
Preventive and Therapeutic Effects of Krill Oil on Obesity and Obesity-Induced Metabolic Syndromes in High-Fat Diet-Fed Mice


 , and
, and
Abstract
:1. Introduction
2. Results
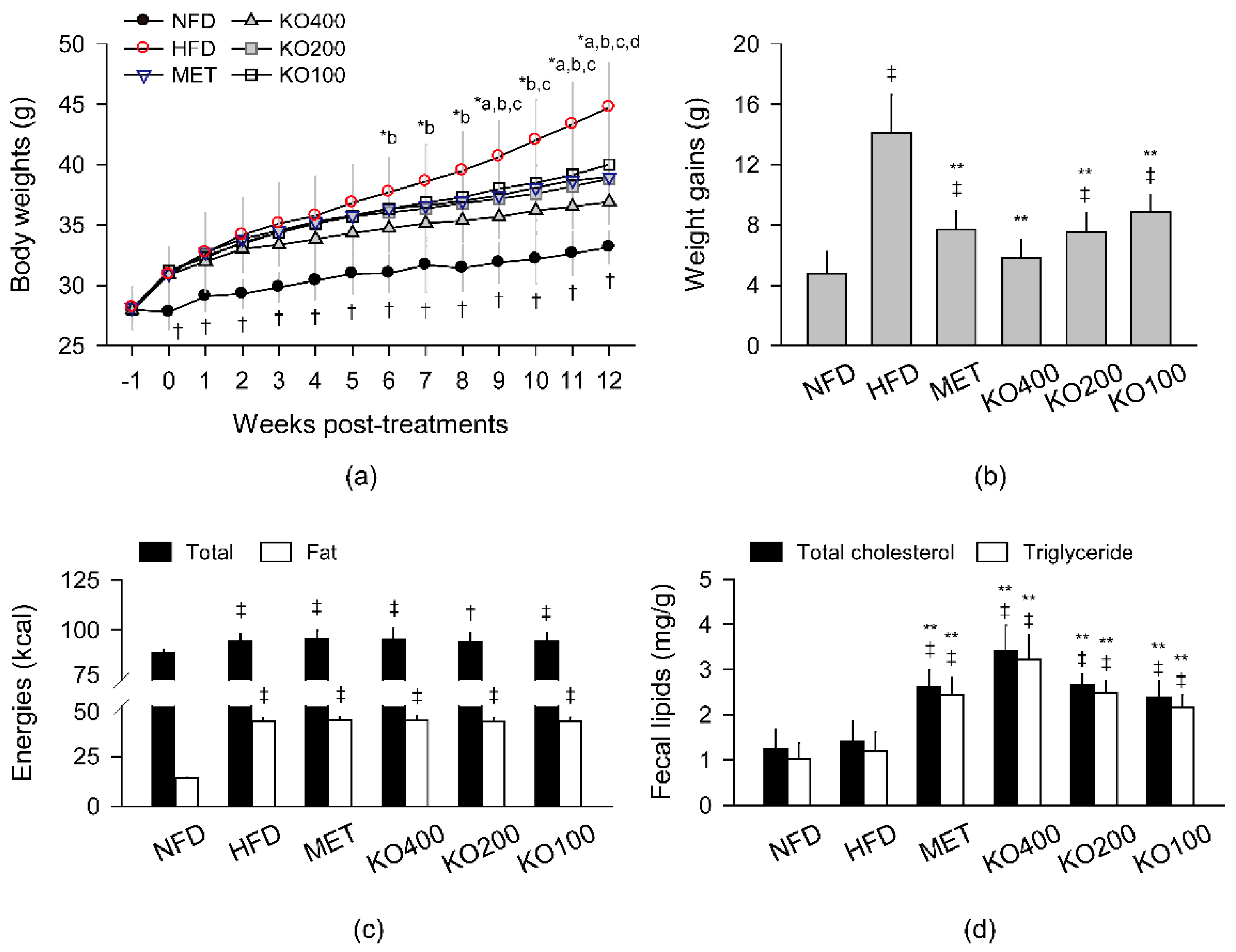
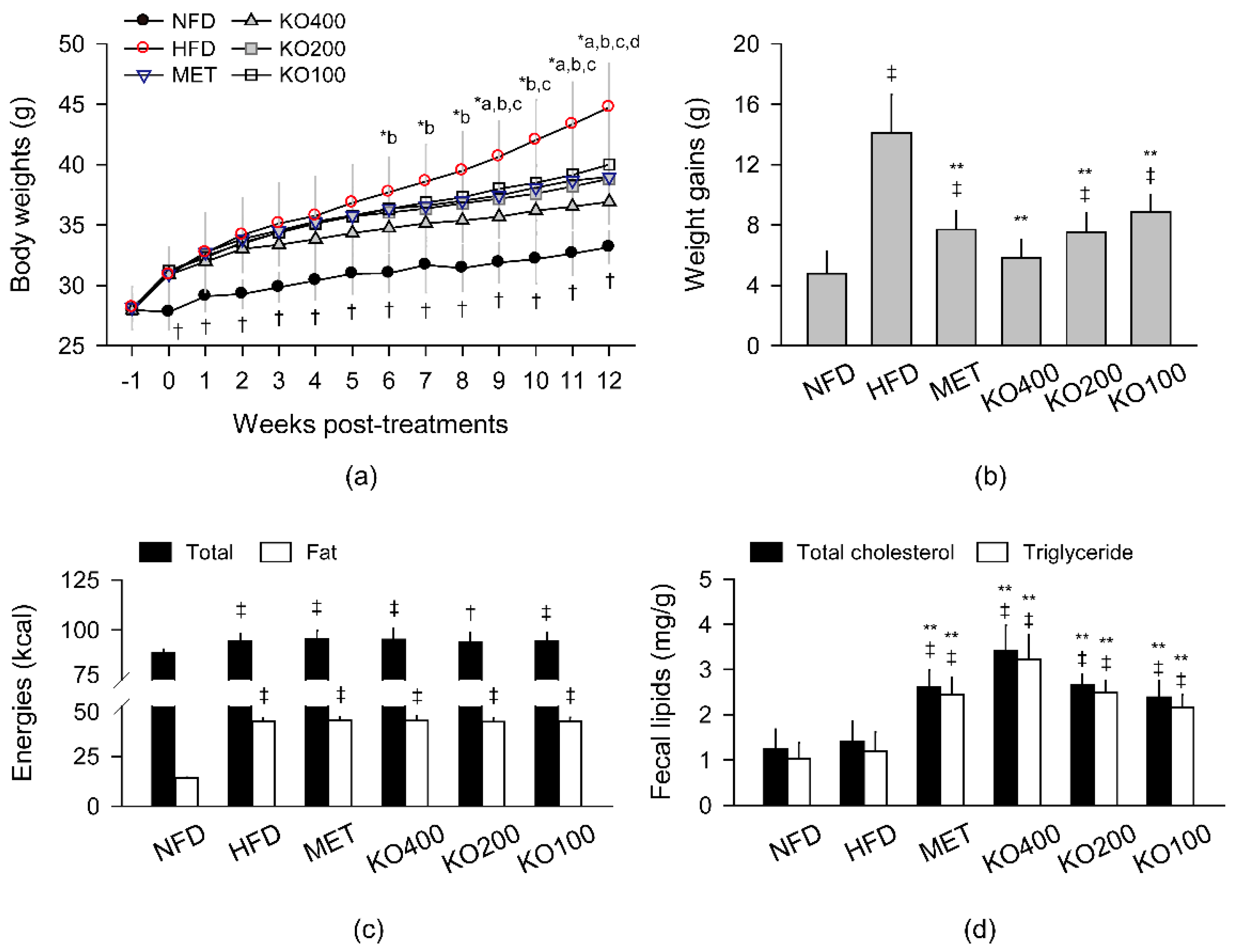
2.1. Inhibition of Body Weight Gain
2.2. Stimulation of Lipid Excretion
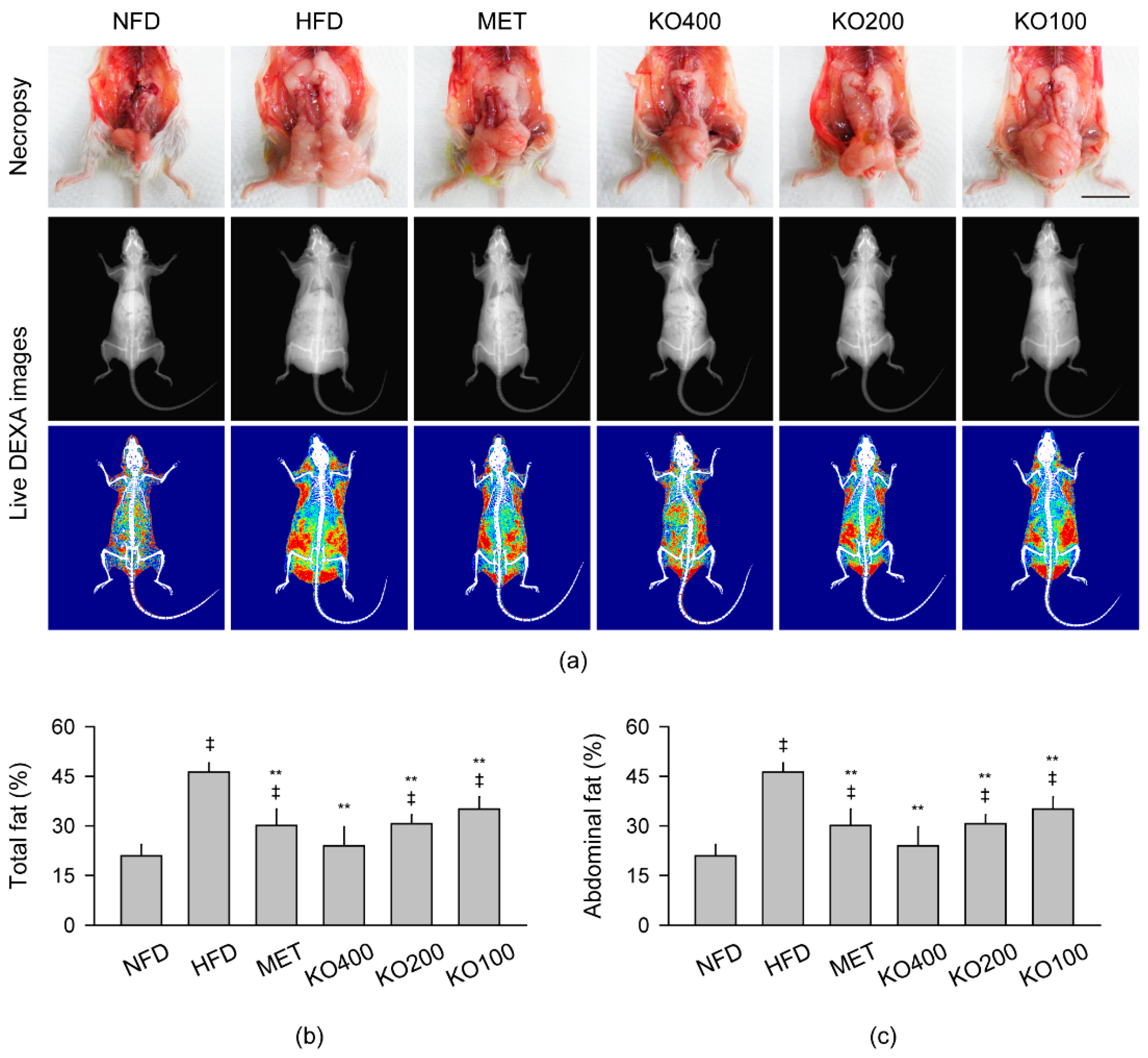
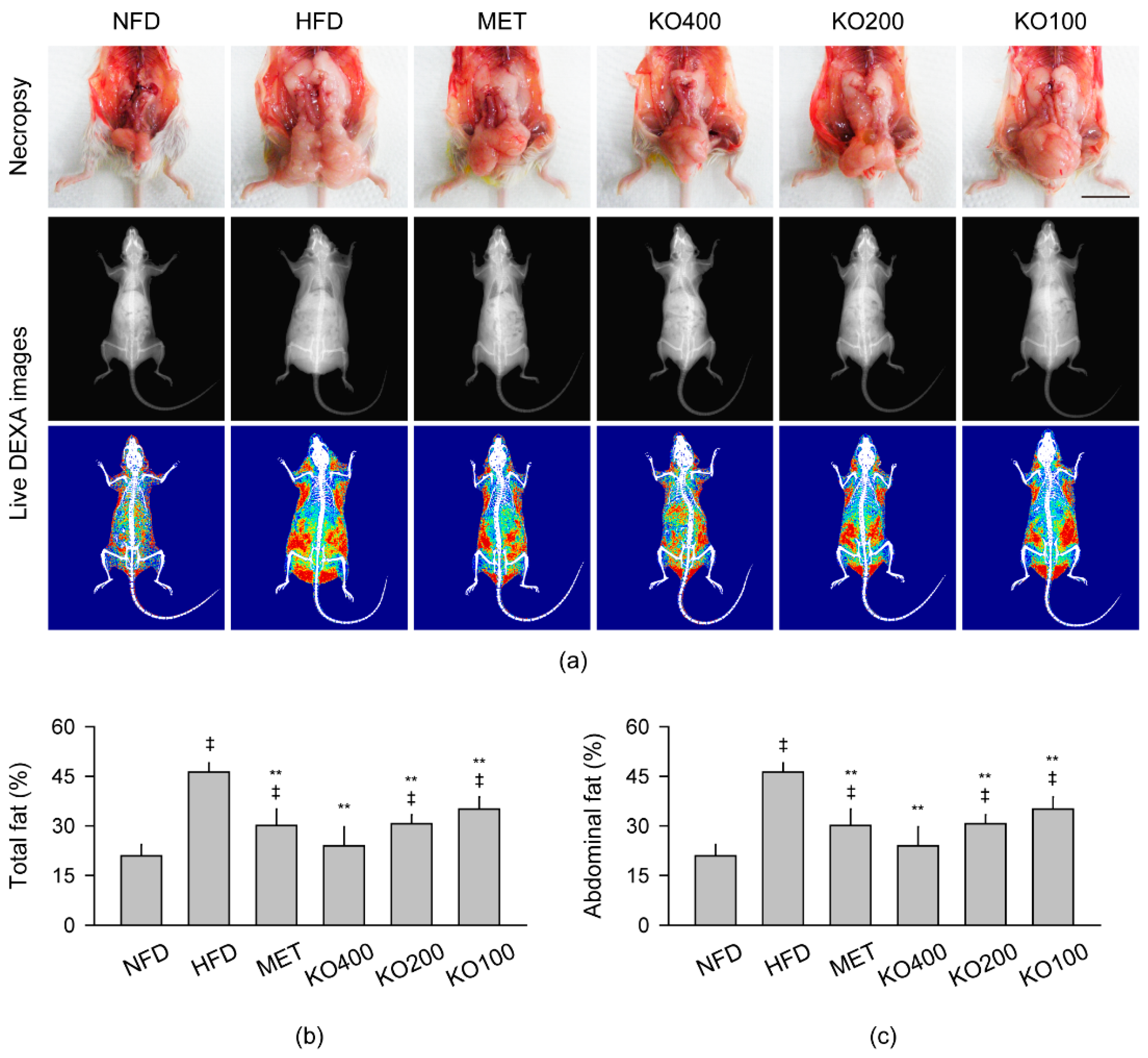
2.3. Inhibition of Body Fat Deposition
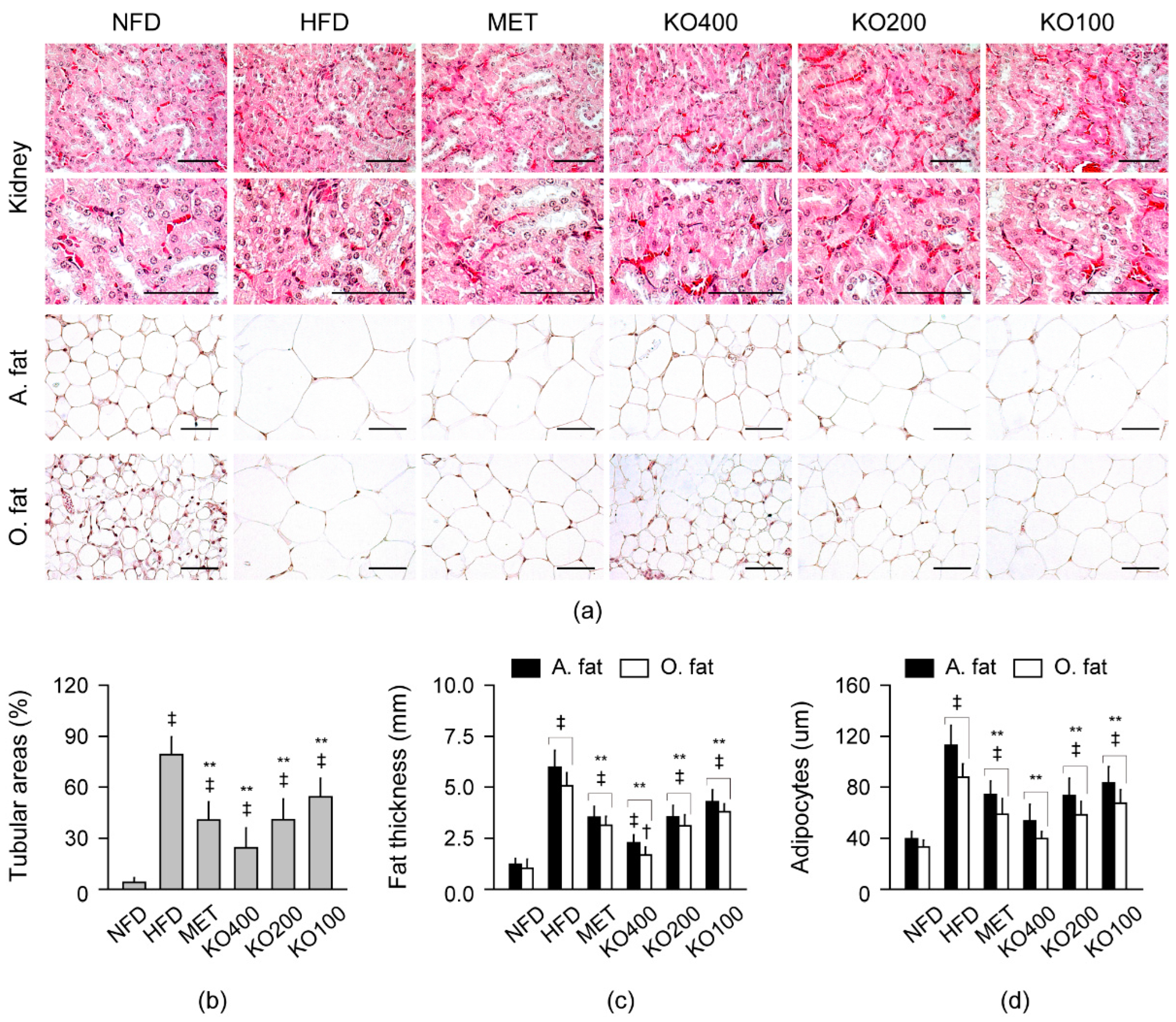
2.4. Improvements in Organ Weights Involved in Metabolic Alteration
2.5. Improvements in Blood Biochemical Parameters Involved in Metabolic Alteration
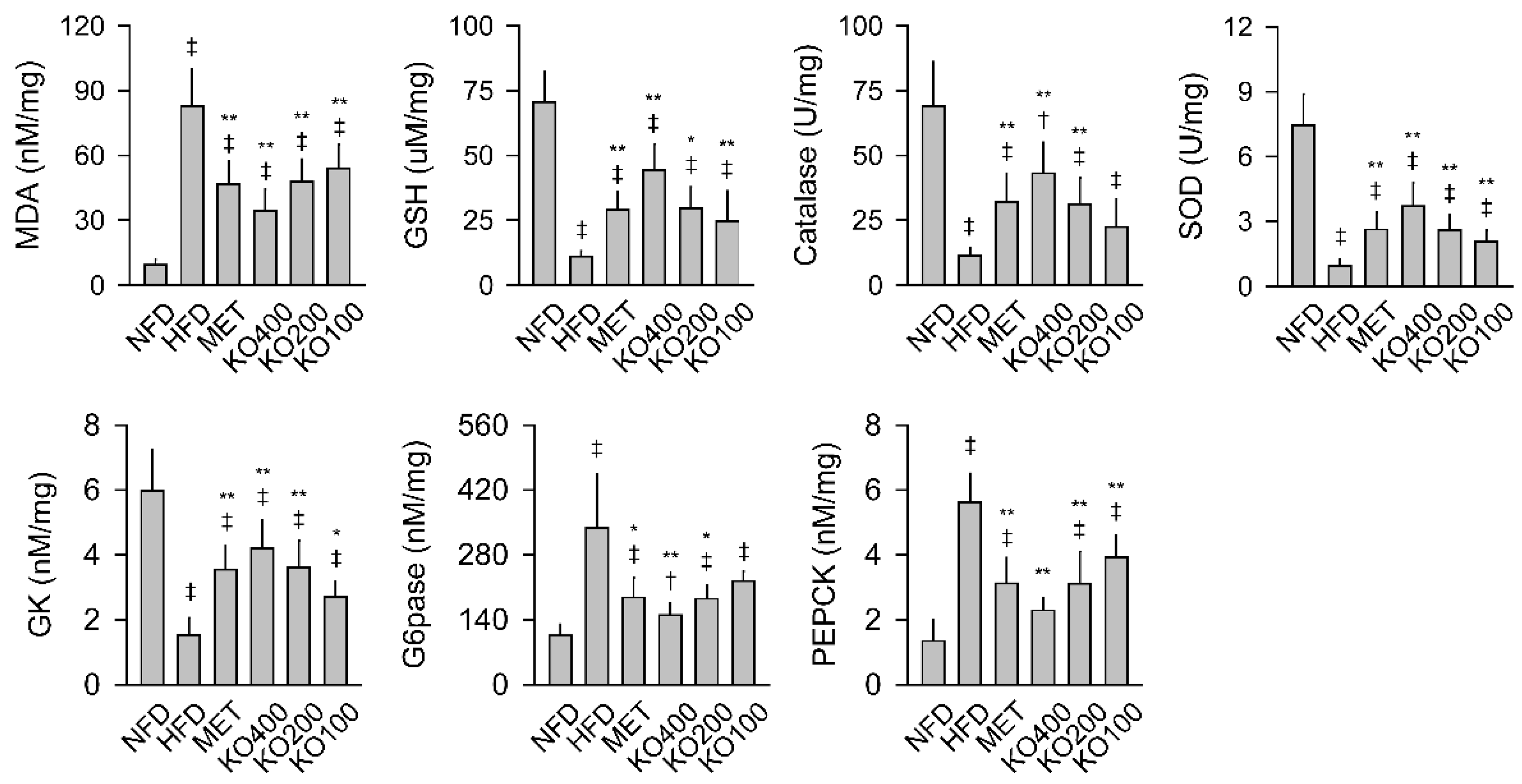
2.6. Enhancement of Hepatic Antioxidant and Glucose-Regulating Enzyme Activities
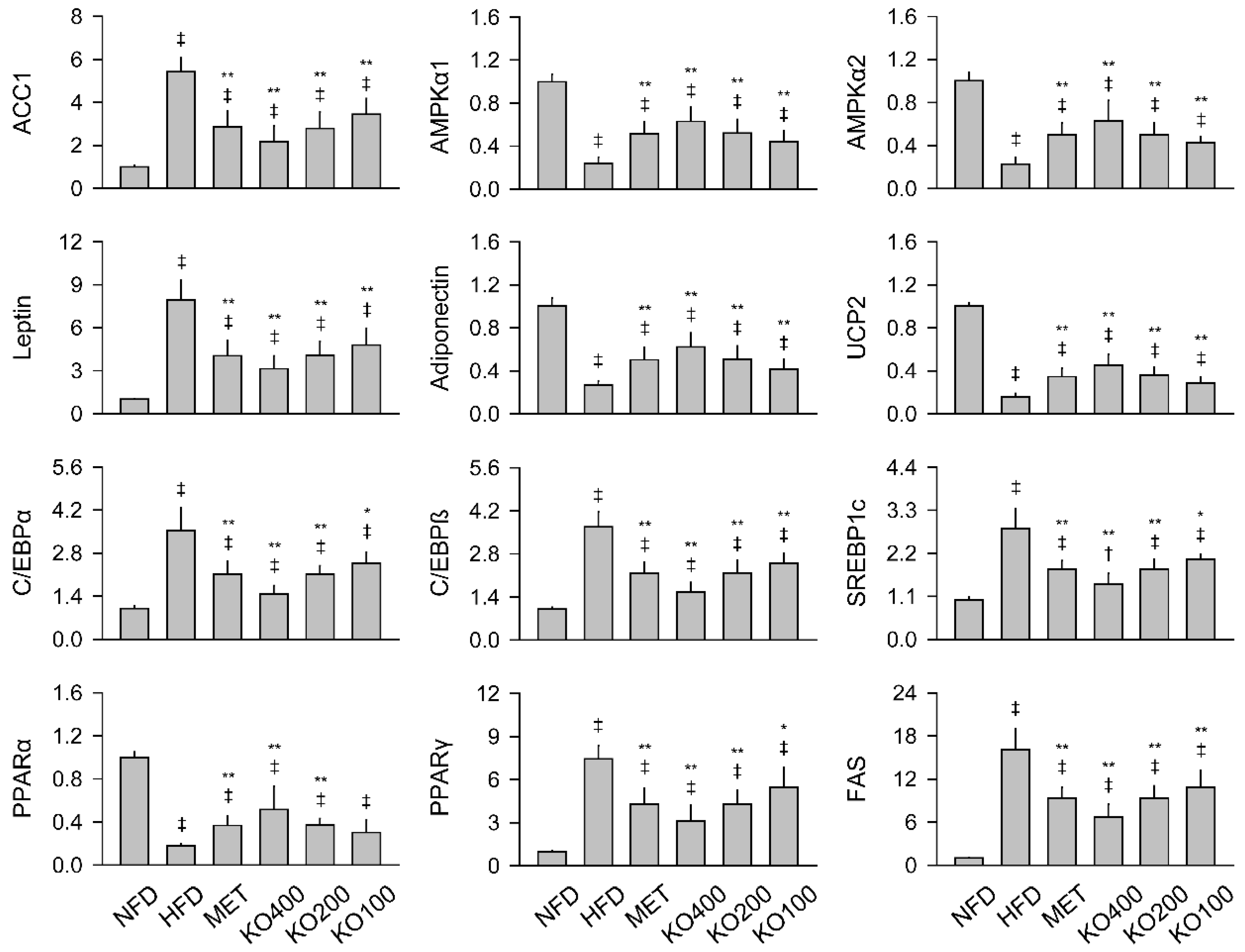
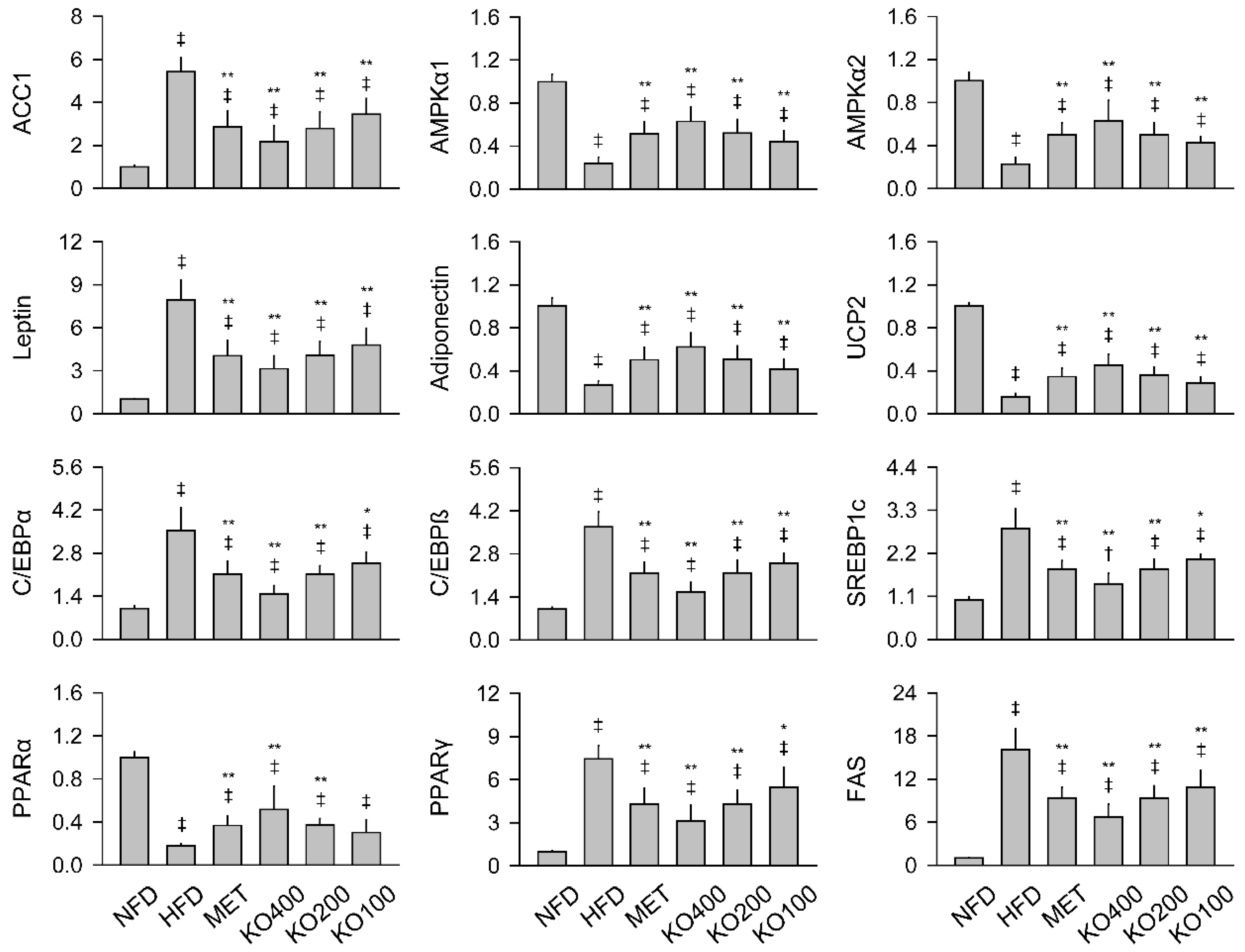
2.7. Modification of Gene Expressions Involved in Metabolic Alteration
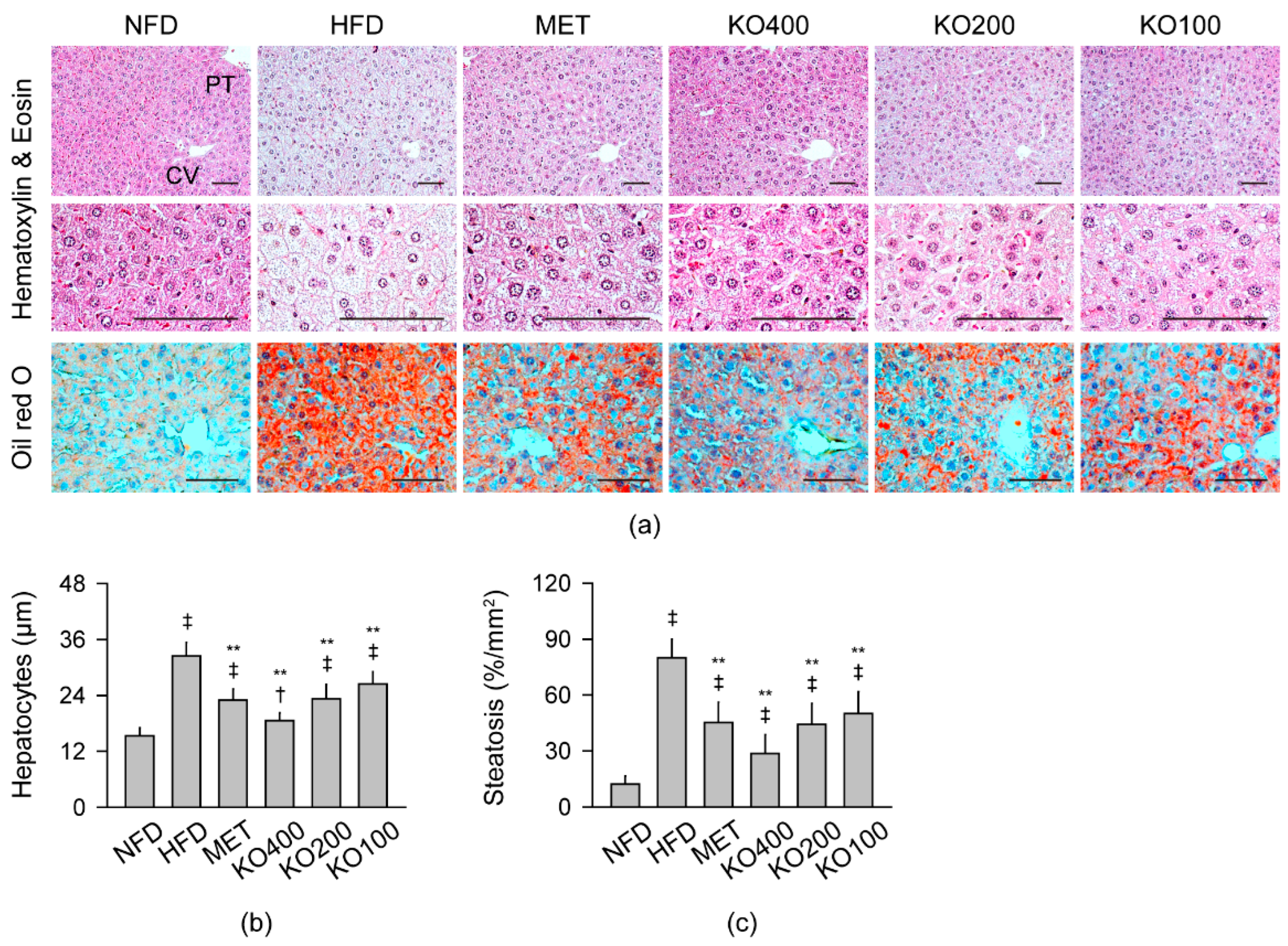
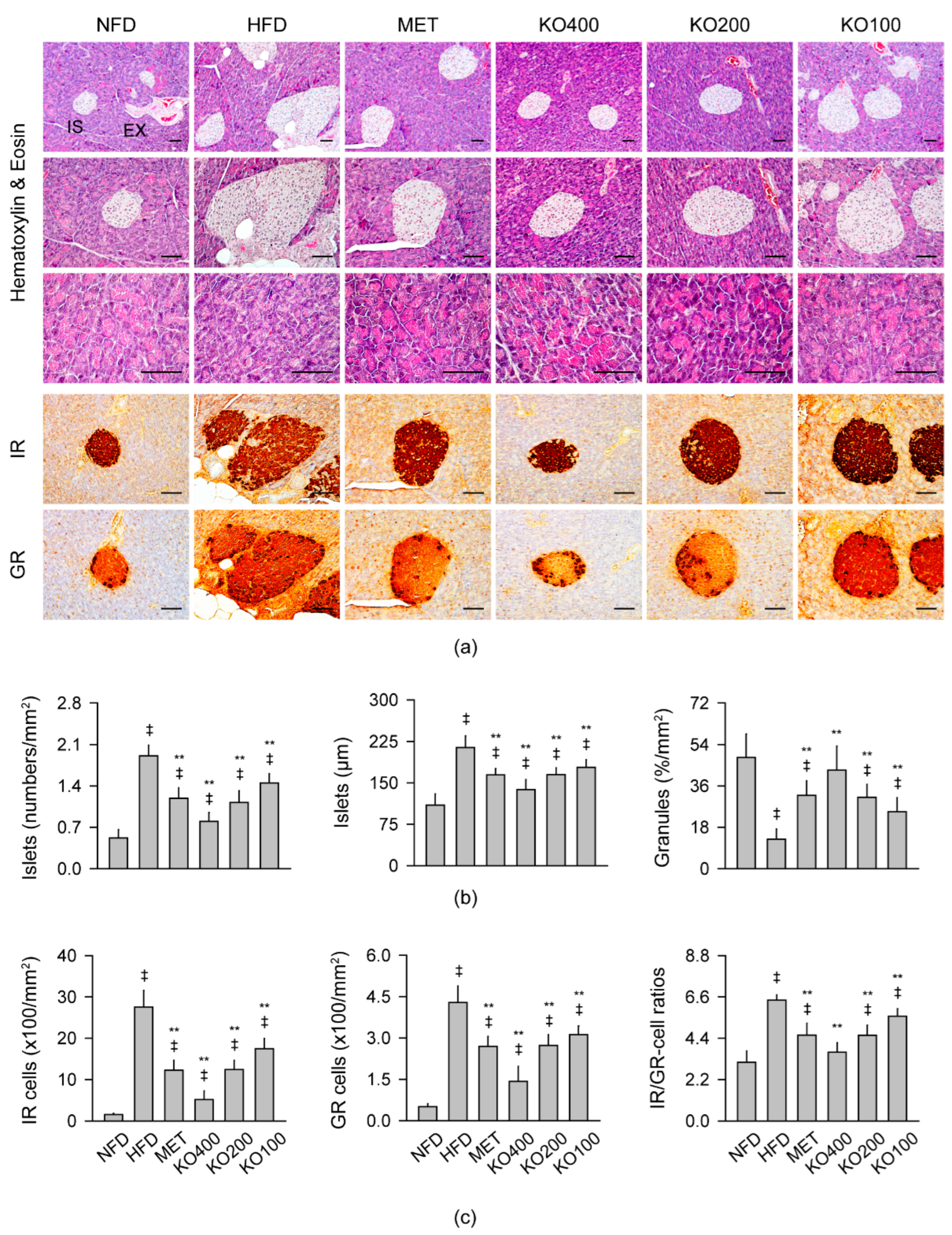
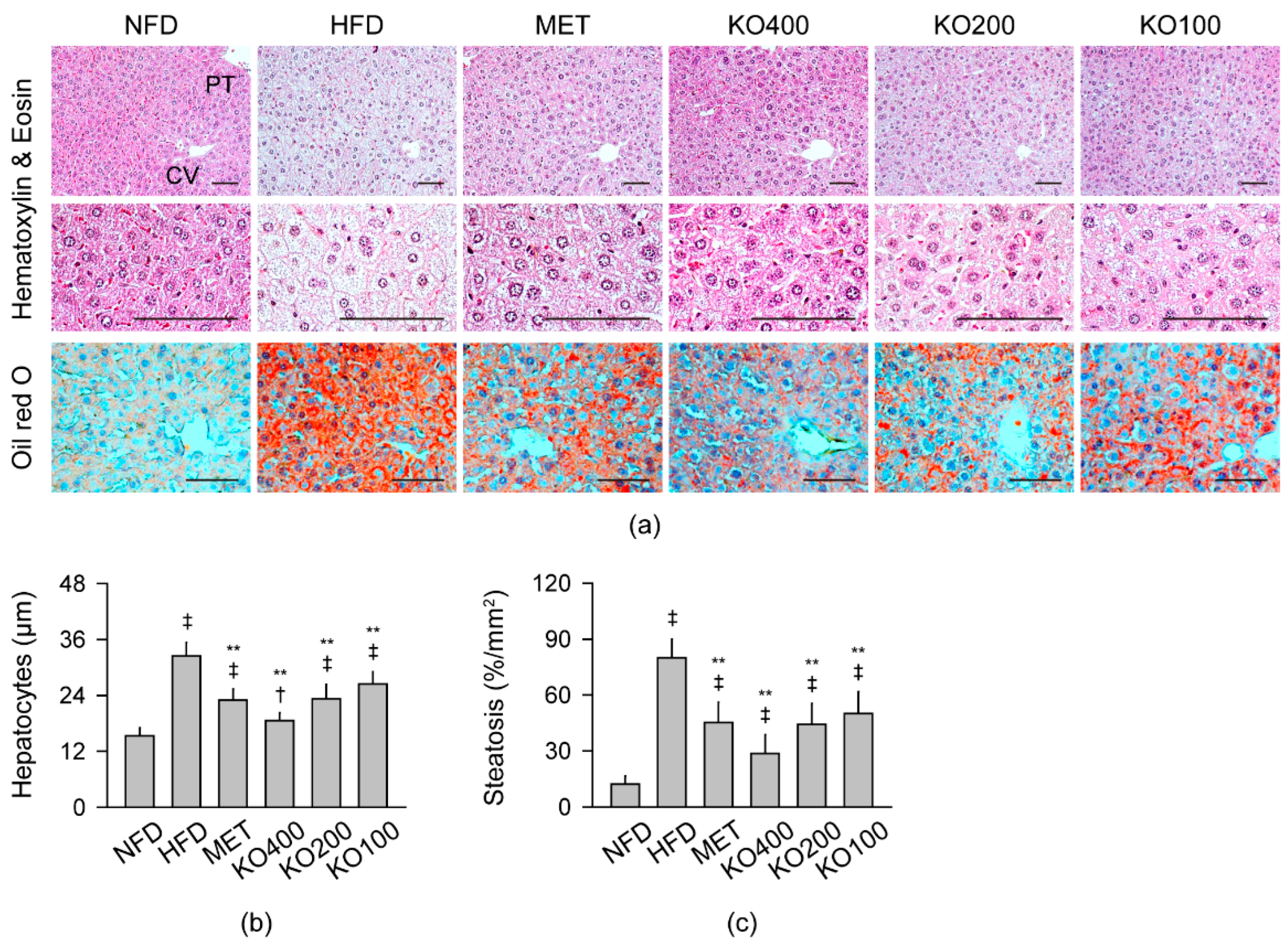
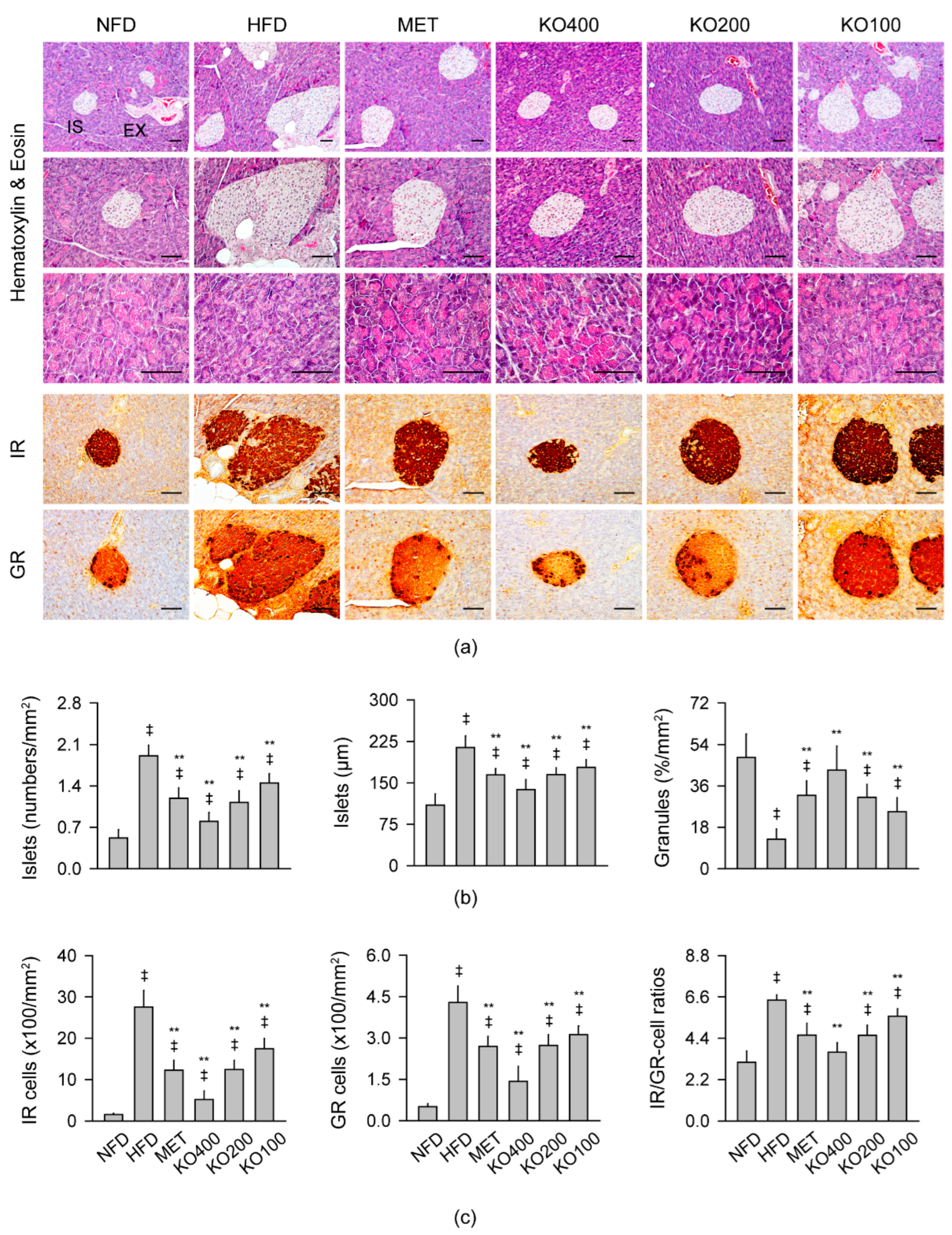
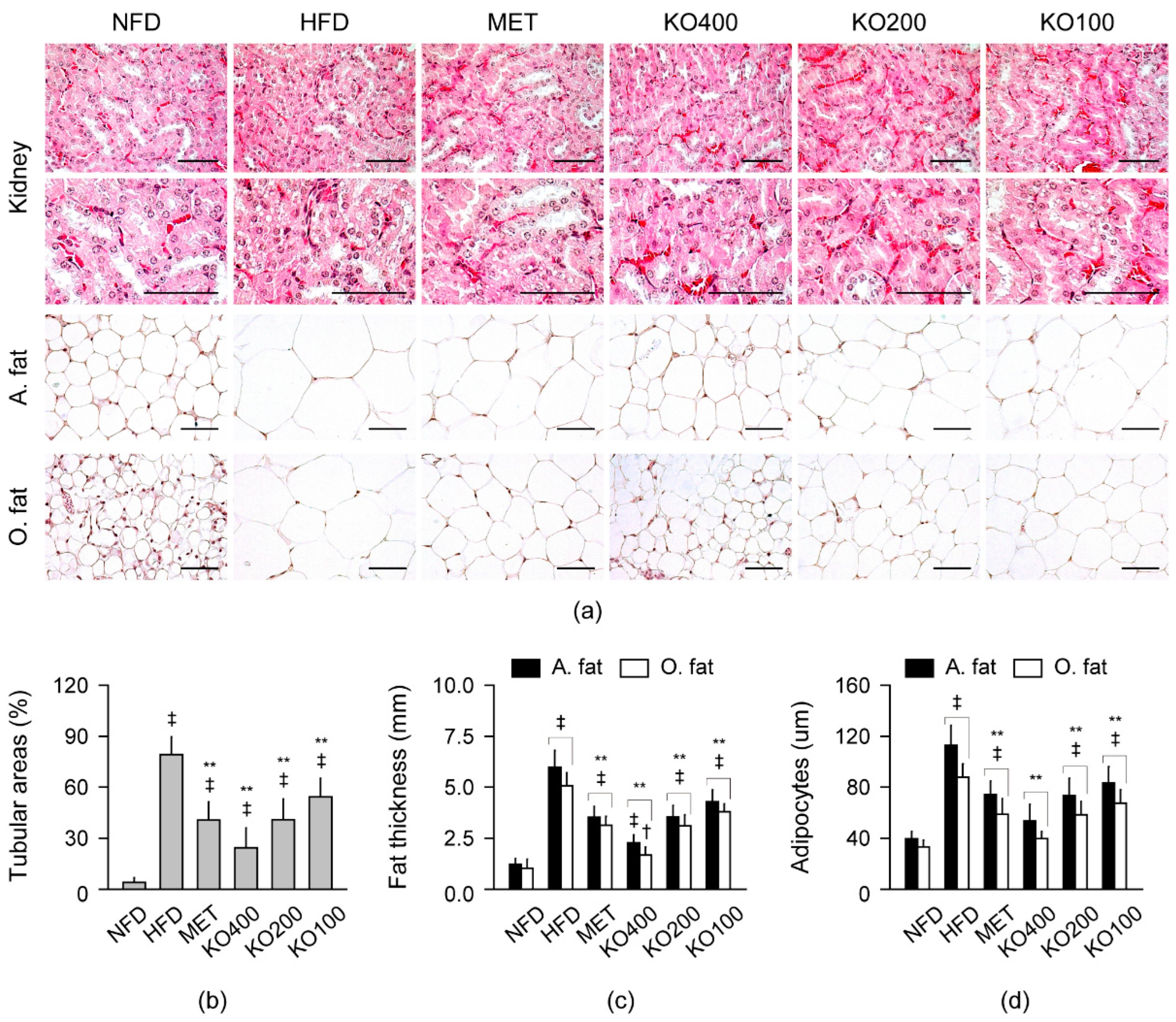
2.8. Improvements in Histopathological Changes Involved in Metabolic Alteration
2.8.1. Histopathological Improvements in the Liver
2.8.2. Histopathological Improvements in the Pancreas
2.8.3. Histopathological Improvements in Kidney and Fat Tissues
3. Discussion
4. Materials and Methods
4.1. Preparation of KO
4.2. HFD-Induced Mice Model and Treatments
4.3. Assessment of Daily Energy Intake, Lipid Excretion, and Body Fat Deposition
4.4. Blood Biochemistry
4.5. Hepatic Antioxidant Activities
4.6. Hepatic Glucose-Regulating Enzyme Activities
4.7. Real-Time Reverse Transcription Polymerase Chain Reaction (RT-PCR) Analysis
4.8. Histopathology
4.9. Immunohistochemistry
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Karczewski, J.; Sledzinska, E.; Baturo, A.; Jonczyk, I.; Maleszko, A.; Samborski, P.; Begier-Krasinska, B.; Dobrowolska, A. Obesity and inflammation. Eur. Cytokine Netw. 2018, 29, 83–94. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration (NCD-RisC). Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization Obesity and Overweight Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 9 June 2021).
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, E.A.; Kahl, S.; Seelig, A.; Begovatz, P.; Irmler, M.; Kupriyanova, Y.; Nowotny, B.; Nowotny, P.; Herder, C.; Barosa, C.; et al. Acute dietary fat intake initiates alterations in energy metabolism and insulin resistance. J. Clin. Investig. 2017, 127, 695–708. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhang, Y.; Graham, S.; Wang, X.; Cai, D.; Huang, M.; Pique-Regi, R.; Dong, X.C.; Chen, Y.E.; Willer, C.; et al. Causal relationships between NAFLD, T2D and obesity have implications for disease subphenotyping. J. Hepatol. 2020, 73, 263–276. [Google Scholar] [CrossRef]
- Panwar, B.; Hanks, L.J.; Tanner, R.M.; Muntner, P.; Kramer, H.; McClellan, W.M.; Warnock, D.G.; Judd, S.E.; Gutierrez, O.M. Obesity, metabolic health, and the risk of end-stage renal disease. Kidney Int. 2015, 87, 1216–1222. [Google Scholar] [CrossRef] [Green Version]
- Tuomilehto, J.; Lindstrom, J.; Eriksson, J.G.; Valle, T.T.; Hamalainen, H.; Ilanne-Parikka, P.; Keinanen-Kiukaanniemi, S.; Laakso, M.; Louheranta, A.; Rastas, M.; et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N. Engl. J. Med. 2001, 344, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- EASL-EASD-EASO. Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. Diabetologia 2016, 59, 1121–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical use in type 2 diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef]
- Coll, A.P.; Chen, M.; Taskar, P.; Rimmington, D.; Patel, S.; Tadross, J.A.; Cimino, I.; Yang, M.; Welsh, P.; Virtue, S.; et al. GDF15 mediates the effects of metformin on body weight and energy balance. Nature 2020, 578, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Makri, E.; Goulas, A.; Polyzos, S.A. Epidemiology, Pathogenesis, Diagnosis and Emerging Treatment of Nonalcoholic Fatty Liver Disease. Arch. Med. Res. 2021, 52, 25–37. [Google Scholar] [CrossRef]
- Jones, G.C.; Macklin, J.P.; Alexander, W.D. Contraindications to the use of metformin. BMJ 2003, 326, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.; Motti, M.L.; Meccariello, R. omega-3 and omega-6 Polyunsaturated Fatty Acids, Obesity and Cancer. Nutrients 2020, 12, 2751. [Google Scholar] [CrossRef]
- Siriwardhana, N.; Kalupahana, N.S.; Cekanova, M.; LeMieux, M.; Greer, B.; Moustaid-Moussa, N. Modulation of adipose tissue inflammation by bioactive food compounds. J. Nutr. Biochem. 2013, 24, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Dewidar, B.; Kahl, S.; Pafili, K.; Roden, M. Metabolic liver disease in diabetes—From mechanisms to clinical trials. Metabolism 2020, 111S, 154299. [Google Scholar] [CrossRef]
- Evert, A.B.; Boucher, J.L.; Cypress, M.; Dunbar, S.A.; Franz, M.J.; Mayer-Davis, E.J.; Neumiller, J.J.; Nwankwo, R.; Verdi, C.L.; Urbanski, P.; et al. Nutrition therapy recommendations for the management of adults with diabetes. Diabetes Care 2014, 37 (Suppl. 1), S120–S143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.G.; Ford, N.A.; Hu, F.B.; Zelman, K.M.; Mozaffarian, D.; Kris-Etherton, P.M. A healthy approach to dietary fats: Understanding the science and taking action to reduce consumer confusion. Nutr J. 2017, 16, 53. [Google Scholar] [CrossRef]
- Li, J.; Li, F.R.; Wei, D.; Jia, W.; Kang, J.X.; Stefanovic-Racic, M.; Dai, Y.; Zhao, A.Z. Endogenous ω-3 Polyunsaturated Fatty Acid Production Confers Resistance to Obesity, Dyslipidemia, and Diabetes in Mice. Mol. Endocrinol. 2014, 28, 1316–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flachs, P.; Rossmeisl, M.; Bryhn, M.; Kopecky, J. Cellular and molecular effects of n-3 polyunsaturated fatty acids on adipose tissue biology and metabolism. Clin. Sci. 2009, 116, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zaloga, G.P. Narrative Review of n-3 Polyunsaturated Fatty Acid Supplementation upon Immune Functions, Resolution Molecules and Lipid Peroxidation. Nutrients 2021, 13, 662. [Google Scholar] [CrossRef]
- Shahidi, F.; Ambigaipalan, P. Omega-3 Polyunsaturated Fatty Acids and Their Health Benefits. Annu. Rev. Food Sci. Technol. 2018, 9, 345–381. [Google Scholar] [CrossRef] [PubMed]
- Ulven, S.M.; Holven, K.B. Comparison of bioavailability of krill oil versus fish oil and health effect. Vasc. Health Risk Manag. 2015, 11, 511–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, G.; Wang, H.; Zhang, M.; Zhang, Y.; Wang, H.; Yu, X.; Wang, J.; Shen, Q. Real-Time Monitoring of the Oxidation Characteristics of Antarctic Krill Oil (Euphausia superba) during Storage by Electric Soldering Iron Ionization Mass Spectrometry-Based Lipidomics. J. Agric. Food Chem. 2020, 68, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Gigliotti, J.C.; Davenport, M.P.; Beamer, S.K.; Tou, J.C.; Jaczynski, J. Extraction and characterisation of lipids from Antarctic krill (Euphausia superba). Food Chem. 2011, 125, 1028–1036. [Google Scholar] [CrossRef]
- Batetta, B.; Griinari, M.; Carta, G.; Murru, E.; Ligresti, A.; Cordeddu, L.; Giordano, E.; Sanna, F.; Bisogno, T.; Uda, S.; et al. Endocannabinoids may mediate the ability of (n−3) fatty acids to reduce ectopic fat and inflammatory mediators in obese Zucker rats. J. Nutr. 2009, 139, 1495–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferramosca, A.; Conte, A.; Zara, V. Krill Oil Ameliorates Mitochondrial Dysfunctions in Rats Treated with High-Fat Diet. BioMed Res. Int. 2015, 2015, 645984. [Google Scholar] [CrossRef] [PubMed]
- Rossmeisl, M.; Pavlisova, J.; Bardova, K.; Kalendova, V.; Buresova, J.; Kuda, O.; Kroupova, P.; Stankova, B.; Tvrzicka, E.; Fiserova, E.; et al. Increased plasma levels of palmitoleic acid may contribute to beneficial effects of Krill oil on glucose homeostasis in dietary obese mice. Biochim. Et Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158732. [Google Scholar] [CrossRef] [PubMed]
- Kroupova, P.; van Schothorst, E.M.; Keijer, J.; Bunschoten, A.; Vodicka, M.; Irodenko, I.; Oseeva, M.; Zacek, P.; Kopecky, J.; Rossmeisl, M.; et al. Omega-3 Phospholipids from Krill Oil Enhance Intestinal Fatty Acid Oxidation More Effectively than Omega-3 Triacylglycerols in High-Fat Diet-Fed Obese Mice. Nutrients 2020, 12, 2037. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Sun, T.; Li, Y.; Zhang, D.; Zhou, J.; Su, X. Modulation of the Gut Microbiota by Krill Oil in Mice Fed a High-Sugar High-Fat Diet. Front. Microbiol. 2017, 8, 905. [Google Scholar] [CrossRef] [Green Version]
- Piscitelli, F.; Carta, G.; Bisogno, T.; Murru, E.; Cordeddu, L.; Berge, K.; Tandy, S.; Cohn, J.S.; Griinari, M.; Banni, S.; et al. Effect of dietary krill oil supplementation on the endocannabinoidome of metabolically relevant tissues from high-fat-fed mice. Nutr. Metab. 2011, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Sistilli, G.; Kalendova, V.; Cajka, T.; Irodenko, I.; Bardova, K.; Oseeva, M.; Zacek, P.; Kroupova, P.; Horakova, O.; Lackner, K.; et al. Krill Oil Supplementation Reduces Exacerbated Hepatic Steatosis Induced by Thermoneutral Housing in Mice with Diet-Induced Obesity. Nutrients 2021, 13, 437. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhang, L.; Chen, H.; Feng, R.; Cao, P.; Liu, Y. Effects of Antarctic krill oil on lipid and glucose metabolism in C57BL/6J mice fed with high fat diet. Lipids Health Dis. 2017, 16, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandy, S.; Chung, R.W.; Wat, E.; Kamili, A.; Berge, K.; Griinari, M.; Cohn, J.S. Dietary krill oil supplementation reduces hepatic steatosis, glycemia, and hypercholesterolemia in high-fat-fed mice. J. Agric. Food Chem. 2009, 57, 9339–9345. [Google Scholar] [CrossRef]
- Yang, G.; Lee, J.; Lee, S.; Kwak, D.; Choe, W.; Kang, I.; Kim, S.S.; Ha, J. Krill oil supplementation improves dyslipidemia and lowers body weight in mice fed a high-fat diet through activation of AMP-activated protein kinase. J. Med. Food 2016, 19, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Burri, L.; Berge, K.; Wibrand, K.; Berge, R.K.; Barger, J.L. Differential effects of krill oil and fish oil on the hepatic transcriptome in mice. Front. Genet. 2011, 2, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillander, V.; Bjorndal, B.; Burri, L.; Bohov, P.; Skorve, J.; Berge, R.K.; Alexson, S.E. Fish oil and krill oil supplementations differentially regulate lipid catabolic and synthetic pathways in mice. Nutr. Metab. 2014, 11, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigerust, N.F.; Bjorndal, B.; Bohov, P.; Brattelid, T.; Svardal, A.; Berge, R.K. Krill oil versus fish oil in modulation of inflammation and lipid metabolism in mice transgenic for TNF-alpha. Eur. J. Nutr. 2013, 52, 1315–1325. [Google Scholar] [CrossRef]
- Aydin Cil, M.; Ghosi Ghareaghaji, A.; Bayir, Y.; Buyuktuncer, Z.; Besler, H.T. Efficacy of krill oil versus fish oil on obesity-related parameters and lipid gene expression in rats: Randomized controlled study. PeerJ 2021, 9, e12009. [Google Scholar] [CrossRef]
- Cui, C.; Li, Y.; Gao, H.; Zhang, H.; Han, J.; Zhang, D.; Li, Y.; Zhou, J.; Lu, C.; Su, X. Modulation of the gut microbiota by the mixture of fish oil and krill oil in high-fat diet-induced obesity mice. PLoS ONE 2017, 12, e0186216. [Google Scholar] [CrossRef]
- Xiang, X.; Zhou, X.; Wang, W.; Zhou, Y.; Zhou, X.; Deng, S.; Zheng, B.; Wen, Z. Effect of Antarctic krill phospholipid (KOPL) on high fat diet-induced obesity in mice. Food Res. Int. 2021, 148, 110456. [Google Scholar] [CrossRef] [PubMed]
- Bunea, R.; El Farrah, K.; Deutsch, L. Evaluation of the effects of Neptune Krill Oil on the clinical course of hyperlipidemia. Altern Med. Rev. 2004, 9, 420–428. [Google Scholar] [PubMed]
- Berge, K.; Piscitelli, F.; Hoem, N.; Silvestri, C.; Meyer, I.; Banni, S.; Di Marzo, V. Chronic treatment with krill powder reduces plasma triglyceride and anandamide levels in mildly obese men. Lipids Health Dis. 2013, 12, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friday, K.E.; Childs, M.T.; Tsunehara, C.H.; Fujimoto, W.Y.; Bierman, E.L.; Ensinck, J.W. Elevated plasma glucose and lowered triglyceride levels from omega-3 fatty acid supplementation in type II diabetes. Diabetes Care 1989, 12, 276–281. [Google Scholar] [CrossRef]
- Choi, J.Y.; Jang, T.W.; Song, P.H.; Choi, S.H.; Ku, S.K.; Song, C.H. Combination Effects of Metformin and a Mixture of Lemon Balm and Dandelion on High-Fat Diet-Induced Metabolic Alterations in Mice. Antioxidants 2022, 11, 580. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.; Westermark, P.O.; Spalding, K.L.; Britton, T.; Ryden, M.; Frisen, J.; Bernard, S.; Arner, P. Adipocyte turnover: Relevance to human adipose tissue morphology. Diabetes 2010, 59, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, W.; Cheng, X.F.; Liu, Y.; Lv, Q.Z.; Liu, G.L.; Zhang, J.G.; Li, X.Y. Potential Nexus of Non-alcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus: Insulin Resistance Between Hepatic and Peripheral Tissues. Front. Pharmacol. 2018, 9, 1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musso, G.; Gambino, R.; Tabibian, J.H.; Ekstedt, M.; Kechagias, S.; Hamaguchi, M.; Hultcrantz, R.; Hagstrom, H.; Yoon, S.K.; Charatcharoenwitthaya, P.; et al. Association of non-alcoholic fatty liver disease with chronic kidney disease: A systematic review and meta-analysis. PLoS Med. 2014, 11, e1001680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suttie, A.W.; Masson, R.; Schutten, M. Chapter 10—Exocrine Pancreas. In Boorman’s Pathology of the Rat, 2nd ed.; Suttie, A.W., Ed.; Academic Press: Boston, MA, USA, 2018; pp. 107–122. [Google Scholar]
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moseti, D.; Regassa, A.; Kim, W.K. Molecular Regulation of Adipogenesis and Potential Anti-Adipogenic Bioactive Molecules. Int. J. Mol. Sci. 2016, 17, 124. [Google Scholar] [CrossRef] [Green Version]
- Tsochatzis, E.; Papatheodoridis, G.V.; Archimandritis, A.J. The evolving role of leptin and adiponectin in chronic liver diseases. Am. J. Gastroenterol. 2006, 101, 2629–2640. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, S.A.; Aronis, K.N.; Kountouras, J.; Raptis, D.D.; Vasiloglou, M.F.; Mantzoros, C.S. Circulating leptin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Diabetologia 2016, 59, 30–43. [Google Scholar] [CrossRef]
- Kim, H.-D.; Lee, S.-B.; Ko, S.-C.; Jung, W.-K.; Kim, Y.-M.; Kim, S.-B. Anti-inflammatory effect of ozonated krill (Euphausia superba) oil in lipopolysaccharide-stimulated RAW 264.7 macrophages. Fish. Aquat. Sci. 2018, 21, 15. [Google Scholar] [CrossRef]
- Li, R.; Chen, L.Z.; Zhao, W.; Zhao, S.P.; Huang, X.S. Metformin ameliorates obesity-associated hypertriglyceridemia in mice partly through the apolipoprotein A5 pathway. Biochem. Biophys. Res. Commun. 2016, 478, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Addy, C.; Kusunoki, J.; Anderson, N.N.; Deja, S.; Fu, X.; Burgess, S.C.; Li, C.; Ruddy, M.; Chakravarthy, M.; et al. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis but Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab. 2017, 26, 394–406.e6. [Google Scholar] [CrossRef] [PubMed]
- Ferramosca, A.; Conte, A.; Burri, L.; Berge, K.; De Nuccio, F.; Giudetti, A.M.; Zara, V. A krill oil supplemented diet suppresses hepatic steatosis in high-fat fed rats. PLoS ONE 2012, 7, e38797. [Google Scholar] [CrossRef] [Green Version]
- Ikeuchi, M.; Koyama, T.; Takahashi, J.; Yazawa, K. Effects of astaxanthin in obese mice fed a high-fat diet. Biosci. Biotechnol. Biochem. 2007, 71, 893–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridges, K.M.; Gigliotti, J.C.; Altman, S.; Jaczynski, J.; Tou, J.C. Determination of digestibility, tissue deposition, and metabolism of the omega-3 fatty acid content of krill protein concentrate in growing rats. J. Agric. Food Chem. 2010, 58, 2830–2837. [Google Scholar] [CrossRef]
- Tou, J.C.; Altman, S.N.; Gigliotti, J.C.; Benedito, V.A.; Cordonier, E.L. Different sources of omega-3 polyunsaturated fatty acids affects apparent digestibility, tissue deposition, and tissue oxidative stability in growing female rats. Lipids Health Dis. 2011, 10, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulven, S.M.; Kirkhus, B.; Lamglait, A.; Basu, S.; Elind, E.; Haider, T.; Berge, K.; Vik, H.; Pedersen, J.I. Metabolic effects of krill oil are essentially similar to those of fish oil but at lower dose of EPA and DHA, in healthy volunteers. Lipids 2011, 46, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyzos, S.A.; Kountouras, J.; Zavos, C. Nonalcoholic fatty liver disease: The pathogenetic roles of insulin resistance and adipocytokines. Curr. Mol. Med. 2009, 9, 299–314. [Google Scholar] [CrossRef]
- Banni, S.; Carta, G.; Murru, E.; Cordeddu, L.; Giordano, E.; Sirigu, A.R.; Berge, K.; Vik, H.; Maki, K.C.; Di Marzo, V.; et al. Krill oil significantly decreases 2-arachidonoylglycerol plasma levels in obese subjects. Nutr. Metab. 2011, 8, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, K.C.; Reeves, M.S.; Farmer, M.; Griinari, M.; Berge, K.; Vik, H.; Hubacher, R.; Rains, T.M. Krill oil supplementation increases plasma concentrations of eicosapentaenoic and docosahexaenoic acids in overweight and obese men and women. Nutr. Res. 2009, 29, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Nicol, S.; James, A.; Pitcher, G. A first record of daytime surface swarming by Euphausia lucens in the Southern Benguela region. Mar. Biol. 1987, 94, 7–10. [Google Scholar] [CrossRef]
- Meyer, B.; Atkinson, A.; Bernard, K.S.; Brierley, A.S.; Driscoll, R.; Hill, S.L.; Marschoff, E.; Maschette, D.; Perry, F.A.; Reiss, C.S.; et al. Successful ecosystem-based management of Antarctic krill should address uncertainties in krill recruitment, behaviour and ecological adaptation. Commun. Earth Environ. 2020, 1, 28. [Google Scholar] [CrossRef]
- Michael, K.; Suberg, L.A.; Wessels, W.; Kawaguchi, S.; Meyer, B. Facing Southern Ocean warming: Temperature effects on whole animal performance of Antarctic krill (Euphausia superba). ZOOLOGY 2021, 146, 125910. [Google Scholar] [CrossRef] [PubMed]
- Ramprasath, V.R.; Eyal, I.; Zchut, S.; Jones, P.J. Enhanced increase of omega-3 index in healthy individuals with response to 4-week n-3 fatty acid supplementation from krill oil versus fish oil. Lipids Health Dis. 2013, 12, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NFD | HFD | MET | KO400 | KO200 | KO100 | |
|---|---|---|---|---|---|---|
| Liver | 1.29 ± 0.07 (4.3 ± 0.2) | 1.90 ± 0.10 ‡ (4.5 ± 0.4) | 1.63 ± 0.11 ‡,** (4.5 ± 0.3) | 1.44 ± 0.10 ‡,** (4.3 ± 0.4) | 1.61 ± 0.10 ‡,** (4.6 ± 0.4) | 1.71 ± 0.06 ‡,** (4.6 ± 0.3) |
| Pancreas | 0.25 ± 0.04 (0.8 ± 0.1) | 0.24 ± 0.03 (0.6 ± 0.1 ‡) | 0.26 ± 0.02 (0.7 ± 0.1 **) | 0.26 ± 0.02 (0.8 ± 0.1 **) | 0.26 ± 0.01 (0.7 ± 0.0 **) | 0.25 ± 0.02 (0.7 ± 0.1 ‡,*) |
| Kidney | 0.21 ± 0.01 (0.7 ± 0.0) | 0.30 ± 0.01 ‡ (0.7 ± 0.1) | 0.26 ± 0.01 ‡,** (0.7 ± 0.0) | 0.24 ± 0.01 ‡,** (0.7 ± 0.0) | 0.25 ± 0.01 ‡,** (0.7 ± 0.0) | 0.27 ± 0.01 ‡,** (0.7 ± 0.0) |
| A. fat | 0.07 ± 0.03 (0.2 ± 0.1) | 0.44 ± 0.08 ‡ (1.1 ± 0.2 ‡) | 0.24 ± 0.05 ‡,** (0.7 ± 0.2 ‡,**) | 0.18 ± 0.05 ‡,** (0.5 ± 0.2 ‡,**) | 0.24 ± 0.05 ‡,** (0.7 ± 0.2 ‡,**) | 0.30 ± 0.03 ‡,** (0.8 ± 0.1 ‡,**) |
| O. fat | 0.08 ± 0.03 (0.3 ± 0.1) | 0.45 ± 0.11 ‡ (1.1 ± 0.3 ‡) | 0.23 ± 0.05 ‡,** (0.6 ± 0.1 ‡,**) | 0.15 ± 0.03 ‡,** (0.4 ± 0.1 †,**) | 0.22 ± 0.03 ‡,** (0.6 ± 0.1 ‡,**) | 0.27 ± 0.03 ‡,** (0.7 ± 0.1 ‡,*) |
| Parameters | NFD | HFD | MET | KO400 | KO200 | KO100 |
|---|---|---|---|---|---|---|
| ALT (IU/dL) | 4.8 ± 1.1 | 15.1 ± 1.3 ‡ | 9.8 ± 1.2 ‡,** | 7.8 ± 1.4 ‡,** | 9.8 ± 1.4 ‡,** | 12.0 ± 1.1 ‡,** |
| AST (IU/dL) | 8.5 ± 2.0 | 20.6 ± 2.3 ‡ | 14.2 ± 1.6 ‡,** | 11.2 ± 1.6 †,** | 14.1 ± 1.3 ‡,** | 16.4 ± 1.4 ‡,** |
| ALP (IU/dL) | 7.7 ± 1.4 | 24.0 ± 5.3 ‡ | 14.0 ± 1.8 ‡,** | 10.0 ± 1.3 †,** | 14.3 ± 1.9 ‡,** | 16.7 ± 1.3 ‡,* |
| LDH (IU/mL) | 0.6 ± 0.2 | 4.1 ± 0.9 ‡ | 2.2 ± 0.5 ‡,** | 1.6 ± 0.5 †,** | 2.2 ± 0.4 ‡,** | 2.8 ± 0.3 ‡,** |
| GGT (IU/dL) | 0.4 ± 0.2 | 1.7 ± 0.2 ‡ | 1.1 ± 0.2 ‡,** | 0.8 ± 0.2 ‡,** | 1.1 ± 0.2 ‡,** | 1.3 ± 0.1 ‡,** |
| Glucose (mg/mL) | 1.0 ± 0.1 | 2.4 ± 0.4 ‡ | 1.5 ± 0.2 ‡,** | 1.2 ± 0.2 ** | 1.5 ± 0.2 ‡,** | 1.7 ± 0.3 ‡,** |
| Insulin (ng/mL) | 0.6 ± 0.1 | 2.7 ± 0.6 ‡ | 1.5 ± 0.3 ‡,** | 1.2 ± 0.2 ‡,** | 1.6 ± 0.3 ‡,** | 1.9 ± 0.2 ‡,* |
| HbA1c (%) | 2.7 ± 1.0 | 9.1 ± 1.3 ‡ | 5.2 ± 1.0 ‡,** | 4.0 ± 1.1 ** | 5.3 ± 1.0 ‡,** | 6.3 ± 1.0 ‡,** |
| BUN (mg/mL) | 0.3 ± 0.1 | 1.3 ± 0.2 ‡ | 0.7 ± 0.1 ‡,** | 0.6 ± 0.2 ‡,** | 0.7 ± 0.1 ‡,** | 0.9 ± 0.2 ‡,** |
| CRE (mg/dL) | 0.6 ± 0.2 | 2.1 ± 0.3 ‡ | 1.4 ± 0.2 ‡,** | 1.0 ± 0.3 †,** | 1.4 ± 0.2 ‡,** | 1.5 ± 0.1 ‡,** |
| TG (mg/mL) | 0.8 ± 0.1 | 2.4 ± 0.2 ‡ | 1.5 ± 0.3 ‡,** | 1.1 ± 0.1 †,** | 1.5 ± 0.2 ‡,** | 1.8 ± 0.2 ‡,** |
| TChol (mg/mL) | 0.8 ± 0.2 | 2.7 ± 0.5 ‡ | 1.6 ± 0.2 ‡,** | 1.3 ± 0.3 †,** | 1.6 ± 0.2 ‡,** | 1.9 ± 0.2 ‡,** |
| LDL (mg/mL) | 0.2 ± 0.1 | 0.8 ± 0.1 ‡ | 0.5 ± 0.1 ‡,** | 0.3 ± 0.1 ‡,** | 0.5 ± 0.1 ‡,** | 0.6 ± 0.1 ‡,* |
| HDL (mg/mL) | 0.9 ± 0.2 | 0.2 ± 0.1 ‡ | 0.5 ± 0.1 ‡,** | 0.7 ± 0.2 ** | 0.5 ± 0.1 ‡,** | 0.4 ± 0.1 ‡,** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, S.-M.; Kim, Y.U.; Kim, J.-K.; Chun, Y.-S.; Kwon, Y.-S.; Ku, S.-K.; Song, C.-H. Preventive and Therapeutic Effects of Krill Oil on Obesity and Obesity-Induced Metabolic Syndromes in High-Fat Diet-Fed Mice. Mar. Drugs 2022, 20, 483. https://doi.org/10.3390/md20080483
Hwang S-M, Kim YU, Kim J-K, Chun Y-S, Kwon Y-S, Ku S-K, Song C-H. Preventive and Therapeutic Effects of Krill Oil on Obesity and Obesity-Induced Metabolic Syndromes in High-Fat Diet-Fed Mice. Marine Drugs. 2022; 20(8):483. https://doi.org/10.3390/md20080483
Chicago/Turabian StyleHwang, Seung-Min, Yeong Uk Kim, Jong-Kyu Kim, Yoon-Seok Chun, Young-Sam Kwon, Sae-Kwang Ku, and Chang-Hyun Song. 2022. "Preventive and Therapeutic Effects of Krill Oil on Obesity and Obesity-Induced Metabolic Syndromes in High-Fat Diet-Fed Mice" Marine Drugs 20, no. 8: 483. https://doi.org/10.3390/md20080483
APA StyleHwang, S.-M., Kim, Y. U., Kim, J.-K., Chun, Y.-S., Kwon, Y.-S., Ku, S.-K., & Song, C.-H. (2022). Preventive and Therapeutic Effects of Krill Oil on Obesity and Obesity-Induced Metabolic Syndromes in High-Fat Diet-Fed Mice. Marine Drugs, 20(8), 483. https://doi.org/10.3390/md20080483