Abstract
Using the OSMAC (One Strain Many Compounds) approach, the actinobacterium Streptomyces griseorubiginosus, derived from an unidentified cnidarian collected from a reef near Pointe de Bellevue in Réunion Island (France), was subjected to cultivation under diverse conditions. This endeavour yielded the isolation of a repertoire of 23 secondary metabolites (1–23), wherein five compounds were unprecedented as natural products (19–23). Specifically, compounds 19 and 20 showcased novel anthrone backbones, while compound 23 displayed a distinctive tetralone structure. Additionally, compounds 21 and 22 presented an unusual naphtho [2,3-c]furan-4(9H)-one chromophore. Interestingly, the detection of all these novel compounds (19–23) was exclusively achieved when the bacterium was cultured in FA-1 liquid medium supplemented with the epigenetic modifier γ-butyrolactone. The elucidation of the structural features of the newfound compounds was accomplished through a combination of HRESIMS, 1D and 2D NMR spectroscopy, as well as QM-NMR (Quantum Mechanical—Nuclear Magnetic Resonance) methods and by comparison with existing literature. Moreover, the determination of the relative configuration of compound 23 was facilitated by employing the mix-J-DP4 computational approach.
1. Introduction
Natural products of microbial origin, especially those from actinobacteria, have been proven to be successful drug leads [1,2,3]. However, standard culture conditions often fail to reveal their full biosynthetic potential and leads to re-isolation of already known metabolites. Strategies to unlock silent biosynthetic gene clusters that are not expressed using conventional fermentation methods include the so-called OSMAC (One Strain Many Compounds) approach [4,5,6,7,8,9]. The OSMAC approach has been shown as a simple and powerful tool to promote the production of different secondary metabolites, potentially bioactive, by altering simple culture parameters, such as variations in medium composition or the addition of epigenetic modifiers (chemical elicitors). The fungus Sphaeropsidales sp. was known to produce cladospirone bisepoxide, a spirobisnaphthalene compound. However, Bode et al. [10], reported that culturing the same strain in altered cultivation conditions, different media, and cultivation vessels led to the isolation of fourteen additional compounds, including eight new cladospirones B–I. Notably, the highest number of metabolites was produced when this strain was subjected to solid-phase cultivation in P-flasks with wet oat grains as a single substrate.
As part of our ongoing research on marine microorganisms, we investigated the actinobacterium Streptomyces griseorubiginosus, derived from an unidentified cnidarian collected at a reef close to Pointe de Bellevue in Réunion Island (France), following an OSMAC approach. Streptomyces griseorubiginosus has been previously reported to produce several antibiotic secondary metabolites, such as the angucyclinone-type rubiginones A1, B1, C1, and C2 [11]; the 15-membered cyclic peptides biphenomycins A-C [12]; the glycosilated anthracyclines cinerubins A and B [13]; and the alkaloid reductiomycin [14].
In this study, the most noteworthy effects with regard to an alteration of the bacterial metabolite pattern were detected following the addition of γ-butyrolactone to the FA-1 liquid medium, compared to the standard culture lacking this chemical elicitor. The investigation of the bacterial extract obtained from fermentation of Streptomyces griseorubiginosus under these conditions led to the isolation of five new aromatic polyketides (19–23), in addition to several known compounds, all of which were not detected when the fungus was grown without this activator. Herein, we report the isolation and the structure elucidation of the new metabolites discovered.
2. Results and Discussion
2.1. Isolation of Secondary Metabolites from S. griseorubiginosus through OSMAC
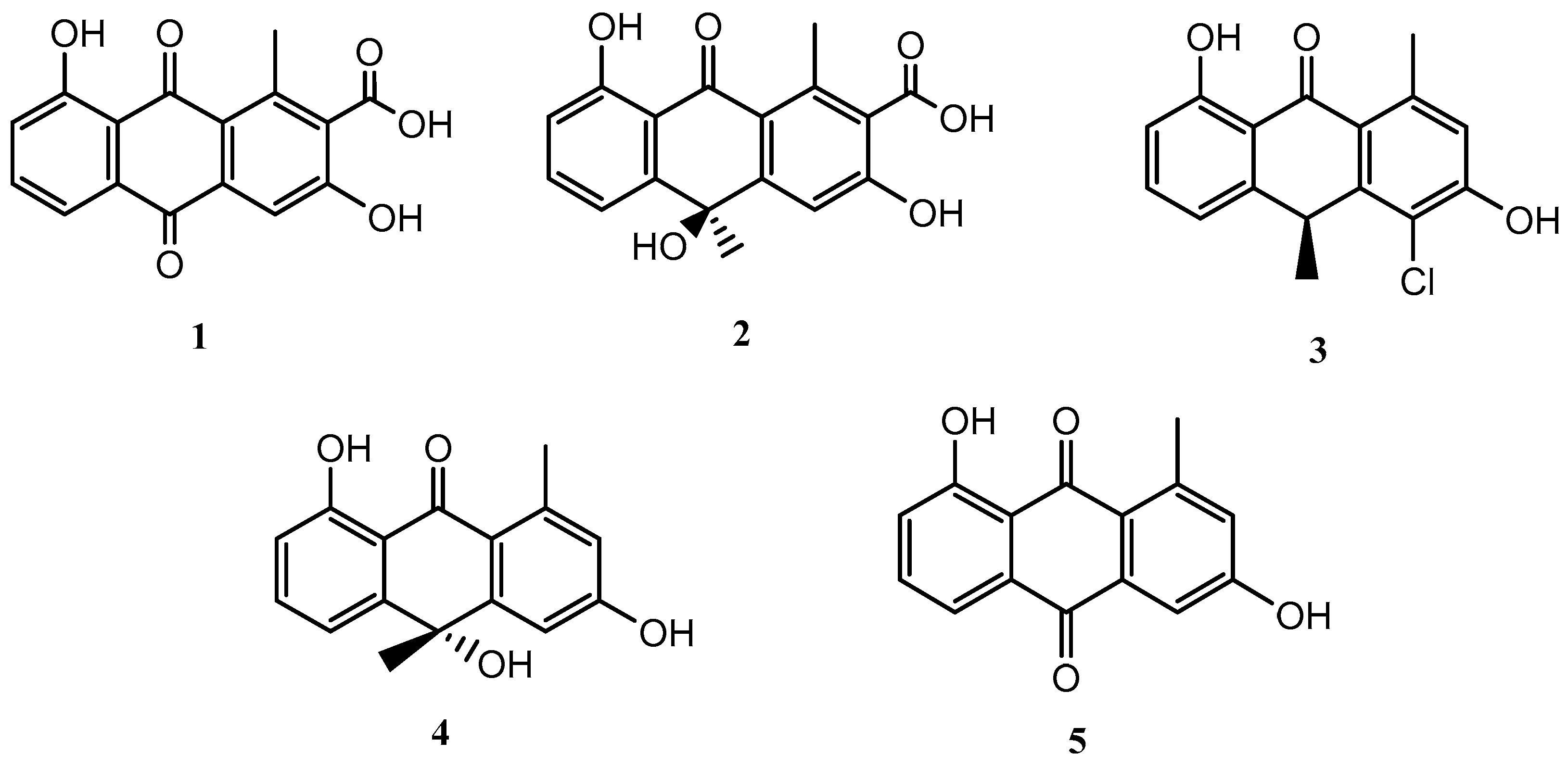
The fermentation of the microorganism Streptomyces griseorubiginosus was carried out under standard conditions (FA-1 medium), and five natural products (1–5) were isolated and identified; all of them were anthraquinone and anthrone derivatives (Figure 1).
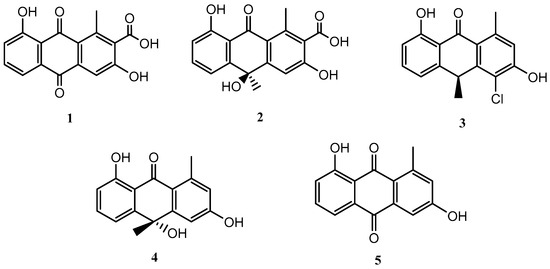
Figure 1.
Secondary metabolites isolated from Streptomyces griseorubiginosus (FA-1 medium culture).
Compounds 1–5 were identified as DMAC (1) [15,16], (–)-oryzanthrone A (2), (+)-chlororyzanthrone B (3), (+)-oryzanthrone B (4) [17], and aloesaponarin II (5) [18], by comparing HRESIMS, 1D and 2D NMR spectra, and their optical rotations with those described in the literature.
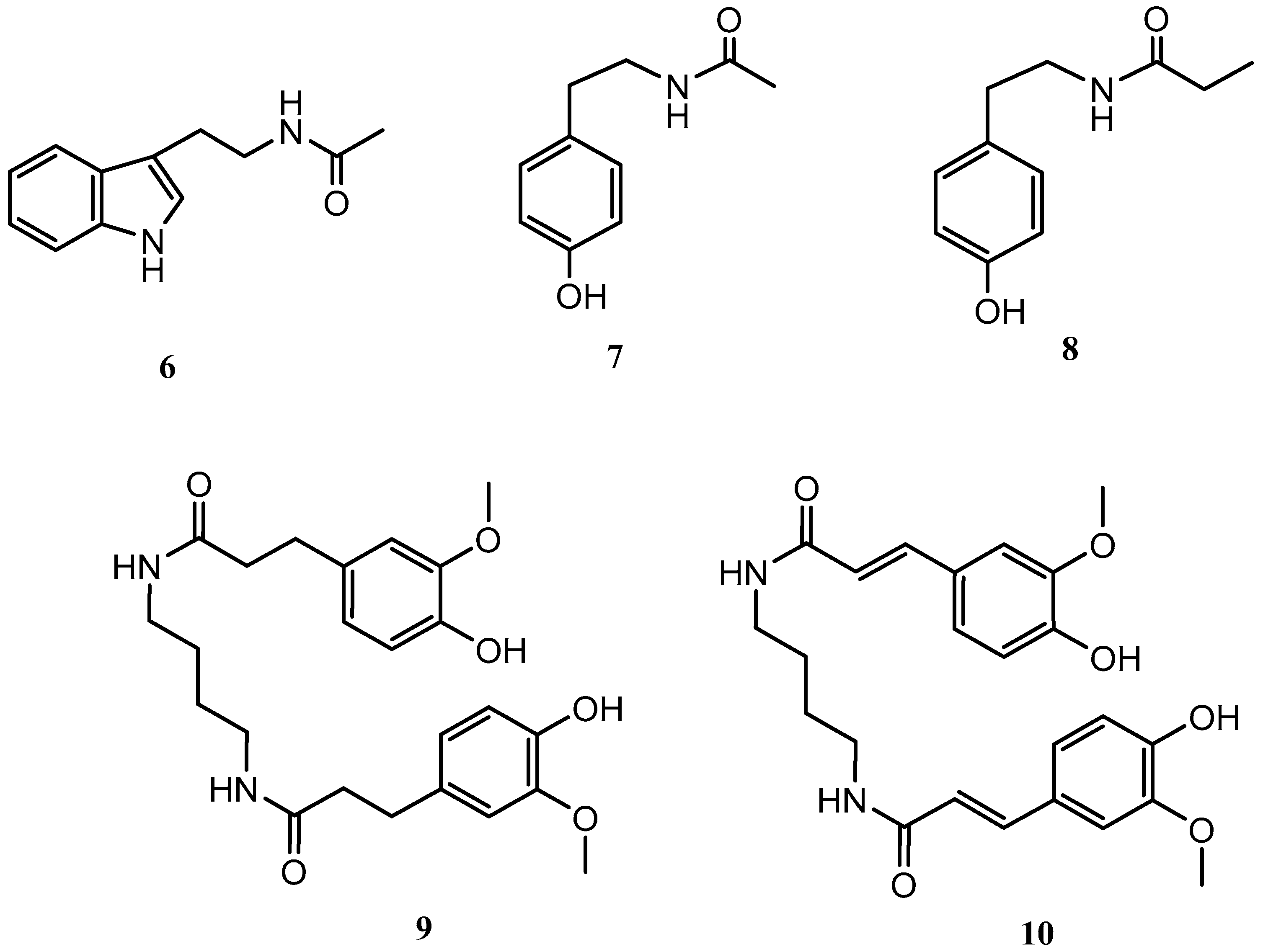
Streptomyces griseorubiginosus was then cultured on SAF medium, yielding five further secondary metabolites (6–10), which were not detected from the standard condition culture.
Compounds 6–10 were identified as N-acetyltryptamine (6), N-acetyltyramine (7), N-(4-hydroxyphenethyl)propionamide (8) [19], JBIR-94 (9) [20] and terrestribisamide (10) [21] by comparison with the data described in the literature (Figure 2).
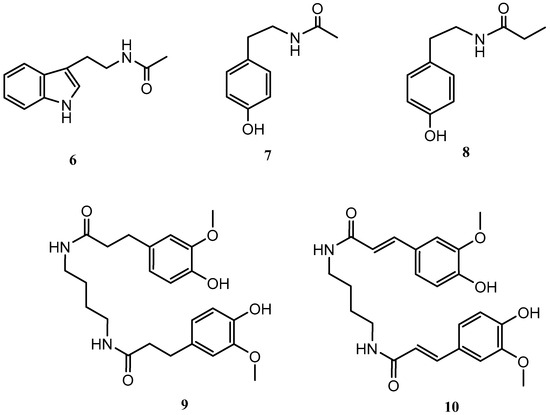
Figure 2.
Secondary metabolites isolated from Streptomyces griseorubiginosus (SAF medium culture).
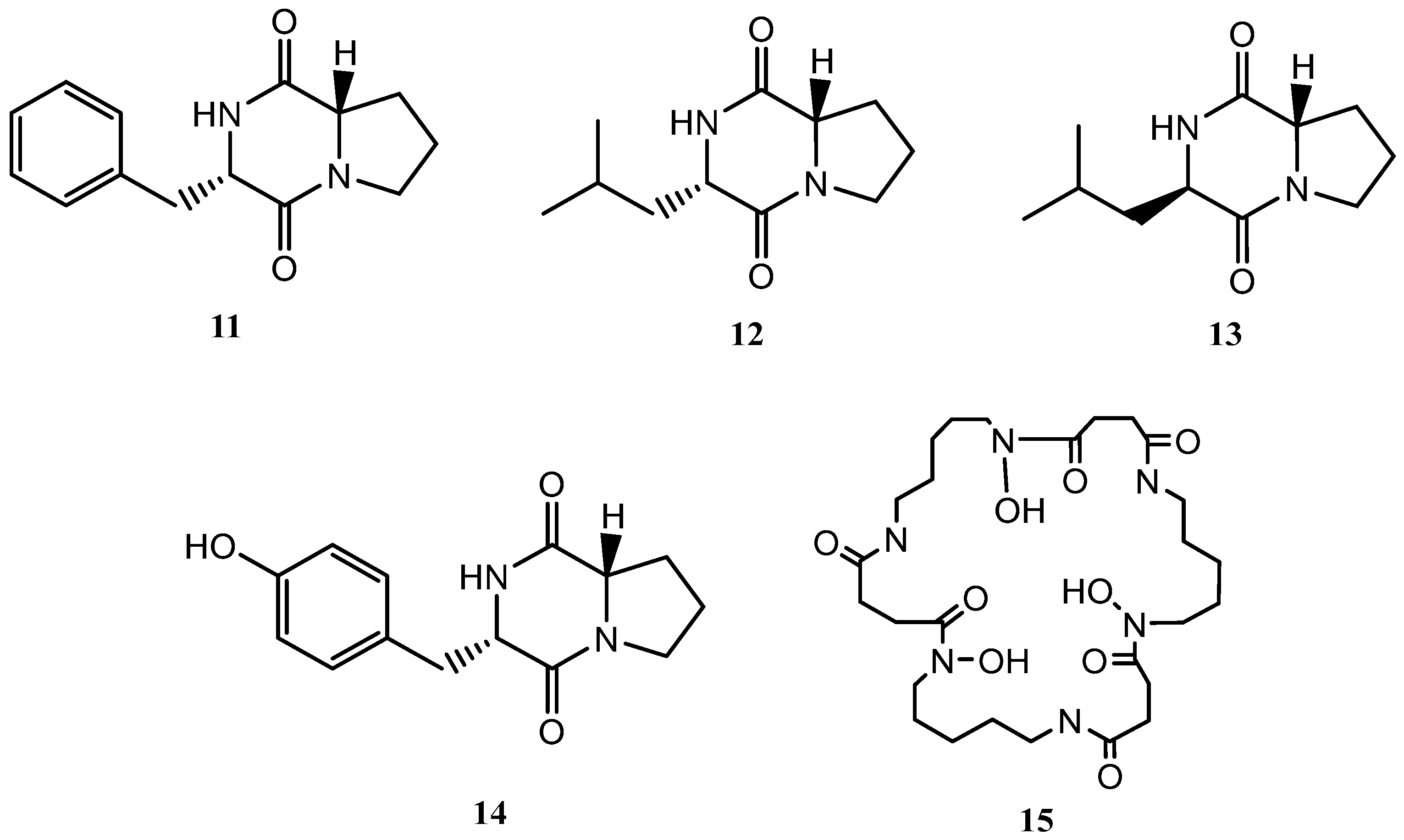
Following the OSMAC approach, the microorganism was fermented in a solid medium (SymPV-21). An analysis of the bacterial culture led to the isolation of the known compounds cyclo(L-prolyl-L-phenylalanine) (11) [22], cyclo(L-leucyl-L-prolyl) (12) [23], cyclo(D-leucyl-L-prolyl) (13) [24], maculosin (14) [24], and nocardamine (15) [25], which were not produced under the previous culture conditions (Figure 3).
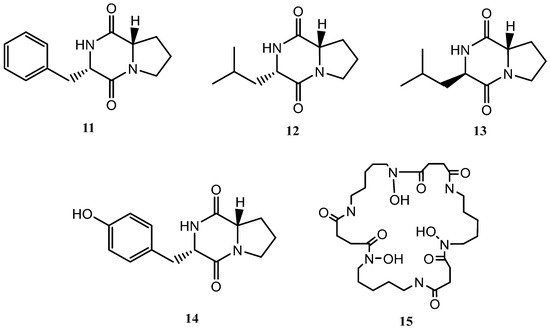
Figure 3.
Secondary metabolites isolated from Streptomyces griseorubiginosus (SymPV-21 solid medium culture).
The addition of epigenetic modifiers to the culture medium was also studied. These molecules can trigger a new metabolic profile, producing new metabolites or increasing their yield.
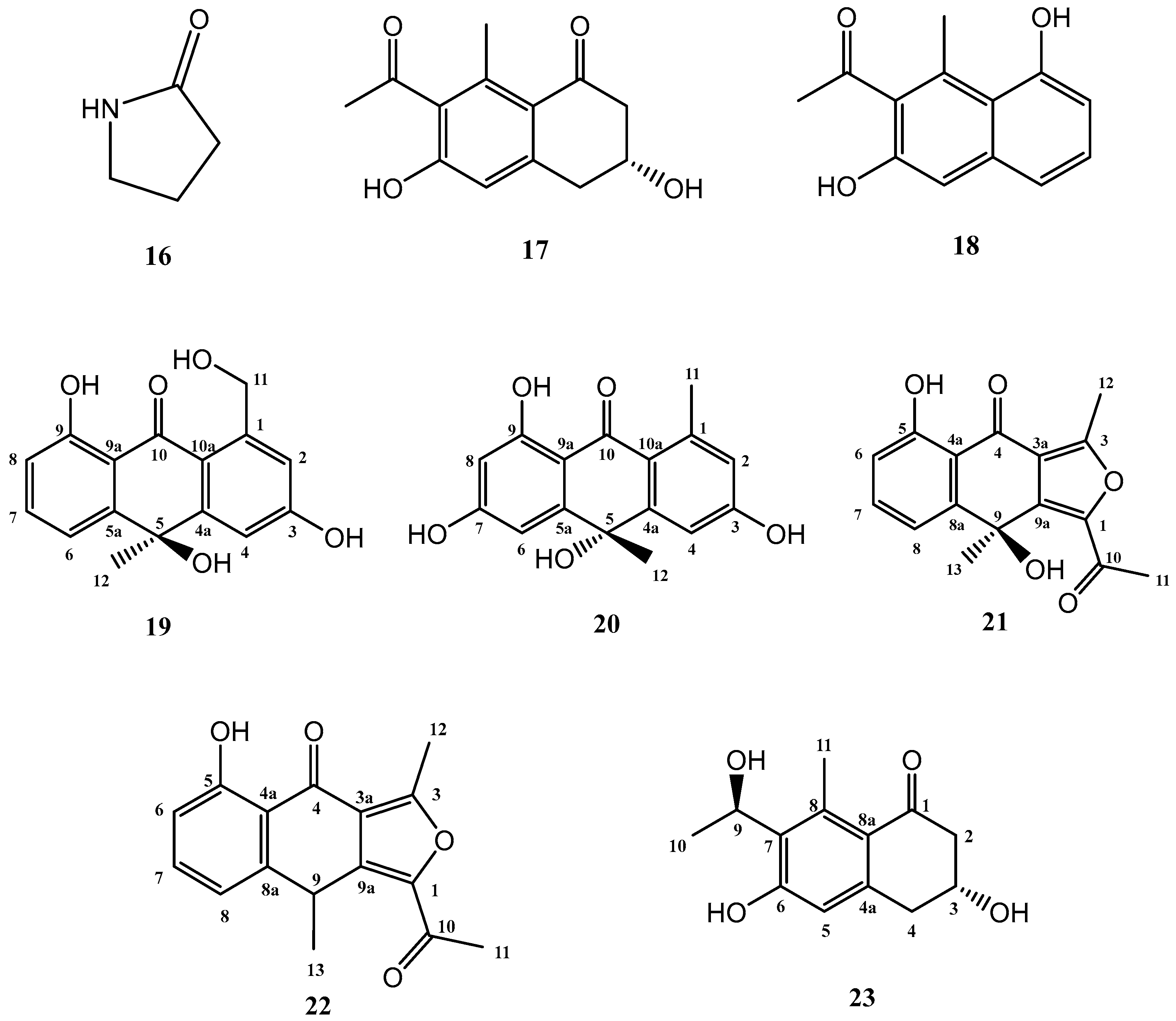
The addition of γ-butyrolactone to the standard culture medium (FA-1), led to the isolation of three known compounds (16–18) and five further secondary metabolites, shown in Figure 4, not previously described in the literature (19–23).
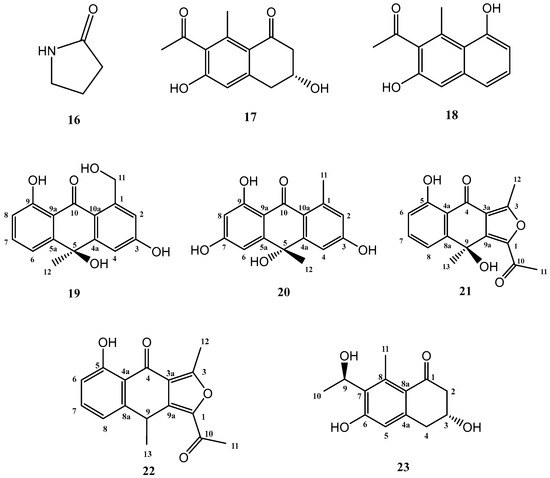
Figure 4.
Secondary metabolites isolated from Streptomyces griseorubiginosus (FA-1 medium culture + γ-butyrolactone).
Known compounds 16–18 were identified as γ-butyrolactam (16) [26], GTRI-02 (17) [27,28], and 6-dehydroxy-GTRI-02 (18) [28] after extensive spectroscopic analysis and comparison with data reported in the literature.
2.2. Structure Elucidation of Novel Compounds
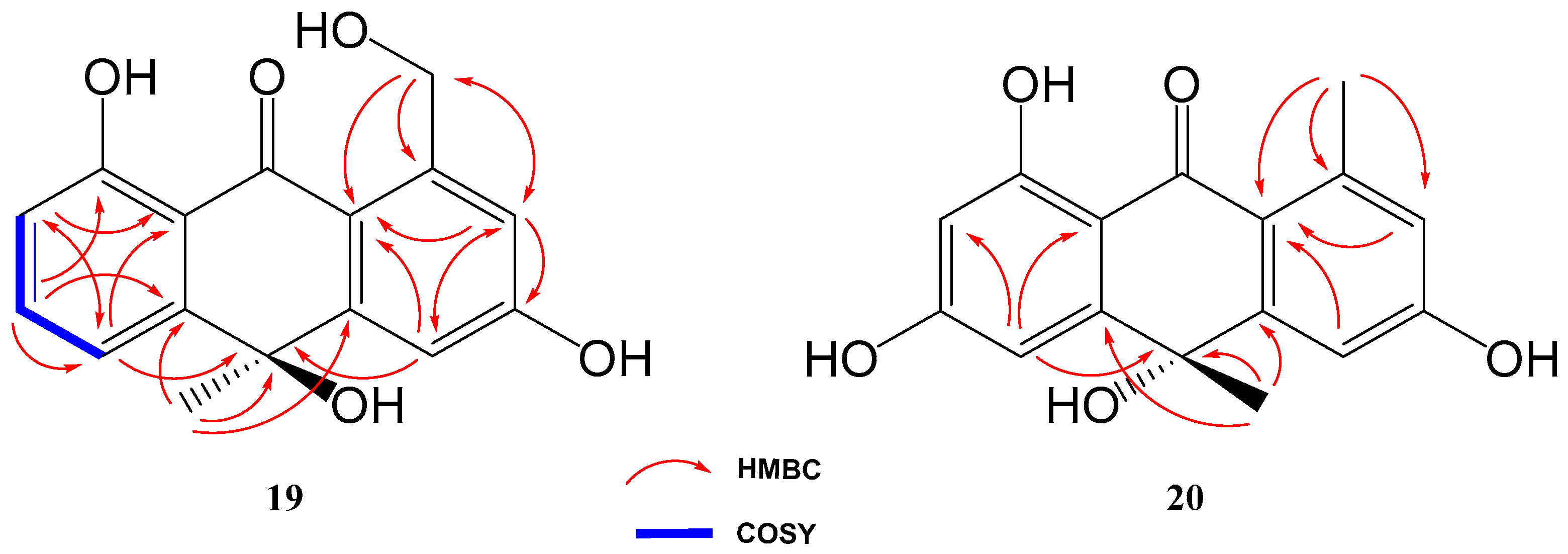
Compound 19 was isolated and purified to obtain a yellowish amorphous solid. Analysis by HRESIMS and NMR data determined its molecular formula to be C16H14O5 (ten degrees of unsaturation). 1D NMR spectra were very similar to those of previously isolated anthrone-derived molecules. In comparison with (+)-Oryzanthrone B (4), the presence of an oxygenated methylene (δH 4.66, 4.92, δC 65.9) was observed instead of the methyl (δH 2.68, δC 24.3) attached to the aromatic ring. The gHMBC correlations observed from H2-11 to C-1, C-2 and C-10a confirmed the replacement of the methyl group by the oxygenated methylene (Figure 5). Further 2D NMR analysis confirmed that the rest of the molecule was identical to 4, determining the structure of 19 as shown in Figure 4.
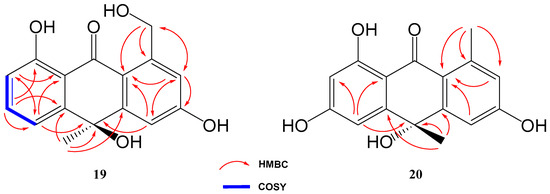
Figure 5.
Key COSY and HMBC correlations of compounds 19 and 20.
Based on the opposite sign of the optical rotation of compound 19 ([α]20D = −133.9°) compared to that published for (+)-Oryzanthrone B ([α]25D = +10.7°), we tentatively assign an absolute configuration R to carbon 5 in this compound.
Compound 20 was a yellowish amorphous solid with the molecular formula C16H14O5 determined by HRESIMS and NMR data (10 degrees of unsaturation). An initial study of the 1D NMR spectra of 20 (Table 1) revealed that this compound was a new anthrone derivative. Comparison of the 1H and 13C NMR spectra with those of (+)-Oryzanthrone B (4) showed the similarity between these two molecules, the only difference being the presence of a hydroxy group attached to C-7. The molecular mass of 20, 16 amu higher than that of 4, supported this suggestion. 1H-NMR and gCOSY experiments suggested the presence of an anthrone scaffold, with two tetrasubstituted aromatic rings with two protons in meta each (J2–4 = 2.5 Hz; J6–8 = 2.4 Hz). These data together with the observed correlations in gHMBC experiments from H-2 and H-4 to C-10a and from H-6 to C-5, C-8 and C-9a confirmed the structure of compound 20 as shown in Figure 4 and Figure 5.

Table 1.
NMR data of compounds 19 and 20.
Moreover, compound 20 showed an optical rotation ([α]20D = +2.2°) similar in sign and magnitude to that of (+)-Oryzanthrone B ([α]25D = +10.7°), in contrast to the negative optical rotation of compound 19 ([α]20D = −133.9°), so we tentatively assign absolute configuration S to carbon 5 in compound 20.
The third novel natural product produced by Streptomyces griseorubiginosus using γ-butyrolactone as an epigenetic modifier on FA-1 medium was a yellowish amorphous solid (21) with the molecular formula C16H14O5, established by HRESIMS and NMR data (10 degrees of unsaturation).
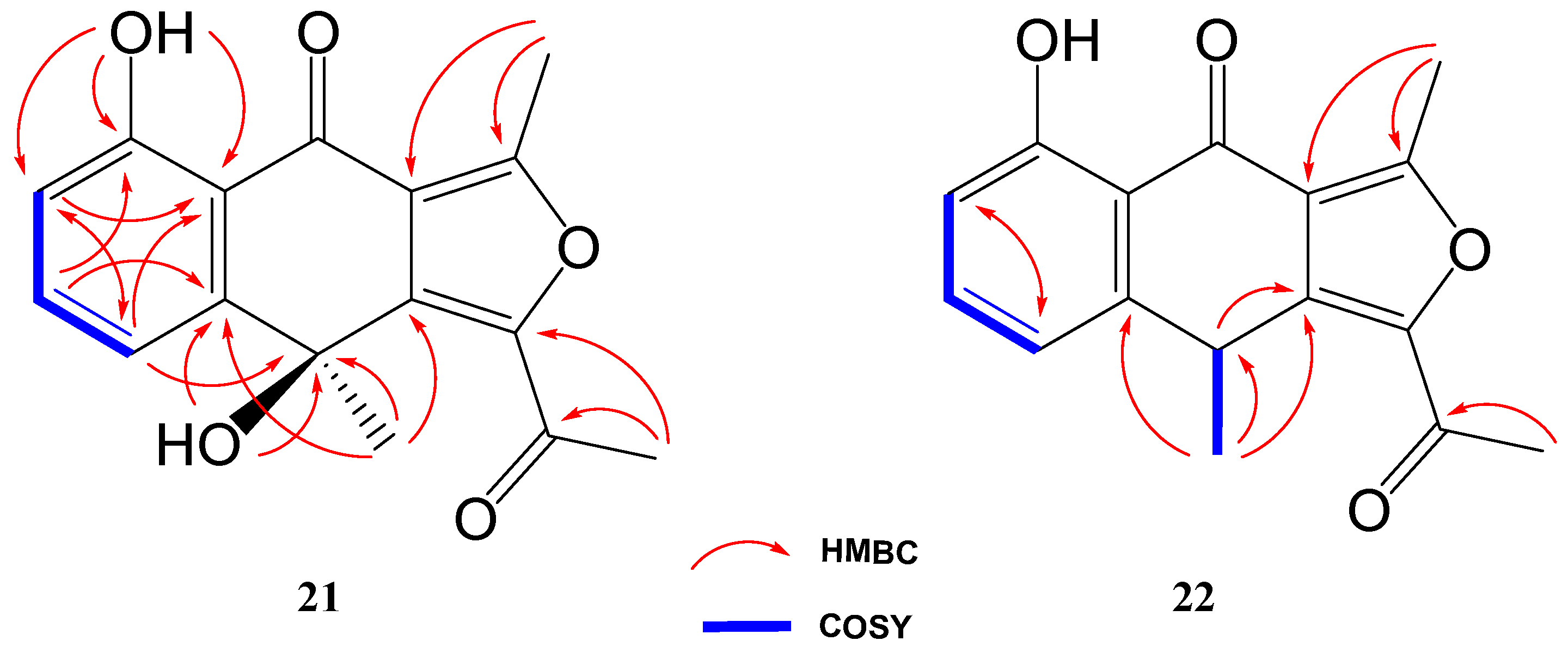
The 1H NMR spectrum of compound 21 was very similar to that of previously elucidated anthrone-derived molecules; however, more detailed analysis of 13C and 2D NMR experiments suggested some differences in the chemical scaffold of 21. The gHMBC correlations (see Table 2) and chemical shifts of H-6, H-7, H-8, and H3-13 confirmed that the naphtho moiety was identical to that of compounds 2, 4, and 19. On the other side, gHMBC correlations from H3-12 to C-3 (δC 161.6) and C-3a (δC 117.5) and from H3-11 to C-1 (δC 145.6) and C-10 (δC 190.4), shown in Figure 6, in conjunction with molecular weight information, suggested that compound 21 was 1-acetyl-5,9-dihydroxy-3,9-dimethylnaphtho[2,3-c]furan-4(9H)-one. The structure of 21 was subsequently confirmed by comparison with the data described in the literature for other polyketides with the naphtho[2,3-c]furan-4(9H)-one backbone [29,30].
Table 2.
NMR data of compounds 21 and 22 in CDCl3.
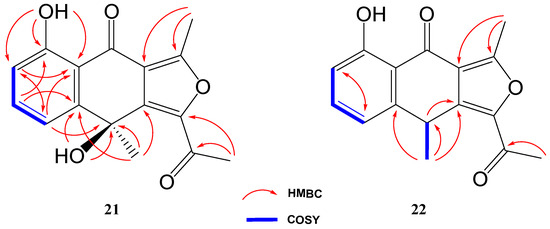
Figure 6.
Key COSY and HMBC correlations of compounds 21 and 22.
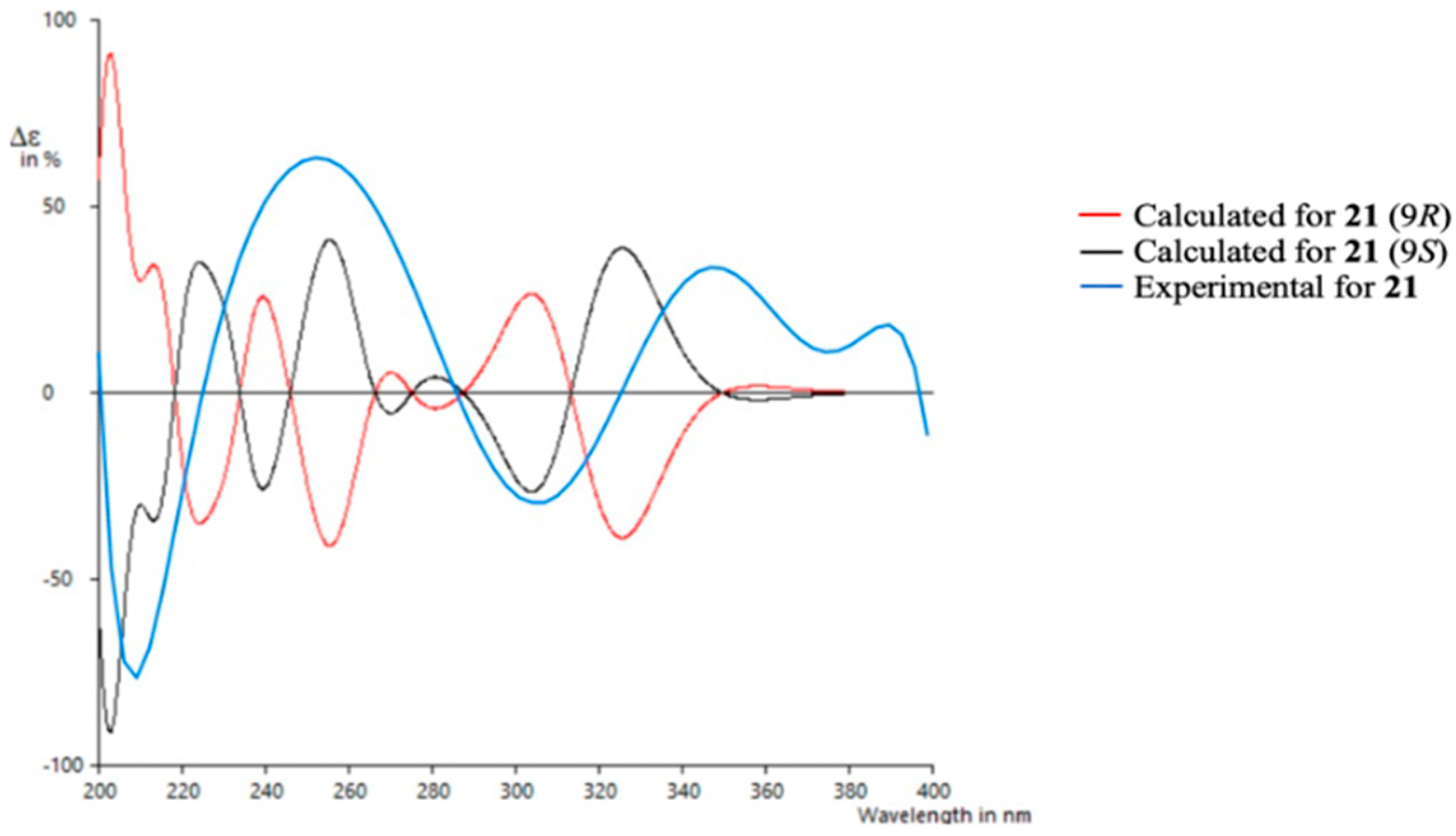
To determine the absolute configuration of 21, TDDFT/ECD (time-dependent density functional theory/electronic circular dichroism) calculations were performed at the PCM/B3LYP/6-31G(d) level of theory using structures previously optimized at the same level. It has to be noted that due to the rigidity of the molecule only one suitable structure was found in a molecular mechanics conformational search within a threshold of 21 kJ/mol. As observed in Figure 7, the experimental spectrum showed negative Cotton effects at ~210 nm and ~305 nm and positive Cotton effects at ~250 nm and ~350 nm. The calculated spectrum for the 9S enantiomer clearly matched better with the experimental data, allowing us to assign the absolute configuration of 21. Similar results were obtained using a different level of theory (ωB97XD) (see Supporting Information).
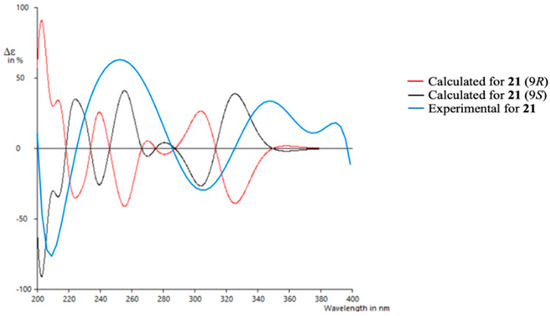
Figure 7.
Experimental and calculated ECD spectra for compound 21.
Compound 22 was isolated as a whitish amorphous solid with the molecular formula C16H14O4 determined based on HRESIMS and NMR data (10 degrees of unsaturation). 1H NMR spectrum of compound 22 suggested a very similar structure to that of 21, the only difference being a quadruplet methine group (δH 4.58, δC 32.2) coupled to H3-13 methyl group (J = 7.1 Hz) in 22, instead of the quaternary carbon (δC 67.7) at C-9 in 21. This suggested the loss of the hydroxy group attached to C-9 in 21, which was also evident from the molecular mass of 22 being 16 amu less than that of 21 (C16H14O5), establishing compound 22 as 1-acetyl-5-hydroxy-3,9-dimethylnaphtho[2,3-c]furan-4(9H)-one (Figure 4).
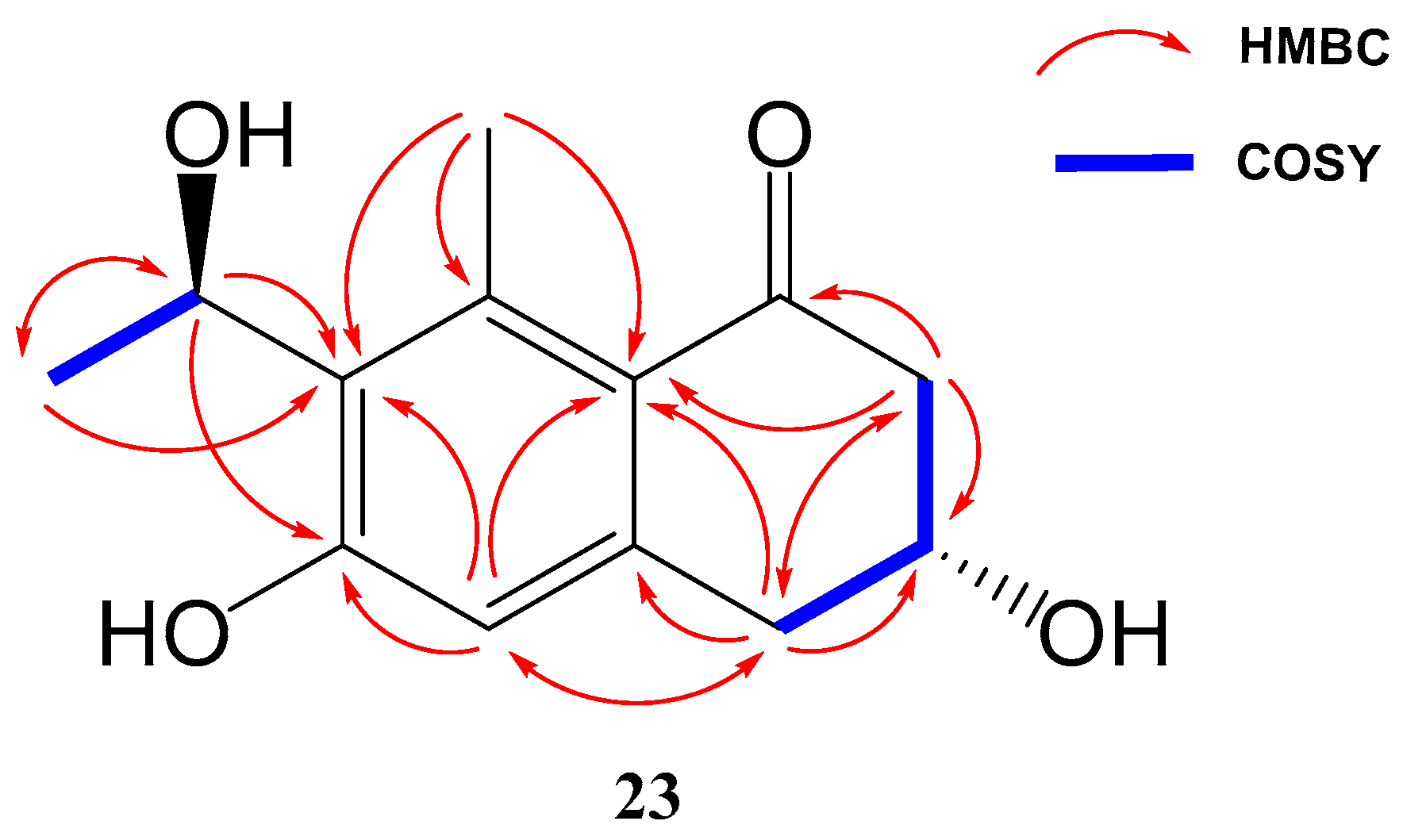
Compound 23 (yellowish amorphous solid) has the molecular formula C13H16O4 (six degrees of unsaturation) based on HRESIMS and NMR data. NMR data (Table 3) were very similar to those of GTRI-02 (17). The presence of a new oxygenated quadruplet methine group (δH 5.31, δC 68.0) coupled to H3-10 methyl group (J = 6.7 Hz) in 23, instead of the ketone group at C-9 in GTRI-02, suggested the reduction of the ketone to alcohol, which was also evident from the molecular formula of 23 (C13H16O4). Further 2D NMR analysis confirmed the structure of 23 as shown in Figure 4 and Figure 8.
Table 3.
NMR data of compound 23 in CDCl3 + MeOH-d4.
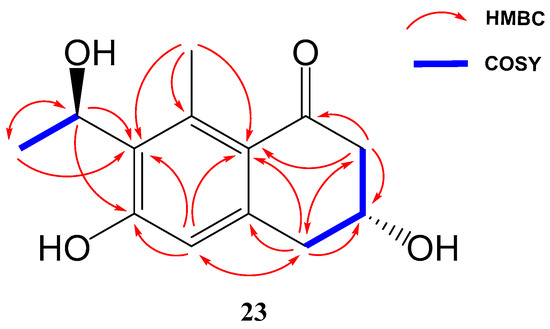
Figure 8.
Key COSY and HMBC correlations of compounds 23.
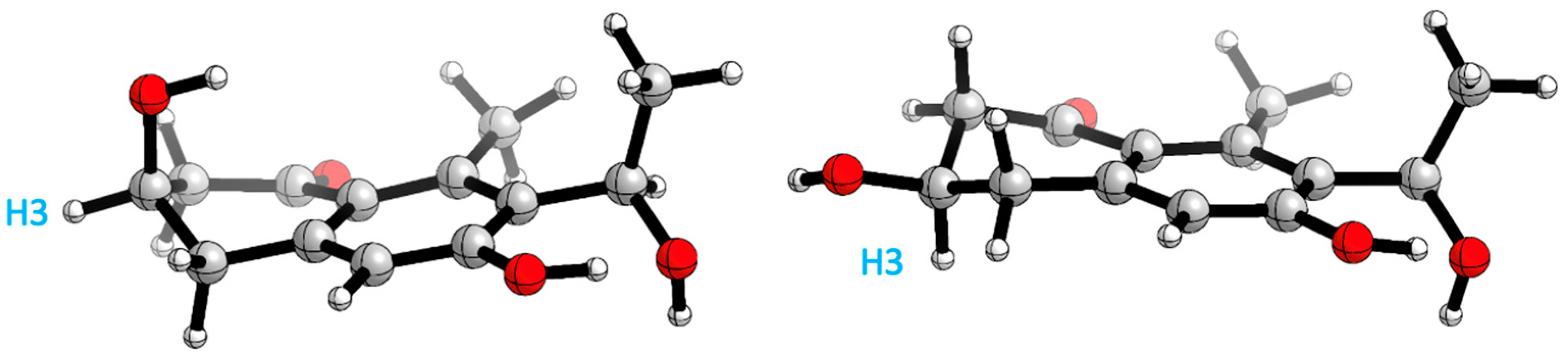
Therefore, two remote chiral centres are present in 23, making it very difficult to determine their relative configurations. The use of quantum mechanical calculations to unravel the information encoded in NMR chemical shifts, also known as the QM-NMR method, is currently one of the most popular strategies for this purpose. There are several methods for this task; however, those based on Bayes’ theorem, such as the original DP4 probability or the more elaborated DP4+ and J-DP4 methods, stand out from the rest [31,32,33,34]. Previous experience with these approaches [35,36,37,38,39,40,41,42] led us to select mix-J-DP4 [43] as the most appropriate method in this case, as it yields high confidence results at modest computational costs by incorporating proton–proton NMR coupling constants. Thereby, a conformational search was achieved using the mixed torsional/low-mode protocol in gas phase by means of MMFF force field; later, the conformers were reoptimized with an AMBER and MM3 force field. The energy cut-off of 12 kJ/mol and geometric criteria of MAD 0.5 Å were used to eliminate duplicate structures; after that, NMR calculations were performed at the J-DP4 recommended level (B3LYP/6-31G**) and 3JHH used only the Fermi Contact term contribution. Finally, a comparison of calculated and experimental NMR data was undertaken as described for the J-DP4 protocol, obtaining an overall probability of >99% for isomer 3R*,9R*. Moreover, the R2s obtained with each force field are: AMBER 0.999 and 0.987, MM3 0.999 and 0.993, MMFF 0.999 and 0.987, for carbon and proton, respectively, and the 13C CMAE value for AMBER and MM3 is 1.5 ppm and for MMFF 1.8 ppm. The 1H CMAE value obtained for all force fields is 0.1 ppm.
A conformational search analysis shows that there exist two major conformations (Figure 9): one conformer where H3 is in an axial position with a contribution of 57–65% (depending on the force field used), and the other conformer which represents 35–43% of the total population that has the same proton in an equatorial position. This hints that there exists a conformational equilibrium between both conformations, which agrees with the experimental results obtained in 3JH3 (8.1 and 4.0 Hz).
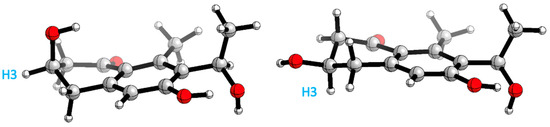
Figure 9.
Main conformations H3 axial (right) and H3 equatorial (left) position.
All isolated compounds were subjected to antibacterial and cytotoxic (against different tumour cell lines) assays, but none of them showed any remarkable bioactivity.
Attempts to obtain more available material of those compounds are currently ongoing, in order to screen them in some relevant crop-protection assays.
In conclusion, the fermentation of Streptomyces griseorubiginosus on FA-1 medium afforded five anthraquinone and anthrone-derivative natural products (1–5). Changing from FA-1 to SAF medium resulted in the production of five further secondary metabolites (6–10), with structures totally different from those observed under the previous conditions. A switch from liquid media to solid SymPV-21 medium yielded five additional compounds (11–15), four cyclodipeptides and the hydroxamate siderophore Nocardamine (15). Finally, the addition of γ-butyrolactone to FA-1 medium caused the accumulation of eight additional metabolites (16–23); five of them (19–23) were novel natural products not previously reported in the literature.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were measured on a JASCO P-2000 polarimeter using MeOH as solvent. CD spectra were recorded on a JASCO J-810 spectropolarimeter. IR data were recorded using a BRUKER VECTOR 22 spectrophotometer. 1H NMR and 13C NMR data were obtained on a Varian “Mercury 400” spectrometer at 400 and 100 MHz, respectively. Chemical shifts are reported in ppm relative to solvent (CDCl3: δ = 7.26/77.16 ppm; [D4] MeOH: δ= 3.31/49.0 ppm). HPLC-MS experiments were carried out with an Agilent UHPLC 1290 Infinity II coupled to an Agilent TOF 6230. HPLC analysis were performed with a Zorbax Eclipse plus C18 column, using a linear gradient from 20% to 100% methanol in 8 min, a post time of 2 min, and a DAD analysis 200–600 nm range signal obtained at 220 nm. Mass spectrometer conditions in ESI-Positive were 100 V Fragmentor, mass range (m/z) = 100–3200. The analysis were performed with Masshunter suite software (Agilent Technologies) version B.08.00. An Agilent 1200 series liquid chromatograph with a photodiode array and evaporative light-scattering detectors was used for HPLC analysis and recording UV spectra and tR values, performed with a Zorbax Eclipse XDB-C18 column (4.6 × 50 mm, 1.8 µm) maintained at 20 °C with a mobile phase flow rate of 0.5 mL/min, using the following linear gradient of MeOH/H2O (+0.04% TFA): Min 0–15: 15–100% MeOH; min 15–20: 100% MeOH; min 20–30: 100–15% MeOH, post-run 5 min, and a DAD analysis 200–600 nm range signal obtained at 220 nm. Semipreparative HPLC purifications were conducted on an Agilent 1100 series (Kinetex 5 µm EVO C18 10 column, 100 × 21.2 mm).
3.2. Bacterial Material
3.2.1. Isolation of the Strain
Strain AA-AR-H-B002 was isolated from an unidentified cnidarian collected at a reef close to Pointe de Bellevue in Réunion Island (France). The strain has been deposited in the Colección Española de Cultivos Tipo, Valencia, Spain, with the accession number CECT 30830.
3.2.2. Identification of the Strain
The strain was identified as Streptomyces griseorubiginosus by partial sequencing of its 16S RNA, showing a 99.78% homology in a 913-nucleotide-length sequence. The DNA of the microorganism was obtained using a Qiagen Dneasy kit following the manufacturer’s instructions. The 16S rDNA gene was amplified using the universal primers 27f (AGAGTTTGATCMTGGCTCAG) and 1492r (TACGGYTACCTTGTTACGACTT). The PCR program used was as follows: 94 °C for 1 min, then 30 cycles of 98 °C for 10 s, 50 °C for 2 min, 172 °C for 10 s, and finally one step of 72 °C for 7.5 min. The amplified gen was purified using the Qiagen QiaQuick kit following the manufacturer’s directions. The purified amplicons were sent to an external service for sequencing. All the different steps were monitored using 1.2% agarose electrophoresis in half-strength TB buffer.
3.3. Fermentations
All the liquid fermentations were carried out in flasks, incubated at 28 °C and with an agitation at 240 rpm. All the fermentations started from a fresh, confluent culture of the actinomycete on 172 solid medium incubated for one week at 28 °C. The inoculum was developed in two stages. The first stage was carried out in Erlenmeyer flasks of 50 mL capacity and containing 10 mL of medium MIM. These flasks were seeded with 3 plugs taken from the lawn of the confluent solid culture. The flasks were incubated as described above for 48 h. The second stage was carried out in Erlenmeyer flasks of 250 mL capacity filled with 40 mL of MIM medium and seeded with 4 mL of the first stage. The flasks were incubated as described for 48 h. The production phase was performed using Erlenmeyer flasks of 2 L capacity and filled with 250 mL of the appropriate medium. These production flasks were seeded with 12 mL of the second-stage inoculum and incubated as described previously for 96 or 120 h depending on the experiment. The epigenetic factor γ-butyrolactone was added to the corresponding medium (FA-1) at a concentration of 50 µM.
All the solid fermentations were carried out in 2 L capacity Erlenmeyer flasks, containing 250 mL of solid medium SymPv-21. The seeded flasks were incubated at 28 ºC in aerobic conditions for 144 h.
Media Compositions
MIM medium per litre: beef extract 3 g, casein peptone 5 g, Hsoy flour 5 g, corn starch 24 g, yeast extract 5 g, glucose 1 g, Na2SO4 7.5 g, NaCl 5.34 g, MgCl2.6H2O 2.4 g, KCl 0.2 g. pH was adjusted to 6.8 and 4 g calcium carbonate was added after adjustment.
FA-1 medium per litre: baker’s yeast 5 g, bacto peptone 1 g, glucidex-12 20 g, soy flour 3 g, glucose 5 g, K2HPO4 0.5 g, MnSO4.H2O 0.0076 g, MgCl2.6H2O 2.4 g, Na2SO4 7.5 g, NaCl 5.34 g, CoCl2.6H2O 0.0013 g, KCl 0.2 g. pH was adjusted to 7 and 4 g calcium carbonate was added after adjustment.
SAF medium per litre: mannitol 40 g, glucose 10 g, atomized corn steep 12.5 g, pharmamedia 16 g, MgSO4.7H2O 2 g, Na2SO4 5.5 g, NH4Cl 2 g, KCl 2 g, NaCl 2.34 g, and MnSO4.H2O 0.0076 g. pH was adjusted to 6.8 and 10 g calcium carbonate was added after adjustment.
SymPv-21 medium per litre: glycerol 15 g, peptone proteose 25 g, pharmamedia 5 g, sodium gluconate 5 g, K2HPO4 2 g, and MgSO4.7H2O 2 g. pH was adjusted to 7.2.
Finally, 172 medium per litre: glucose 10 g, glucidex-12 20 g, yeast extract 5 g, casein peptone 5 g, and agar 15 g. pH was adjusted to 6.8 and 1 g calcium carbonate was added after adjustment.
3.4. Extraction and Isolation
Fermentation broth (8 L) of Streptomyces griseorubiginosus under standard conditions (FA-1 liquid medium) was stirred with Amberlite XAD1180N™ resin (10% v/v) for 1 h. Then, culture broth was filtered through Celite®, and the mycelial cake and the resin were extracted with 4 L of a mixture of EtOAc/MeOH (3:1). The resultant suspension was filtered and partitioned between EtOAc and water. The organic layer was taken to dryness, and the crude extract (3.9 g) was fractionated by VFC (vacuum-flash chromatography) on silica gel, eluting with a stepwise gradient of hexane/EtOAc/MeOH to give 12 fractions (1F1–1F12). Fractions 1F11–12 were further fractionated by normal-phase liquid chromatography using a stepwise gradient of CH2Cl2 and MeOH to yield 12 subfractions (2F1–2F12). Subfractions 2F7–10 and 2F5 were purified by reversed-phase liquid chromatography using a stepwise gradient of MeOH/H2O to give 1 (3.7 mg, tR 14.2 min) and 2 (13.2 mg, tR 12.3 min), respectively. Compounds 3 (12.5 mg, tR 16.1 min) and 5 (27.4 mg, tR 15.8 min) were isolated from fraction 1F5 by normal-phase liquid chromatography with a gradient of hexane/EtOAc followed by a further step of normal-phase liquid chromatography with a gradient of hexane/ether, while compound 4 (74.5 mg, tR 13.9 min) was obtained from fractions 1F6–7, following a similar procedure, with the difference that the last step of purification was carried out by normal-phase liquid chromatography with a gradient of CH2Cl2/MeOH.
The same extraction procedure was followed for all liquid medium fermentations. The chromatographic work up of the OSMAC extracts followed the same procedure as described for the standard-condition culture. Silica gel vacuum-flash chromatography using a stepwise gradient of hexane/EtOAc/MeOH was used for separation of the crude extracts obtained from SAF medium (4.55 g) and FA-1 medium with the addition of the chemical elicitor γ-butyrolactone (4.94 g). A total of twelve fractions (SA1F1–SA1F12) were obtained from the SAF culture extract. Fractions SA1F9–10 were further fractionated by normal-phase liquid chromatography using a stepwise gradient of CH2Cl2/MeOH to yield 10 subfractions (SA2F1–SA2F10). Subfraction SA2F5 was purified by reversed-phase liquid chromatography using a stepwise gradient of MeOH/H2O to give 6 (8.4 mg, tR 9.0 min), while subfractions SA2F7 and SA2F8 were also purified by MeOH/H2O reversed-phase liquid chromatography, yielding 7 (15.1 mg, tR 5.8 min), 8 (5.6 mg, tR 7.3 min), 9 (4.6 mg, tR 9.7 min), and 10 (3 mg, tR 10.9 min). Fraction B1F11, obtained by VFC from FA-1 culture after the addition of γ-butyrolactone extract, was fractionated by normal-phase liquid chromatography using a stepwise gradient of CH2Cl2/MeOH to yield 12 subfractions (B2F1–B2F12). Compound 16 (13.9 mg, tR 1.2 min) was isolated from subfractions B2F5–7 after purification by reversed-phase liquid chromatography using a stepwise gradient of MeOH/H2O. Fractions B1F9–10 were also fractionated following the same procedure described for compound 16 to give 17 (46.9 mg, tR 10.9 min), 18 (4.6 mg, tR 14.2 min), and 23 (3.2 mg, tR 9.3 min). Compounds 19 (2 mg, tR 11.3 min), 20 (3 mg, tR 14.8 min), 21 (3.1 mg, tR 17.3 min), and 22 (1.1 mg, tR 12.6 min) were isolated from B1F5 (21 and 22), B1F7 (19), and B1F8 (20), respectively, after purification by normal-phase liquid chromatography using a stepwise gradient of hexane/EtOAc, followed by semi-preparative HPLC purification with a gradient of MeOH/H2O as an eluting system.
In the case of the solid-state fermentation (SymPV-21 medium), the contents of the flasks were collected and homogenized with the aid of an electric mixer, followed by extraction with 100% v/v of a mixture of EtOAc/MeOH (3:1). Twelve fractions were obtained from the SymPV-21 solid culture extract (4.26 g) after VFC (Sy1F1–Sy1F12). Compounds 11 (41.1 mg, tR 8.6 min), 12 (56 mg, tR 8.0 min), and 13 (7.8 mg, tR 7.7 min) were isolated from Sy1F10 after purification by normal-phase liquid chromatography using a stepwise gradient of CH2Cl2/MeOH, followed by a further step of normal-phase liquid chromatography with a gradient of CH2Cl2/acetone. Finally, fraction Sy1F11–12 was also fractionated by normal-phase liquid chromatography, using a gradient of CH2Cl2/MeOH as mobile phase, followed by reversed-phase liquid chromatography using a stepwise gradient of MeOH/H2O to afford 14 (27.5 mg, tR 5.0 min) and 15 (8.4 mg, tR 9.7 min).
3.5. Spectroscopic Data of Compounds 19–23
Compound 19. Yellowish amorphous solid; [α]20D = −133.9° (c 0.2, MeOH); UV (MeOH) λmax (log ε) 210 (2.41), 244 (2.12), 270 (1.95), 320 (2.13), 340 (2.09) nm; IR (ATR) νmax 3282 (br), 2977, 2928, 1624, 1594, 1458, 1332, 1269, 1242, 1172 cm−1; for 1H and 13C NMR data, see Table 1; HRESIMS m/z 287.0907 [M + H]+ (calcd for C16H15O5, 287.0919).
Compound 20. Yellowish amorphous solid; [α]20D = +2.2° (c 0.136, MeOH); UV (MeOH) λmax (log ε) 210 (2.16), 242 (1.89), 280 (1.73), 350 (1.98) nm; IR (ATR) νmax 3397 (br), 2969, 2926, 1627, 1608, 1577, 1473, 1316, 1270, 1176 cm−1; for 1H and 13C NMR data, see Table 1; HRESIMS m/z 287.0911 [M + H]+ (calcd for C16H15O5, 287.0919).
Compound 21. Yellowish amorphous solid; [α]20D = +0.35° (c 0.173, MeOH); UV (MeOH) λmax (log ε) 210 (2.22), 232 (2.11), 276 (2.22), 308 (1.95), 344 (1.80) nm; IR (ATR) νmax 3365 (br), 2918, 2850, 1650, 1635, 1580, 1542, 1456, 1418, 1345, 1248, 1175 cm−1; for 1H and 13C NMR data, see Table 2; HRESIMS m/z 287.0920 [M + H]+ (calcd for C16H15O5, 287.0919).
Compound 22. Whitish amorphous solid; UV (MeOH) λmax (log ε) 218 (2.50), 234 (2.43), 274 (2.53), 308 (2.21) nm; IR (ATR) νmax 3388 (br), 2917, 2849, 1653, 1635, 1588, 1541, 1456, 1354, 1244 cm−1; for 1H and 13C NMR data, see Table 2; HRESIMS m/z 271.0960 [M + H]+ (calcd for C16H15O4, 271.0970).
Compound 23. Yellowish amorphous solid; [α]20D = −47.5° (c 0.226, MeOH); UV (MeOH) λmax (log ε) 216 (2.28), 232 (2.21), 280 (2.20) nm; IR (ATR) νmax 3382 (br), 2973, 2926, 1715, 1650, 1572, 1457, 1369, 1305, 1243, 1158, 1059 cm−1; for 1H and 13C NMR data, see Table 3; HRESIMS m/z 237.1140 [M + H]+ (calcd for C13H17O4, 237.1127).
3.6. Computational Methods
Calculations were performed following the general workflow described in the mix-J-DP4 method [43]. As previously described, all possible isomers were created, and conformational searches achieved using the mixed torsional/low-mode conformational sampling protocol in gas phase with the MMFF force field and reoptimizing the conformers with an AMBER and MM3 force field. NMR calculations were achieved using the conformations under 12 kJ/mol energy cut-off and MAD 0.5 Å. The DFT levels of theory as recommended for J-DP4 (B3LYP/6-31G**) in Gaussian 16 [44], were used to calculate 3JHH, employing exclusively the Fermi Contact term contribution and magnetic shielding constants (σ) by means of the gauge, including the atomic orbital method [45,46,47,48]. Unscaled chemical shifts (δu) were calculated using TMS as reference standard according to the following expression: δu = σ0 − σx, where σx is the Boltzmann-averaged shielding tensor (over all significantly populated conformations) and σ0 is the shielding tensor of TMS computed at the same level of theory used to calculate σx. Boltzmann averaging was performed according to the following equation:
where σxi is the shielding constant for nucleus x in conformer i, R is the molar gas constant (8.3145 J /(K mol)), T is the temperature used for the calculation (298 K), and Ei is the relative energy of conformer i (to the lowest energy conformer) obtained from a single-point NMR calculation at the corresponding level of theory. The scaled chemical shifts (δs) were computed as δs = (δu − b)/m, where m and b are the slope and intercept, respectively, resulting from a linear regression calculation on a plot of δu against δexp. All molecular models figures were performed using CYLview20 [49].
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/md21100526/s1. Figures S1–S35: NMR spectra of known compounds 1–18. Figures S36–S72: NMR, HRESIMS, UV and IR spectra of novel compounds 19–23. Figures S73–S75: Correlation of compounds 23; Figures S76 and S77: ECD spectra for compound 21, Tables S1–S12: Computational section.
Author Contributions
All listed authors contributed to this work. V.R.M.-A.: Performed most of the isolation, purification, and structural elucidation of the compounds described, and wrote this paper. F.R.M.: Bacterial fermentation. C.C. and A.H.D.: QM-NMR studies to stablish relative configuration of compound 23 and contribution to the final manuscript. A.F.M.: Advised, assisted, and shared the tasks of the manuscript revision. J.M.S.L.: Project leader organizing and guiding the experiments and manuscript writing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Ministerio de Ciencia, Innovación y Universidades (DIN2018-009716A24330292; PID2019-109476RB-C21 and PID2022-143235NB-I00) of Spanish Government, Agencia Canaria de Investigación, Innovación y Sociedad de la Información (ACIISI-FEDER) ProID2021010118 and Fondo Social Europeo (Programa Operativo Integrado de Canarias FSE 2014–2020, Eje 3, Tema Prioritario 74–85%). Víctor Rodríguez Martín-Aragón thanks Ministerio de Ciencia e Innovación (MICINN) of the Spanish Government for financial support (State Plan for Scientific and Technical Research and Innovation 2021–2023) for a predoctoral fellowship. C. Cuadrado thanks Agencia Canaria de Investigación, Innovación y Sociedad de la Información (ACIISI) and Fondo Social Europeo (Programa Operativo Integrado de Canarias FSE 2014–2020, Eje 3, Tema Prioritario 74–85%) for a predoctoral fellowship (TESIS 2021010030).
Data Availability Statement
The data used to prepare this manuscript are contained within the article and its corresponding Supplementary Material.
Acknowledgments
This study made use of Galicia Supercomputing Center (CESGA) facilities as provided by CSIC. Authors also wish to thank Joaquin Altarejos, from the University of Jaen, Spain, for measuring optical rotations of the isolated compounds.
Conflicts of Interest
The authors declare no conflict of interest.
References
- van Bergeijk, D.A.; Terlouw, B.R.; Medema, M.H.; van Wezel, G.P. Ecology and genomics of Actinobacteria: New concepts for natural product discovery. Nat. Rev. Microbiol. 2020, 18, 546–558. [Google Scholar] [CrossRef]
- Bahrami, Y.; Bouk, S.; Kakaei, E.; Taheri, M. Natural Products from Actinobacteria as a Potential Source of New Therapies Against Colorectal Cancer: A Review. Front. Pharmacol. 2022, 13, 929161. [Google Scholar] [CrossRef]
- Jose, P.A.; Maharshi, A.; Jha, B. Actinobacteria in natural products research: Progress and prospects. Microbiol. Res. 2021, 246, 126708. [Google Scholar] [CrossRef]
- Tran-Cong, N.M.; Mándi, A.; Kurtán, T.; Müller, W.E.G.; Kalscheuer, R.; Lin, W.; Liu, Z.; Proksch, P. Induction of cryptic metabolites of the endophytic fungus Trichocladium sp. through OSMAC and co-cultivation. RSC Adv. 2019, 9, 27279–27288. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Bai, X.; Chen, J.; Zhang, H.; Wang, H. Exploring Structural Diversity of Microbe Secondary Metabolites Using OSMAC Strategy: A Literature Review. Front. Microbiol. 2019, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Serrano, R.; González-Menéndez, V.; Rodríguez, L.; Martín, J.; Tormo, J.R.; Genilloud, O. Co-culturing of Fungal Strains Against Botrytis cinerea as a Model for the Induction of Chemical Diversity and Therapeutic Agents. Front. Microbiol. 2017, 8, 649. [Google Scholar] [CrossRef] [PubMed]
- Pinedo-Rivilla, C.; Aleu, J.; Durán-Patrón, R. Cryptic Metabolites from Marine-Derived Microorganisms Using OSMAC and Epigenetic Approaches. Mar. Drugs 2022, 20, 84. [Google Scholar] [CrossRef]
- Romano, S.; Jackson, S.A.; Patry, S.; Dobson, A.D.W. Extending the “One Strain Many Compounds” (OSMAC) Principle to Marine Microorganisms. Mar. Drugs 2018, 16, 244. [Google Scholar] [CrossRef]
- Liu, M.; Grkovic, T.; Liu, X.; Han, J.; Zhang, L.; Quinn, R.J. A systems approach using OSMAC, Log P and NMR fingerprinting: An approach to novelty. Synth. Syst. Biotechnol. 2017, 2, 276–286. [Google Scholar] [CrossRef]
- Bode, H.; Walker, M.; Zeeck, A. Cladospirones B to I from Sphaeropsidales sp. F-24′707 by Variation of Culture Conditions. Eur. J. Org. Chem. 2000, 2000, 3185–3193. [Google Scholar] [CrossRef]
- Oka, M.; Kamei, H.; Hamagishi, Y.; Tomita, K.; Miyaki, T.; Konishi, M.; Oki, T. Chemical and Biological Properties of Rubiginone, a Complex of New Antibiotics with Vincristine-Cytotoxicity. J. Antibiot. 1990, 43, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Paintner, F.F.; Görler, K.; Voelter, W. A new convergent approach to biphenomycin antibiotics. Synlett 2003, 2003, 0522–0526. [Google Scholar] [CrossRef]
- David, L.; Duteurtre, M.; Kergomard, A.; Kergomard, G.; Scanzi, E.; Staron, T. Production of cinerubins by a Streptomyces griseorubiginosus strain. J. Antibiot. 1980, 33, 49–53. [Google Scholar] [CrossRef]
- Shimizu, K.-I.; Tamura, G. Reductiomycin, a new antibiotic I. Taxonomy, fermentation, isolation, characterization and biological activities. J. Antibiot. 1981, 34, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Hawas, U.W.; El-Ansari, M.A.; Laatsch, H. A new alpha-methylanthraquinone glucoside from Emex spinosus. Nat. Prod. Res. 2006, 20, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Krupa, J.; Lessmann, H.; Lackner, H. Ein α-Methylanthrachinon aus Streptomyceten. Liebigs Ann. Chem. 1989, 1989, 699–701. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, C.; Zhang, L. Investigation of the Molecular Landscape of Bacterial Aromatic Polyketides by Global Analysis of Type II Polyketide Synthases. Angew. Chem. Int. Ed. 2022, 61, e202202286. [Google Scholar] [CrossRef]
- Cui, H.; Shaaban, K.A.; Qin, S. Two Anthraquinone Compounds from a Marine Actinomycete Isolate M097 Isolated from Jiaozhou Bay. World J. Microbiol. Biotechnol. 2006, 22, 1377–1379. [Google Scholar] [CrossRef]
- Lin, Z.J.; Lu, X.M.; Zhu, T.J.; Fang, Y.C.; Gu, Q.Q.; Zhu, W. GPR12 selections of the metabolites from an endophytic Streptomyces sp. associated with Cistanches deserticola. Arch. Pharm. Res. 2008, 31, 1108–1114. [Google Scholar] [CrossRef]
- Kawahara, T.; Izumikawa, M.; Otoguro, M.; Yamamura, H.; Hayakawa, M.; Takagi, M.; Shin-ya, K. JBIR-94 and JBIR-125, antioxidative phenolic compounds from Streptomyces sp. R56-07. J. Nat. Prod. 2012, 75, 107–110. [Google Scholar] [CrossRef]
- Wu, T.-S.; Shi, L.-S.; Kuo, S.-C. Alkaloids and other constituents from Tribulus terrestris. Phytochemistry 1999, 50, 1411–1415. [Google Scholar] [CrossRef]
- Fdhila, F.; Vázquez, V.; Sánchez, J.L.; Riguera, R. dd-diketopiperazines: Antibiotics active against Vibrio anguillarum isolated from marine bacteria associated with cultures of Pecten maximus. J. Nat. Prod. 2003, 66, 1299–1301. [Google Scholar] [CrossRef] [PubMed]
- Selvakumar, S.; Sivasankaran, D.; Singh, V.K. Enantioselective Henry reaction catalyzed by C2-symmetric chiral diamine–copper(II) complex. Org. Biomol. Chem. 2009, 7, 3156–3162. [Google Scholar] [CrossRef]
- Campbell, J.; Lin, Q.; Geske, G.D.; Blackwell, H.E. New and unexpected insights into the modulation of LuxR-type quorum sensing by cyclic dipeptides. ACS Chem. Biol. 2009, 4, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Maehr, H.; Benz, W.; Smallheer, J.; Williams, T. Mikrobielle Produkte, I NMR-Spektren von Nocardamin und Massenspektrum des Tri-O-methyl-nocardamins/Microbiological Products, I NMR Spectra of Nocardamine and Mass Spectra of Tri-O-methyl-nocardamine. Z. Für Naturforschung B 2014, 32, 937–942. [Google Scholar] [CrossRef]
- Stavila, E.; Loos, K. Synthesis of lactams using enzyme-catalyzed aminolysis. Tetrahedron Lett. 2013, 54, 370–372. [Google Scholar] [CrossRef]
- Yeo, W.H.; Yun, B.S.; Kim, S.S.; Park, E.K.; Kim, Y.H.; Yoo, I.D.; Yu, S.H. GTRI-02, a new lipid peroxidation inhibitor from Micromonospora sp. SA246. J. Antibiot. 1998, 51, 952–953. [Google Scholar] [CrossRef]
- Wu, C.; Ichinose, K.; Choi, Y.H.; van Wezel, G.P. Aromatic Polyketide GTRI-02 is a Previously Unidentified Product of the act Gene Cluster in Streptomyces coelicolor A3(2). Chembiochem 2017, 18, 1428–1434. [Google Scholar] [CrossRef]
- Koyama, J.; Ogura, T.; Tagahara, K. Naphtho [2,3-c]furan-4,9-dione and its derivatives from Aloe ferox. Phytochemistry 1994, 37, 1147–1148. [Google Scholar] [CrossRef]
- Fotso, S.; Maskey, R.P.; Grün-Wollny, I.; Schulz, K.-P.; Munk, M.; Laatsch, H. Bhimamycin AE and bhimanone: Isolation, structure elucidation and biological activity of novel quinone antibiotics from a terrestrial streptomycete. J. Antibiot. 2003, 56, 931–941. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An Improved Probability for the Stereochemical Assignment of Isomeric Compounds using Quantum Chemical Calculations of NMR Shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Gavín, J.A.; Hernández Daranas, A.; Sarotti, A.M. Combining the Power of J Coupling and DP4 Analysis on Stereochemical Assignments: The J-DP4 Methods. Org. Lett. 2019, 21, 4003–4007. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.H.; Amichetti, M.; Zanardi, M.M.; Grimson, R.; Daranas, A.H.; Sarotti, A.M. ML-J-DP4: An Integrated Quantum Mechanics-Machine Learning Approach for Ultrafast NMR Structural Elucidation. Org. Lett. 2022, 24, 7487–7491. [Google Scholar] [CrossRef]
- Daranas, A.H.; Sarotti, A.M. Are Computational Methods Useful for Structure Elucidation of Large and Flexible Molecules? Belizentrin as a Case Study. Org. Lett. 2021, 23, 503–507. [Google Scholar] [CrossRef]
- Rodríguez-Expósito, R.; Nicolás-Hernández, D.; Sifaoui, I.; Cuadrado, C.; Salazar, L.; Reyes Batlle, M.; Hernández-Daranas, A.; Omaña-Molina, M.; Fernández, J.; Diaz-Marrero, A.; et al. Gongolarones as antiamoeboid chemical scaffold. Biomed. Pharmacother. 2023, 158, 114185. [Google Scholar] [CrossRef]
- Sosa-Rueda, J.; Domínguez-Meléndez, V.; Ortiz-Celiseo, A.; López-Fentanes, F.C.; Cuadrado, C.; Fernández, J.J.; Daranas, A.H.; Cen-Pacheco, F. Squamins C-F, four cyclopeptides from the seeds of Annona globiflora. Phytochemistry 2022, 194, 112839. [Google Scholar] [CrossRef]
- Li, S.W.; Mudianta, I.W.; Cuadrado, C.; Li, G.; Yudasmara, G.A.; Setiabudi, G.I.; Daranas, A.H.; Guo, Y.W. Litosetoenins A-E, Diterpenoids from the Soft Coral Litophyton setoensis, Backbone-Rearranged through Divergent Cyclization Achieved by Epoxide Reactivity Inversion. J. Org. Chem. 2021, 86, 11771–11781. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.-W.; Cuadrado, C.; Gao, C.-L.; Wu, Q.; Li, X.; Pang, T.; Daranas, A.; Guo, Y.-W.; Li, X.-W. Polyoxygenated anti-inflammatory biscembranoids from the soft coral Sarcophyton tortuosum and their stereochemistry. Chin. Chem. Lett. 2020, 32, 271–276. [Google Scholar] [CrossRef]
- Domínguez, H.J.; Cabrera-García, D.; Cuadrado, C.; Novelli, A.; Fernández-Sánchez, M.T.; Fernández, J.J.; Daranas, A.H. Prorocentroic Acid, a Neuroactive Super-Carbon-Chain Compound from the Dinoflagellate Prorocentrum hoffmannianum. Org. Lett. 2021, 23, 13–18. [Google Scholar] [CrossRef]
- Li, S.W.; Cuadrado, C.; Huan, X.J.; Yao, L.G.; Miao, Z.H.; Hernandez Daranas, A.; Guo, Y.W. Rare new bicyclic cembranoid ethers and a novel trihydroxy prenylated guaiane from the Xisha soft coral Lobophytum sp. Bioorg. Chem. 2020, 103, 104223. [Google Scholar] [CrossRef] [PubMed]
- Li, S.W.; Cuadrado, C.; Yao, L.G.; Daranas, A.H.; Guo, Y.W. Quantum Mechanical-NMR-Aided Configuration and Conformation of Two Unreported Macrocycles Isolated from the Soft Coral Lobophytum sp.: Energy Calculations versus Coupling Constants. Org. Lett. 2020, 22, 4093–4096. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, C.; Daranas, A.H.; Sarotti, A.M. May the Force (Field) Be with You: On the Importance of Conformational Searches in the Prediction of NMR Chemical Shifts. Mar. Drugs 2022, 20, 699. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01, Gaussian: Wallingford, CT, USA, 2016.
- Ditchfield, R. Molecular Orbital Theory of Magnetic Shielding and Magnetic Susceptibility. J. Chem. Phys. 2003, 56, 5688–5691. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- McMichael Rohlfing, C.; Allen, L.C.; Ditchfield, R. Proton and carbon-13 chemical shifts: Comparison between theory and experiment. Chem. Phys. 1984, 87, 9–15. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Legault, C.Y. CYLview, 1.0b; Université de Sherbrooke: Sherbrooke, QC, Canada, 2009. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).