J.; Otte, K.; Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; et al. The marine triterpene glycoside frondoside A exhibits activity in vitro and in vivo in prostate cancer. Int. J. Cancer. 2016, 138, 2450–2465. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, Q.; Li, C.; Sun, S.; Li, Z.; Wu, J. Quantitative determination of autophagy flux by probes. Methods Cell Biol. 2021, 164, 157–165. [Google Scholar] [PubMed]
- Korolchuk, V.I.; Mansilla, A.; Menzies, F.M.; Rubinsztein, D.C. Autophagy Inhibition Compromises Degradation of Ubiquitin-Proteasome Pathway Substrates. Mol. Cell 2009, 33, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Tao, M.; Liu, T.; You, Q.; Jiang, Z. p62 as a therapeutic target for tumor. Eur. J. Med. Chem. 2020, 193, 112231. [Google Scholar] [CrossRef]
- Teramachi, J.; Silbermann, R.; Yang, P.; Zhao, W.; Mohammad, K.S.; Guo, J.; Anderson, J.L.; Zhou, D.; Feng, R.; Myint, K.Z.; et al. Blocking the ZZ domain of sequestosome1/p62 suppresses myeloma growth and osteoclast formation in vitro and induces dramatic bone formation in myeloma-bearing bones in vivo. Leukemia 2016, 30, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef]
- Jiang, Z.; Lu, M.-C.; Xu, L.; Yang, T.-T.; Xi, M.-Y.; Xu, X.-L.; Guo, X.-K.; Zhang, X.-J.; You, Q.-D.; Sun, H.-P. Discovery of Potent Keap1–Nrf2 Protein–Protein Interaction Inhibitor Based on Molecular Binding Determinants Analysis. J. Med. Chem. 2014, 57, 2736–2745. [Google Scholar] [CrossRef]
- Yasuda, D.; Nakajima, M.; Yuasa, A.; Obata, R.; Takahashi, K.; Ohe, T.; Ichimura, Y.; Komatsu, M.; Yamamoto, M.; Imamura, R.; et al. Synthesis of Keap1-phosphorylated p62 and Keap1-Nrf2 protein-protein interaction inhibitors and their inhibitory activity. Bioorganic Med. Chem. Lett. 2016, 26, 5956–5959. [Google Scholar] [CrossRef]
- Jiang, M.; Qi, L.; Li, L.; Li, Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. 2020, 6, 112. [Google Scholar] [CrossRef]
- Cao, W.; Chen, G.; Wu, L.; Yu, K.; Sun, M.; Yang, M.; Jiang, Y.; Jiang, Y.; Xu, Y.; Peng, S.; et al. Ionizing Radiation Triggers the Antitumor Immunity by Inducing Gasdermin E-Mediated Pyroptosis in Tumor Cells. Int. J. Radiat. Oncol. 2022, 115, 440–452. [Google Scholar] [CrossRef]
- Sansone, C.; Bruno, A.; Piscitelli, C.; Baci, D.; Fontana, A.; Brunet, C.; Noonan, D.; Albini, A. Natural Compounds of Marine Origin as Inducers of Immunogenic Cell Death (ICD): Potential Role for Cancer Interception and Therapy. Cells 2021, 10, 231. [Google Scholar] [CrossRef]
- Volynets, G.P.; Chekanov, M.O.; Synyugin, A.R.; Golub, A.G.; Kukharenko, O.P.; Bdzhola, V.G.; Yarmoluk, S.M. Identification of 3H-Naphtho[1,2,3-de]quinoline-2,7-diones as Inhibitors of Apoptosis Signal-Regulating Kinase 1 (ASK1). J. Med. Chem. 2011, 54, 2680–2686. [Google Scholar] [CrossRef] [PubMed]
- Dziadulewicz, E.K.; Bevan, S.J.; Brain, C.T.; Coote, P.R.; Culshaw, A.J.; Davis, A.J.; Edwards, L.J.; Fisher, A.J.; Fox, A.J.; Gentry, C.; et al. Naphthalen-1-yl-(4-pentyloxynaphthalen-1-yl)methanone: A potent, orally bioavailable human CB1/CB2 dual agonist with antihyperalgesic properties and restricted central nervous system penetration. J. Med. Chem. 2007, 50, 3851–3856. [Google Scholar] [CrossRef] [PubMed]
- Meegan, M.J.; Hughes, R.B.; Lloyd, D.G.; Williams, D.C.; Zisterer, D.M. Flexible estrogen receptor modulators: Design, synthesis, and antagonistic effects in human MCF-7 breast cancer cells. J. Med. Chem. 2001, 44, 1072–1084. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| LC3B knock-in sgRNA | ACGTTCGGCTAAGATGCCGTCGG |
| Puc19-hindIII-EGFP-LC3-Foward | GACCATGATTACGCCAAGCTTCCGACGGCATGGTGCAGGGATCTGTGAG-CAAGGGCGAGGAGC |
| Puc19-EcoRI-GFP-LC3-Reverse | AAAACGACGGCCAGTGAATTCAGATCCCTGCACCATGCCGTCGGTCTTGTACAGCTCGTCCATGCC |
| Genome sequencing Forward1 | CTATCGCCAGAGTCGGATTCGC |
| Genome sequencing Reverse1 | TCACGGCGTCCCAGGCCCTG |
| Genome sequencing Forward2 | TGGCTATCGCCAGAGTCGGA |
| Genome sequencing Reverse2 | CTCCTCTCGACCGAGGCACT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Fan, J.; Chen, Y.; Liu, Y.; Liu, H.; Jiang, W.; Li, D.; Dang, Y. Marine-Derived Natural Product HDYL-GQQ-495 Targets P62 to Inhibit Autophagy. Mar. Drugs 2023, 21, 68. https://doi.org/10.3390/md21020068
Li Q, Fan J, Chen Y, Liu Y, Liu H, Jiang W, Li D, Dang Y. Marine-Derived Natural Product HDYL-GQQ-495 Targets P62 to Inhibit Autophagy. Marine Drugs. 2023; 21(2):68. https://doi.org/10.3390/md21020068
Chicago/Turabian StyleLi, Quanfu, Jianjun Fan, Yinghan Chen, Yiyang Liu, Hang Liu, Wei Jiang, Dehai Li, and Yongjun Dang. 2023. "Marine-Derived Natural Product HDYL-GQQ-495 Targets P62 to Inhibit Autophagy" Marine Drugs 21, no. 2: 68. https://doi.org/10.3390/md21020068
APA StyleLi, Q., Fan, J., Chen, Y., Liu, Y., Liu, H., Jiang, W., Li, D., & Dang, Y. (2023). Marine-Derived Natural Product HDYL-GQQ-495 Targets P62 to Inhibit Autophagy. Marine Drugs, 21(2), 68. https://doi.org/10.3390/md21020068