
Marine Cyclic Peptides: Antimicrobial Activity and Synthetic Strategies





Abstract
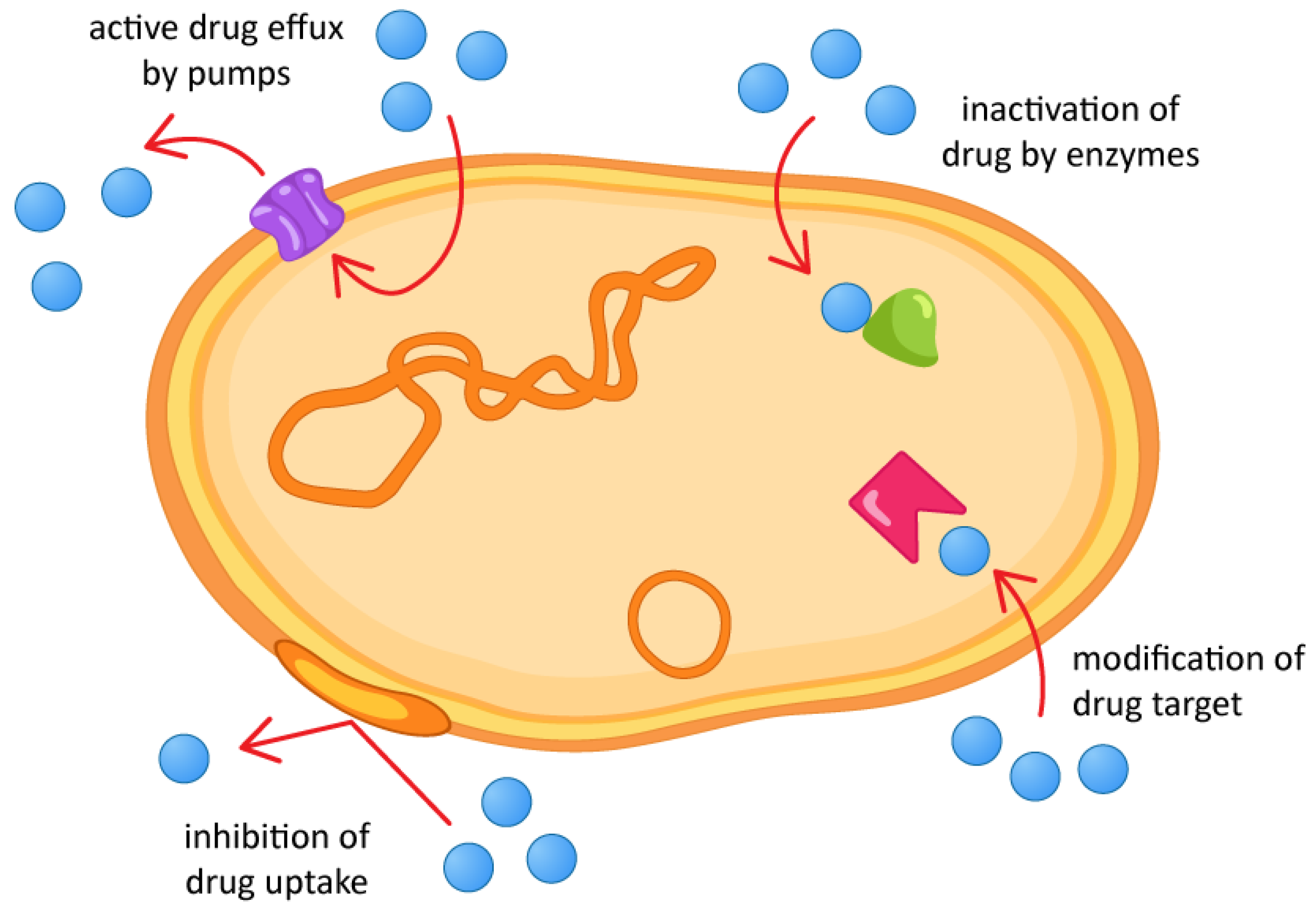
1. Introduction
2. Marine Cyclic Peptides with Antimicrobial Activities
2.1. Sponge-Produced Cyclic Peptides

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | Source | Antimicrobial Activity | Synthesis | References |
|---|---|---|---|---|---|
| Aciculitins A-C (1–3) | Bicyclic octa-peptides | Aciculites orientalis | C. albicans (2.5 µg/disk, standard disk assay) | Semi-synthesis | [134,135] |
| Callipeltin A (4) | Cyclic deca-depsipeptide | Callipelta sp. | HIV-1 infection inhibition (CD50 = 0.29 µg/mL, ED50 = 0.01 µg/mL), C. albicans (100 µg/disk) | Total synthesis of analogues | [136,137,138,139,140] |
| Callyaerins A (5) and B (6) | Cyclic undeca-peptides | Callyspongia aerizusa | IC90: M. tuberculosis (2 μM and 5 μM, respectively), isoniazide (0.625 μM) | Total synthesis | [141,142] |
| Celebeside A (7) | Cyclic penta-depsipeptide | Siliquaria-spongia mirabilis | IC50: Neutralized HIV-1 (1.9 µg/mL) | - | [133] |
| Cyclolithistide A (8) | Cyclic deca-despipeptide | Bacteria symbiosis Theonella swinhoei | C. albicans (20 µg/disk) | - | [143] |
| Geodiamolides A (9) and B (10) | Cyclic depsipeptides | Geodia sp. | MIC: C. albicans (31.3 µg/mL) | Total synthesis | [144,145,146] |
| Guangomides A (11) and B (12) | Cyclic tetra-depsipeptides | Unidentifiable sponge derived fungus | MIC: S. epidermidis (100 µg/mL), E. durans (100 µg/mL) | - | [147] |
| Halicylindramides A-C (13–15) | Cyclic tetra-decapeptides | Halichondria cylindruta | M. ramanniana (7.5 µg/disk) | Total synthesis and analogues | [118,148,149] |
| Homophymine A (16) | Cyclic undeca-depsipeptide | Homophymia sp. | IC50: HIV-1 infection cytoprotective (75 nM) | Semi-synthesis | [119,150,151] |
| Hymenamides A (17), B (18), C (19), and E (20) | Cyclic hepta-peptides | Hymeniacidon sp. | MIC: C. albicans (33–66 µg/mL), C. neoformans (33–133 µg/mL) | Total synthesis and analogues | [152,153,154] |
| Jasplakinolide (or jaspamide) (21) | Cyclic depsipeptide | Jaspis sp. | H. virescens (LC50 = 4 ppm), N. brasiliensis (LD50 < 1 µg/mL), C. albicans (MIC > 25 µg/mL), in vivo murine vaginal C. albicans infection (2% jasplakinolide was equivalent in efficacy to administration of miconazole nitrate at 2%) | Total synthesis and analogues | [120,121,122,155,156,157] |
| Koshikamides F (22) and H (23) | Cyclic heptadeca-peptides | Theonella swinhoei and T. cupola | IC50: HIV-1 neutralization (2.3–5.5 µM) | - | [45,123] |
| Microcionamides A (24) and B (25) | Cyclic hexapeptides | Clathria abietina | MIC: M. tuberculosis (5.7 µM) | - | [158] |
| Microsclero-dermins A–K (26–36) and anhydromicros-clerodermin C (37) | Cyclic hexapeptides | Cyanobacteria simbiosis Microsclero-derma herdmani sp. and Theonella sp. | C. albicans (2.5–100 µg/disk, standard disk assay) | Total synthesis and analogues | [159,160,161,162,163] |
| Microspinosamide (38) | Cyclic trideca-depsipeptide | Sidonops microspinosa | EC50: HIV-1 infection inhibition (0.2 µg/mL) | Semi-synthesis | [124,164] |
| Mirabamides A–H (39–46) | Cyclic glyco-depsipeptides | Siliquarias-pongia mirabilis and Stelletta clavosa | IC50: neutralized and fusion HIV-1 (40 nM–3.9 µM), B. subtilis, C. albicans (1–5 µg/disk) | Semi-synthesis | [125,126,165] |
| Nagahamide A (47) | Cyclic hexa-depsipeptide | Theonella swinhoei | E. coli or S. aureus (50 µg/disk, inhibition zone 7 mm) | Semi-synthesis | [166,167] |
| Neamphamide A (48) | Cyclic undeca-depsipeptide | Neamphius huxleyi | EC50: HIV-1 infection cytoprotective (28 nM) | - | [128] |
| Neamphamide B (49) | Cyclic undeca-depsipeptide | Neamphius sp. | MIC: M. smegmatis (1.56 µg/mL), M. bovis (6.2–12.5 µg/mL) | - | [129] |
| Neosiphoniamolide A (50) | Cyclic tetra-depsipeptide | Neosiphonia suprtes | P. oryzae (IC90 = 5 ppm) H. gramineum (MIC ≤ 2 µg/mL) | - | [168] |
| Papuamides A (51) and B (52) | Cyclic depsipeptides | Bacteria symbiosis Theonella mirabilis and Theonella swinhoei | EC50: HIV-1 infection inhibition (1–74 ng/mL) | Total synthesis and analogues | [130,165,169,170,171,172] |
| Polydiscamide A (53) | Cyclic tridecapeptide | Discodermia sp. | MIC: B. subtilis (3.1 µg/mL) | Total synthesis and analogues | [173,174] |
| Stellettapeptins A (54) and B (55) | Cyclic undecadepsi-peptides | Microorganisms symbiosis Stelletta sp. | EC50: infection of human T-lymphoblastoid cells by HIV-1RF (23 and 27 nM, respectively) | - | [175] |
| Stylissamide G (56) | Cyclic heptapeptide | Stylissa caribica | MIC: M. audouinii, T. mentagrophytes, C. albicans (6 μg/mL) | Total Synthesis | [176] |
| Theonegramide (57) | Bicyclic glycododecapeptide | Bacteria symbiosis Theonella swinhoei | C. albicans (10 µg/disk) | - | [177] |
| Theonellamide G (58) | Bicyclic glyco-depsipeptide | Bacteria symbiosis Theonella swinhoei | IC50: Wild and amphotericin B-resistant strains of C. albicans (2.0–4.49 μM), amphotericin-B (1.48 μM) | Semi-synthesis | [131,178] |
| Theonellapeptolide congeners 1 (59) and 2 (60) | Cyclic trideca-depsipeptides | Theonella sp. | MIC: S. aureus (8.0–16 µg/mL), M. luteus (8.0 µg/mL), B. subtilis (8.0–16 µg/mL), M. smegmatis (16–66 µg/mL), T. mentagrophytes (4.0–8.0 µg/mL), A. niger (8.0–66 µg/mL) | Total synthesis and analogues | [179,180] |
| Theopapuamide A-C (61–63) | Cyclic undeca-depsipeptides | Bacteria symbiosis Theonella swinhoei and Siliquarias-pongia mirabilis | Wild type and amphotericin B-resistant strains of C. albicans (1–5 µg/disk); in vitro HIV-1 infectivity assay IC50 = 0.8 μg/mL | - | [133] |
2.2. Bacteria-Produced Cyclic Peptides
| Compound | Structure | Source | Antimicrobial Activity | Synthesis | References |
|---|---|---|---|---|---|
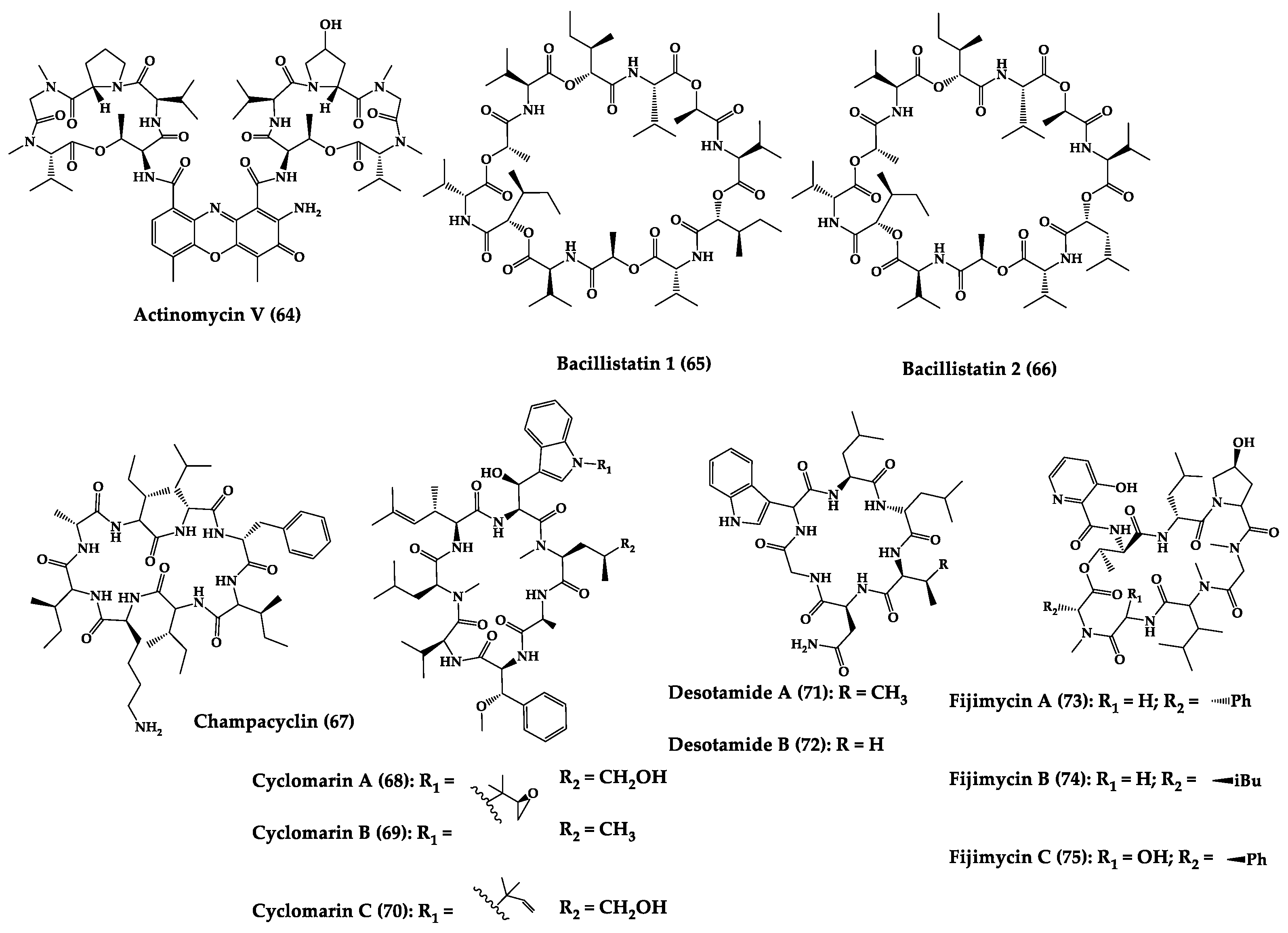
| Actinomycin V (64) | Cyclic pentapep-tide | Streptomyces sp. | MIC: MRSA (0.10–0.39 μg/mL), S. epidermidis (0.20–0.39 μg/mL), E. faecium (0.05–0.4 μg/mL), E. faecalis (0.20–0.39 μg/mL) | - | [203] |
| Bacillistatins 1 (65) and 2 (66) | Cyclic dodeca-despsipeptide | Bacillus silvestris | MIC: S. pneumoniae (1–2 μg/mL), PRSP (1 μg/mL), MDRSP (<0.5 μg/mL), S. pyogenes (1–8 μg/mL) | Total synthesis | [204,205] |
| Champacyclin (67) | Cyclic octapeptide | Streptomyces champavatii | 40% inhibition of E. amylovora at 25 μM | - | [206] |
| Cyclomarins A-C (68–70) | Cyclic heptapepti-des | Streptomyces sp. | IC50: multidrug-resistant Plasmodium falciparum strains (0.25 μM), MIC: anti-tuberculosis activity (0.1 μM) | Total synthesis and analogues | [192,207] |
| Desotamide A (71) and desotamide B (72) | Cyclic hexapep-tide | Streptomyces scopuliridis | MIC: S. pneumoniae (13 µg/mL), S. aureus (16 µg/mL), MRSE (32 µg/mL) | Total synthesis | [208,209,210] |
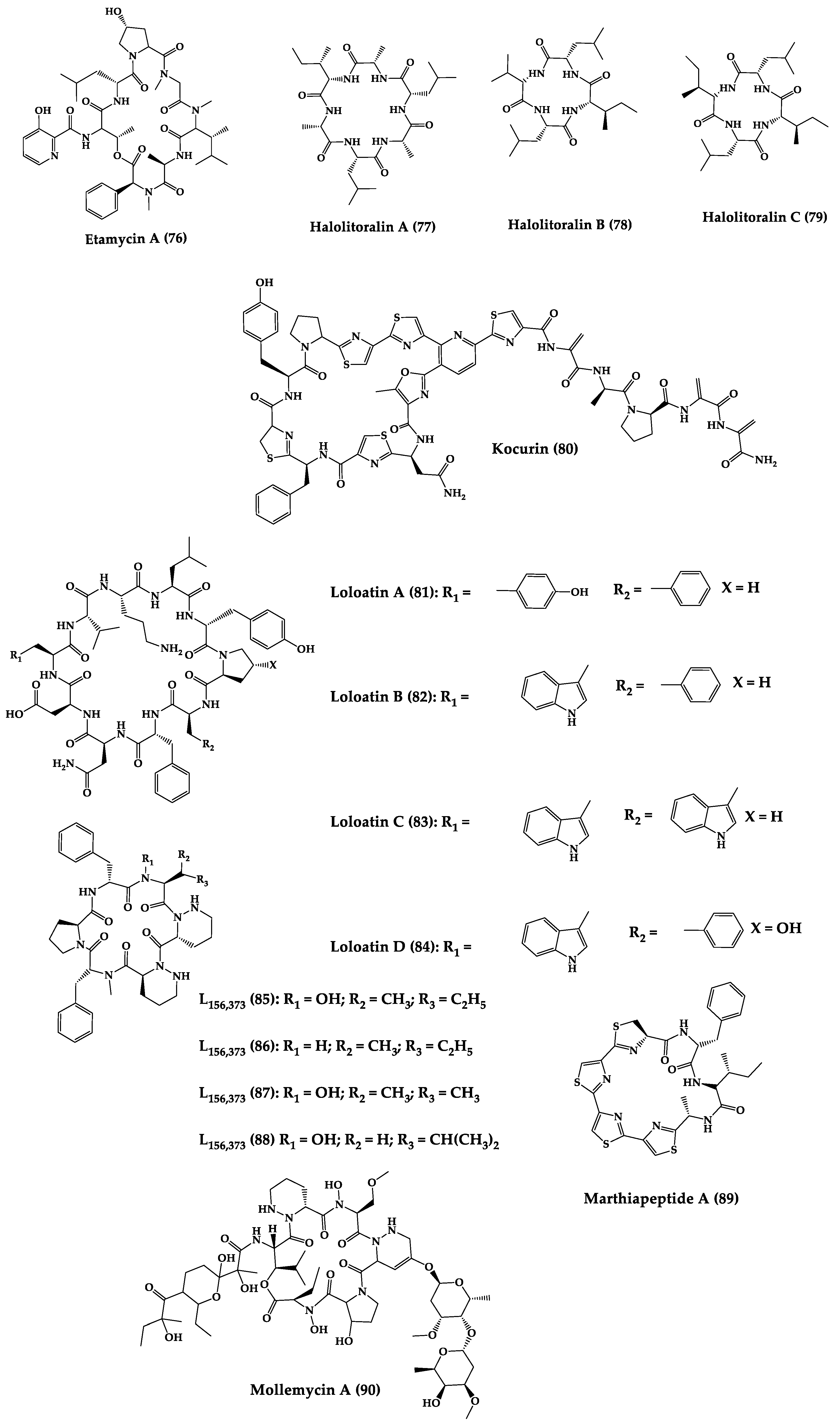
| Fijimycins A–C (73–75) and etamycin A (76) | Cyclic octadepsi-peptides | Streptomyces sp. | MIC: three MRSA strains (4–32 µg/mL) | - | [211] |
| Halolitoralin A–C (77–79) | Cyclic tetrapepti-des | Halobacillus litoralis | MIC: C. albicans (20–30 µg/mL), and T. rubrum (25–40 µg/mL) | Total synthesis | [212,213] |
| Kocurin (80) | Cyclic thiazolyl heptadecapeptide | Kocuria palustris | MIC: MRSA (0.25 μg/mL) | - | [214] |
| Loloatins A-D (81–84) | Cyclic decapepti-des | Unknown bacteria from the Great Barrier Reef in Papua New Guinea | MIC: MRSA, VRE, PRSP (0.25–8 μg/mL) | Total synthesis | [215,216] |
| L-156,373 and three derivatives (85–88) | Cyclic heptapep-tides | Streptomyces sp. | MIC: S. aureus, MRSA, B. subtilis (0.025 to 1.25 μg/mL. Control vancomycin = 0.2 μg/mL, 0.625 μg/mL, 0.2 μg/mL, for each strain respectively), Bacillus Calmette-Guérin (1.25–12.5 μg/mL, Isoniazid (0.05 μg/mL) C. albicans (12.5 μg/mL), ketoconazole (0.016 μg/mL) | Total synthesis and analogues | [217,218] |
| Marthiapeptide A (89) | Tristhiazole-thiazoline cyclic peptide | Marinactinospora thermotole-rans | MIC: panel of Gram-positive bacteria (2.0–8.0 μg/mL) | Total synthesis | [219,220] |
| Mollemycin A (90) | Cyclic glycohexadepsipeptide-polyketide | Streptomyces sp. | IC50: S. aureus (10–50 nM), S. epidermidis (50 nM), and B. subtilis (10 nM), E. coli (10 nM), P. aeruginosa (50 nM), M. bovis (3.2 μM), antimalarial properties against drug sensitive strains (9 nM), MRPFC (7 nM) | - | [221] |
| Nocathiacins I (91), II (92), and III (93) | Cyclic thiazolyl peptides | Nocardia sp. or the fungi Amicolaptosis sp. | MIC: MRSA, MREF, FPRSP (0.01–0.1 μg/mL), vancomycin (0.25–4.0 μg/mL), in vivo efficacy of a systemic S. aureus infection mice model (PD50 = 0.62–0.89 mg/kg/day) | Semi-synthesis and analogues | [186,188,222,223,224,225,226] |
| Ohmyungsamycins A (94) and B (95) | Cyclic dodecapep-tides | Streptomyces sp. | MIC: Gram-positive and Gram-negative bacteria (8.50–34.0 μM) | Total synthesis | [227,228] |
| Pedein A (96) | Cyclic hexapeptide | Chondromyces pediculatus | MIC: R. glutinis (0.6 µg/mL), S. cerevisiae, C. albicans (1.6 µg/mL), and U. maydis (3.1 µg/mL) | - | [229] |
| Rhodopeptin C1 (97), C2 (98), C3 (99), C4 (100), and B5 (101) | Cyclic lipotetra-peptides | Rhodococcus sp. | MIC: C. albicans (1.25–5 µg/mL) and C. neoformans (0.63–1.25 µg/mL) | Total synthesis and analogues | [189,190,230] |
| Rufomycins A (102), B (103) and NBZ8 (104) | Cyclic heptapepti-des | Streptomyces sp. | MIC: M. smegmatis (0.2–5 μg/mL), M. tuberculosis (0.1–5 μg/mL), no toxicity by intraperitoneal injection 102 | Total synthesis and analogues | [192,193,194,195,196,197,198] |
| Salinamides A (105), B (106), and F (107) | Bicyclic polidepsi-peptides | Streptomyces sp. | MIC: S. pneumoniae, S. pyogenes (2–4 µg/mL, 105 and 106) S. aureus (4 μM), MIC for compound 107: E. faecalis (12.5 μg/mL), H. influenzae (12.5 μg/mL), N. gonorrhoeae (25 μg/mL), E. cloacae (50 μg/mL), and E. coli (0.20 μg/mL) | Total synthesis | [231,232,233] |
| Streptocidins C (108) and D (109) | Cyclic homodeca-peptide | Streptomyces sp. | MIC: B. subtilis (3 µg/mL), S. aureus (3–10 µg/mL), S. viridochromogenes (1–3 µg/mL), and Streptomyces (3–10 µg/mL) | Total synthesis | [234,235] |
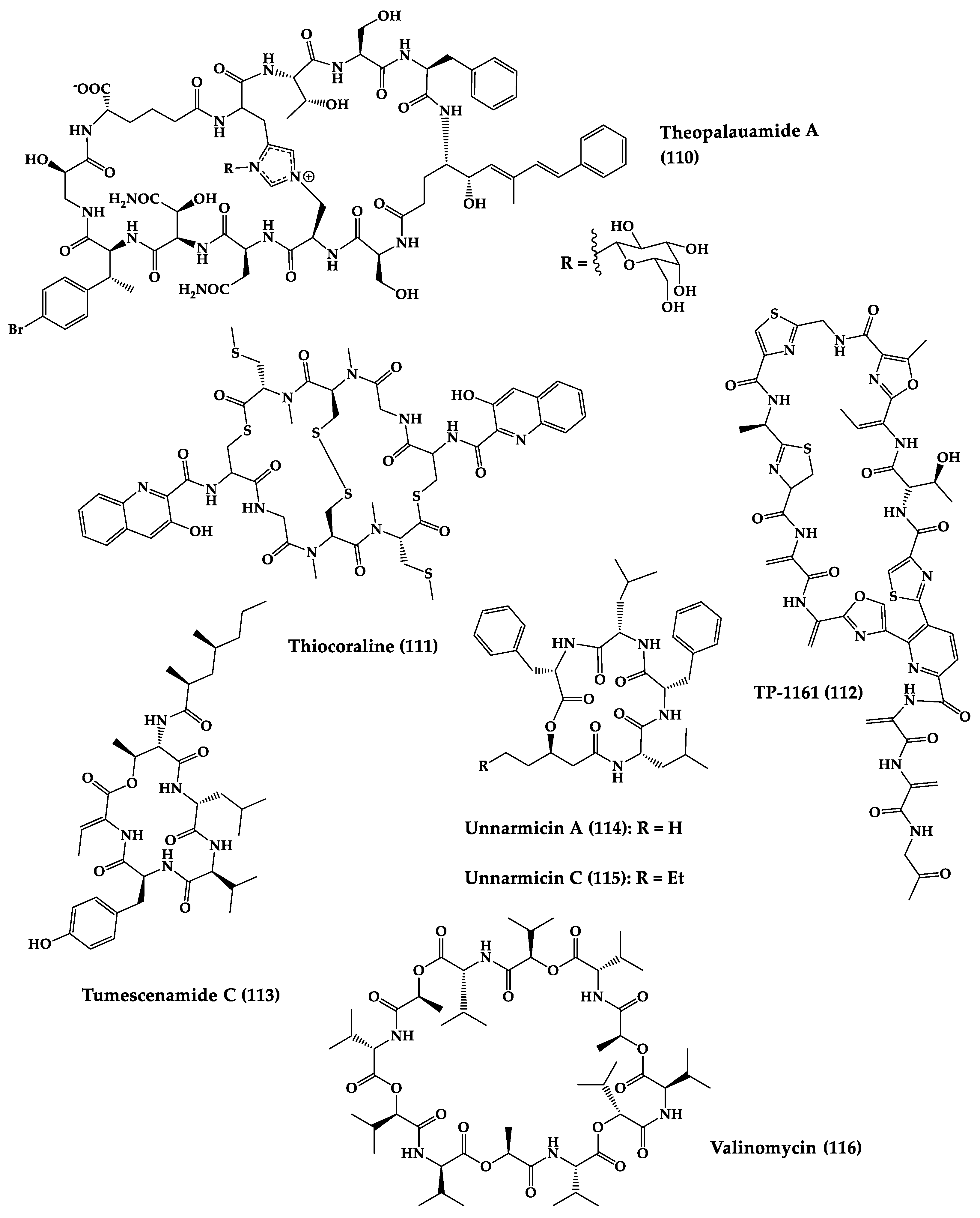
| Theopalauamide A (110) | Bicyclic glycodode-capeptide | Eubacteria symbiosis sponge Theonella swinhoei | C. albicans (10 µg/disk) | - | [236] |
| Thiocoraline (111) | Bicyclic octadepsipeptide | Actinomycete | MIC: S. aureus (0.05 µg/mL), B. subtills (0.05 µg/mL), M. luteus (0.03 µg/mL). | Total synthesis and analogues | [237,238,239] |
| TP-1161 (112) | Cyclic thiopeptide | Nocardiopsis sp. | MIC: S. aureus (0.5–32 μg/mL), S. haemolyticus (0.5–1 μg/mL), S. epidermidis (0.5–4 μg/mL), E. faecalis (1 μg/mL), E. faecium (0.5 μg/mL), VREF (1 μg/mL), S. pneumoniae (0.5 μg/mL), S. agalactiae (0.5 μg/mL) | - | [240] |
| Tumescenamide C (113) | Cyclic lipopenta- depsipeptide | Streptomyces sp. | S. coelicolor, S. lividans (inhibition zone 3.0 mg/paper disk) | Total synthesis and analogues | [241,242] |
| Unnarmicin A (114) and C (115) | Cyclic tetradepsi-peptides | Photobacte-rium sp. | IC50: Fluconazole-resistant C. albicans isolates (0.495–0.688 μM) | Total synthesis of analogue | [201] |
| Valinomycin (116) | Cyclic dodecadep-sipeptide | Streptomyces sp. | IC50: T. brucei (0.0032 μM) and L. major (<0.11 μM) | Total synthesis and analogues | [202,243,244,245] |
2.3. Cyanobacteria-Produced Cyclic Peptides
| Compound | Structure | Source | Antimicrobial Activity | Synthesis | References |
|---|---|---|---|---|---|
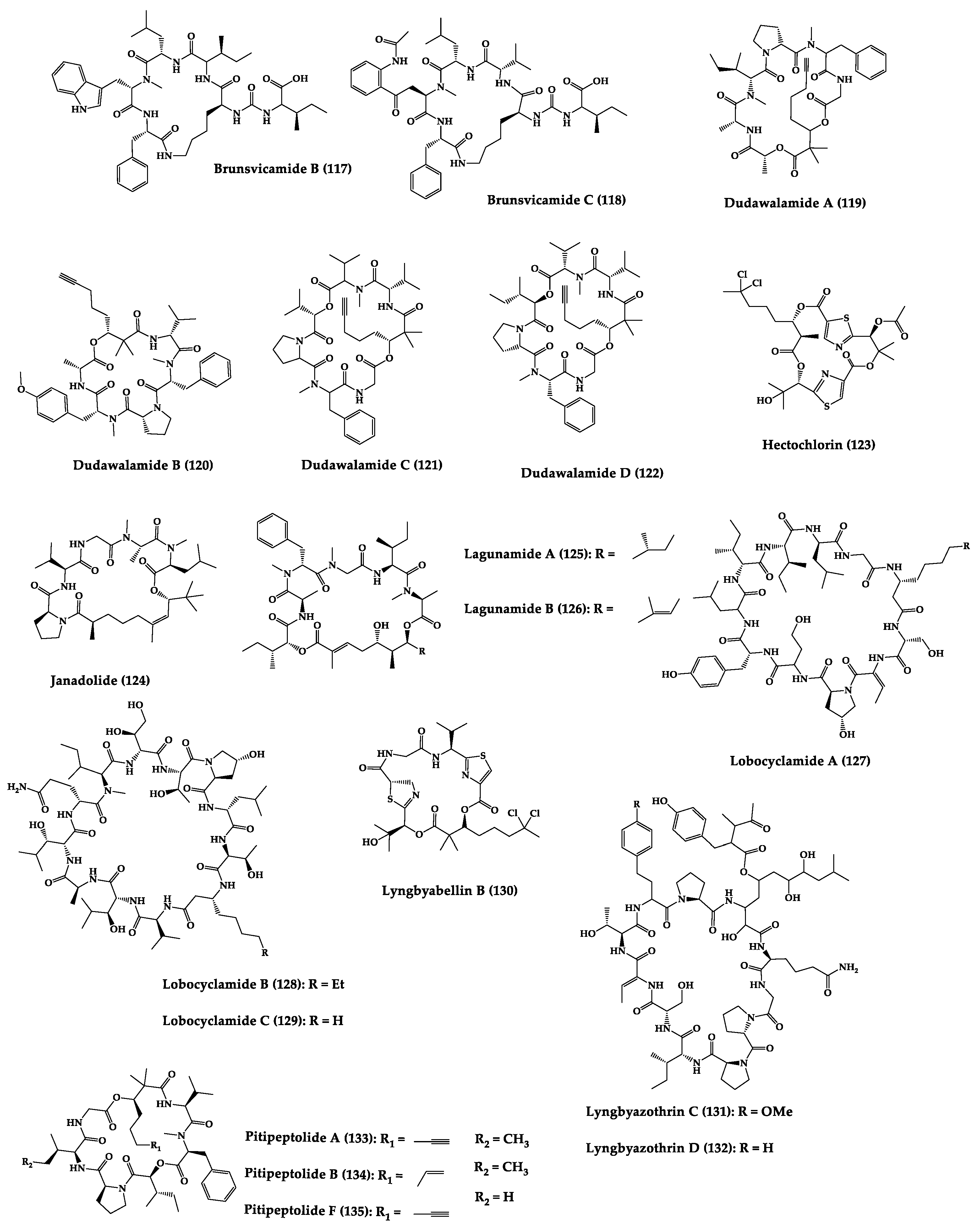
| Brunsvica-mide B (117) and C (118) | Cyclic hexapep-tides | Sponges symbiosis Tychonema sp. | IC50: M. tuberculosis protein tyrosine phosphatase B (7.3–8.0 µM) | Total synthesis of analogues | [254,255,256,257] |
| Dudawala-mides A-D (119–122) | Cyclic depsipep-tides | Lyngbya sp. | IC50: P. falciparum (2.7–7.7 μM), L. donovani (2.6–25.9 μM), and 116 against T. cruzi (7.3 μM) | - | [249] |
| Hectochlorin (123) | Cyclic depsipep-tide | Lyngbya majuscula | C. albicans (10 µg/disk: 11 mm) | Total synthesis | [258,259] |
| Janadolide (124) | Cyclic polyketi-depeptide hybrid | Okeania sp. | IC50: Antitrypanosomal activity (47 nM) | Total synthesis | [250,260] |
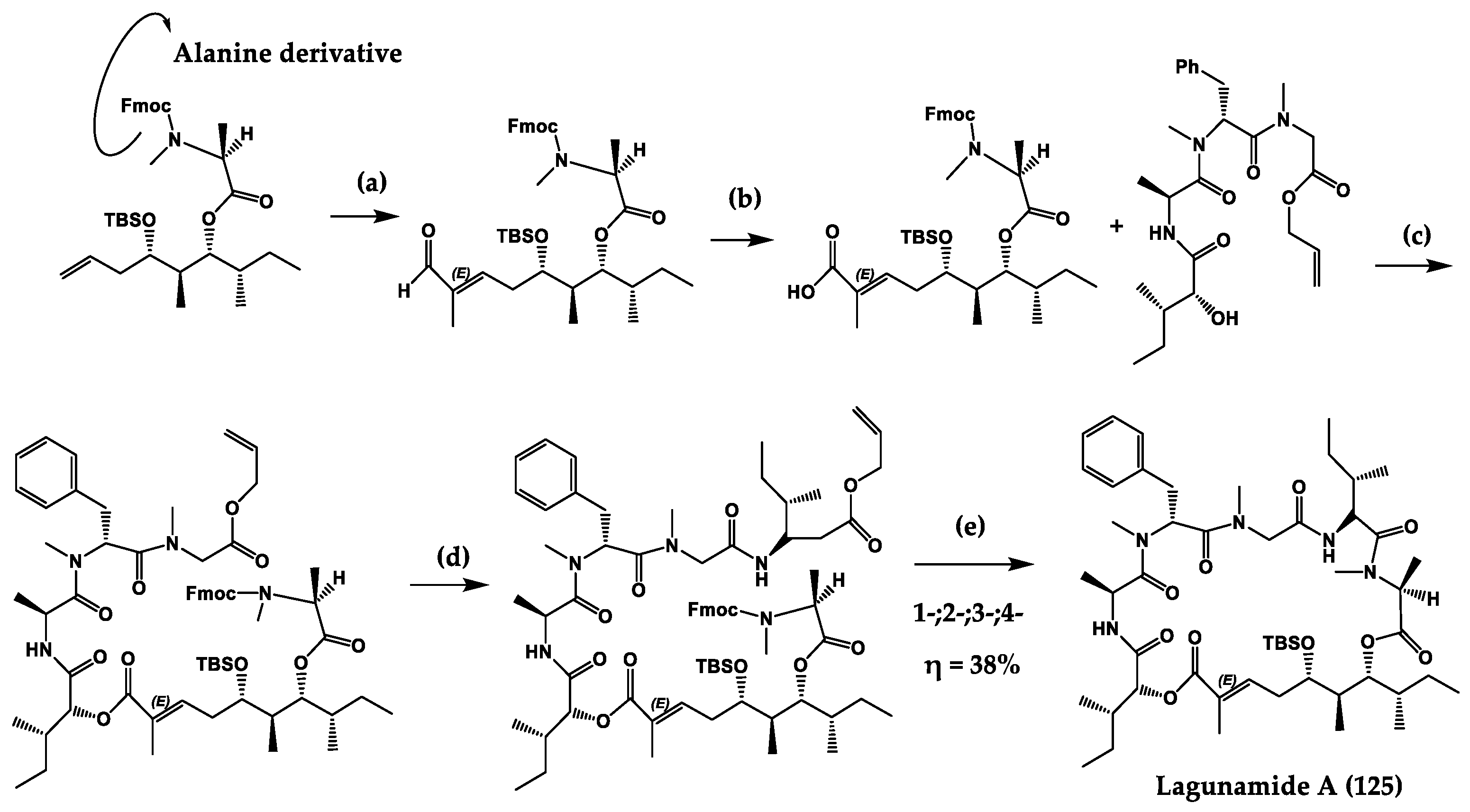
| Lagunamides A (125) and B (126) | Cyclic penta-depsipep-tides | Lyngbya majuscula | IC50: P. falciparum (0.19–0.91 µM), P. aeruginosa (antiswarming activity at 100 ppm, exerted 62% for 119 and 56% for 120) | Total synthesis and analogues | [251,261,262,263,264,265] |
| Lobocycla-mides A-C (127–129) | Cyclic dodeca-peptide | Sponges symbiosis Lyngbya confervoides | Antifungal activity: FRFCA (150 µg/disk: 121 = 7 mm inhibition zone diameters; 122 = 8 mm; 121 = 10 mm) and C. glabrata (150 µg/disk: 122 = 6 mm; 123 = 8 mm) | - | [266,267] |
| Lyngbya- bellin B (130) | Cyclic hexa- depsipeptide | Sponges symbiosis Lyngbya majuscula | C. albicans (100 µg/disk: 10.5 mm) | Total synthesis and analogues | [268,269,270,271] |
| Lyngbyazo-thrins C (131) and D (132) | Cyclic undeca-peptides | Sponges symbiosis Lyngbya sp. | B. subtilis (25 µg/disk: 18 mm), E. coli (100 µg/disk: 15 mm), P. aeruginosa (100 µg/disk: 8 mm), S. marcescens (200 µg/disk: 8 mm) | - | [272] |
| Pitipeptolides A (133), B (134) and F (135) | Cyclic hexa-depsipeptides | Sponges symbiosis Lyngbya majuscula | M. tuberculosis (10 µg/disk: 9–14 mm), streptomycin (10 µg/disk: 40 mm) | Semi-synthesis | [252,253,273,274] |
| Symplocamide A (136) | Cyclic lipodepsi-peptide | Symploca sp. | IC50: P. falciparum (0.95 µM), T. cruzi (>9.5 µM), L. donovani (>9.5 µM) | Total synthesis | [275,276] |
| Tolybyssidin A (137) | Cyclic trideca-peptides | Tolypothrix byssoidea | MIC: C. albicans (32 µg/mL), miconazole (8 µg/mL) | - | [277] |
| Venturamides A (138) and B (139) | Cyclic hexa-peptides | Sponges symbiosis Oscillatoria sp. | IC50: P. falciparum (5.6–8.2 µM), T. cruzi (14.6–15.8 µM), L. donovani (>19–20 µM) | Total synthesis | [278,279] |
2.4. Fungi-Produced Cyclic Peptides
| Compound | Structure | Source | Antimicrobial Activity | Synthesis | References |
|---|---|---|---|---|---|
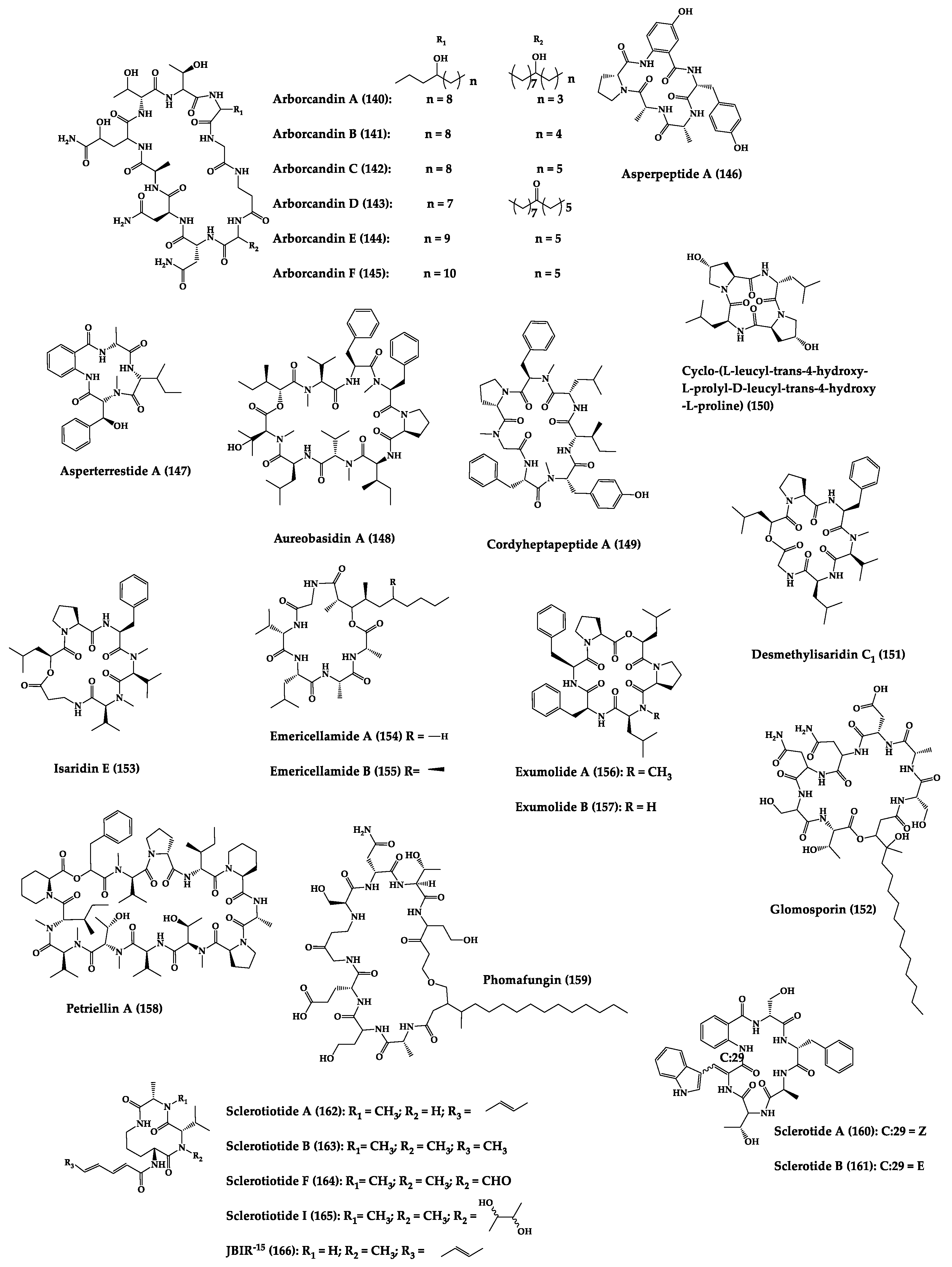
| Arborcandins A–F (140–145) | Cyclic lipopentapep-tides | Unknown filamentous fungi | MIC: Candida spp. (0.25–8 µg/mL), A. fumigatus (0.063–4 µg/mL) | - | [283] |
| Asperpeptide A (146) | Cyclic pentapeptide | Aspergillus sp. | MIC: B. cereus, S. epidermidis (12.5 μM) | - | [287] |
| Asperterrestide A (147) | Cyclic tetrapeptide | Aspergillus terreus | IC50: H1N1, H3N2 influenza strains (8.1–15 μM), ribavirin (0.41–20.2 μM) | Total synthesis | [288,289] |
| Aureobasidin A (148) | Cyclic octadepsipep-tide | Aureobasidium pullulans | MIC: C. albicans (0.05 µg/mL) and C. neoformans (0.78 µg/mL) | Total synthesis and analogues | [284,285,290,291] |
| Cordyhep- tapeptide A (149) | Cyclic heptapeptide | Cordyceps sp. | IC50: Antimalarial activity (3.8 μM) | Total synthesis | [292,293,294] |
| Cyclo-(L-leucyl-trans-4-hydroxy-L-prolyl-D-leucyl-trans-4- hydroxy-L-proline) (150) | Cyclic tetrapeptide | Phomopsis sp. and Alternaria sp. | MIC: G. graminis (220 µg/mL), R. cerealis (160 µg/mL), H. sativum (130 µg/mL), F. graminearum (250 µg/mL) | - | [295] |
| Desmethyl- isaridin C1 (151) and isaridin E (152) | Cyclic hexadepsipeptides | Bryozoan-derived fungus Beauveria felina | E. coli (MIC = 8–16 µg/mL) | - | [296] |
| Emericellamides A (153) and B (154) | Cyclic pentadepsi-peptide | Emericella sp. | MIC: MRSA (3.8 and 6.0 µM, respectively) | Total synthesis | [297,298,299,300] |
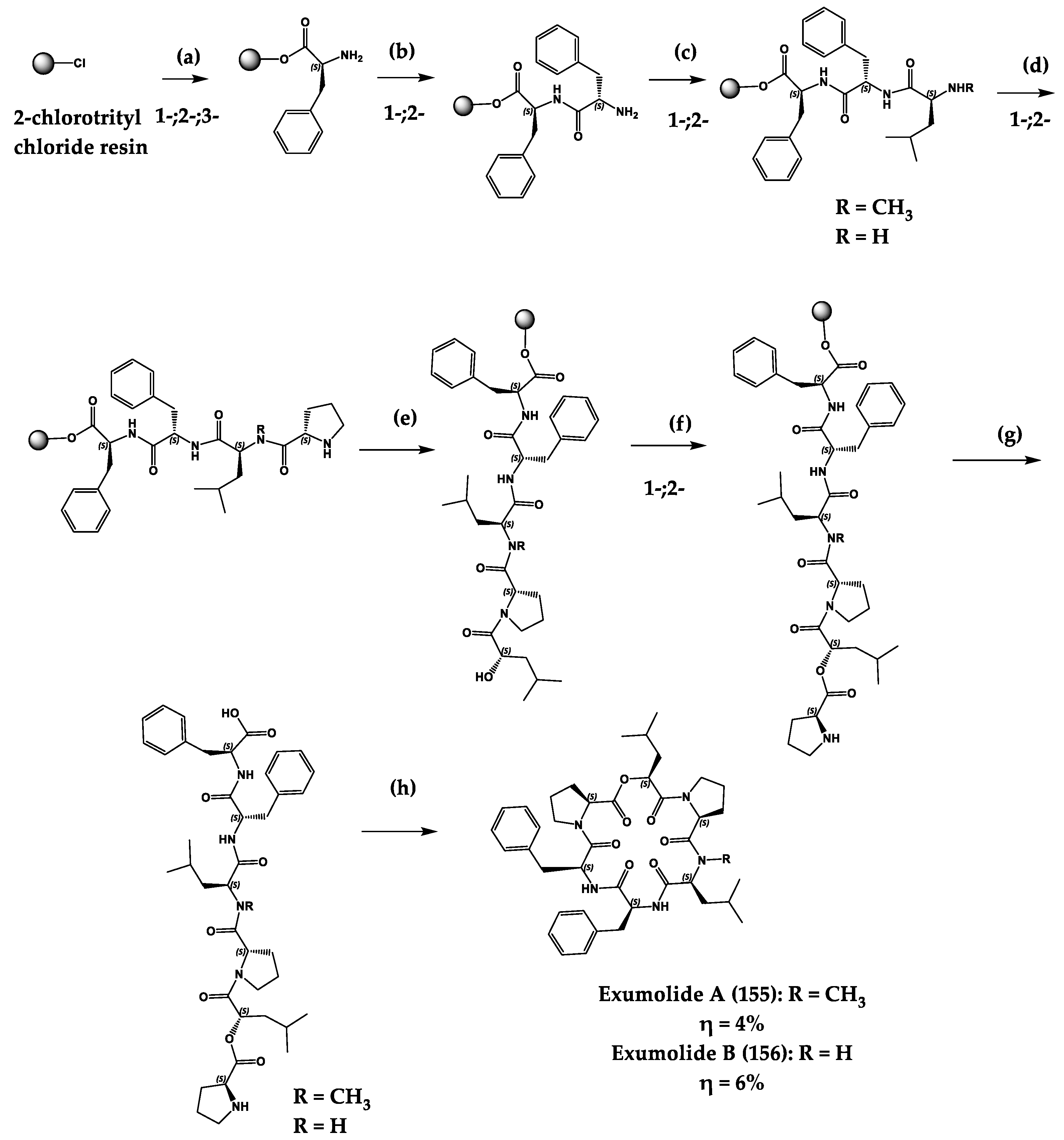
| Exumolides A (155) and B (156) | Cyclic hexadepsipeptides | Scytalidium sp. | Antimicroalgal activity against chlorophyte Dunaliella (reduction in growth of 27–33% at 20 µg/mL) | Total synthesis | [301,302] |
| Glomosporin (157) | Cyclic lipohepta-depsipeptide | Glomospora sp. | MIC: A. fumigatus (16 µg/mL) | - | [303] |
| Petriellin A (158) | Cyclic dodecadepsi-peptide | Petriella sordida | MIC: A. furfuraceus (5 µg/mL), S. fimicola (52 µg/mL) | Total synthesis | [304,305] |
| Phomafungin (159) | Cyclic lipoocta-depsipeptide | Phoma sp. | MIC: Candida spp., A. fumigatus, T. mentagrophytes (2–8 µg/mL) | - | [286] |
| Sclerotides A (160) and B (161) | Cyclic hexapeptides | Aspergillus sclerotiorum | MIC: C. albicans (7.0 µM and 3.5 µM, respectively), P. aeruginosa (35.3 µM for 156) | Total synthesis | [306,307] |
| Sclerotiotides A (162), B (163), F (164), I (165) and JBIR-15 (166) | Cyclic tripeptides | Aspergillus sclerotiorum | MIC: C. albicans (3.8–30 µM) | - | [308] |
2.5. Other Marine Invertebrate-Produced Cyclic Peptides
| Compound | Structure | Source | Antimicrobial Activity | Synthesis | References |
|---|---|---|---|---|---|
| Kahalalides A (167), E (168), F (169), and R1 (170) | Cyclic depsipep-tides | Green alga metabolites Sacoglossan mollusk Elysia rufescens | M. tuberculosis (inhibited 83% at 12.5 μg/mL), C. albicans (IC50 = 3.02 μM), C. neoformans (IC50 = 1.53 μM), A. fumigatus (IC50 = 3.21 μM), C. herbarum and C. cucumerinum at 5 μg/disk with inhibition zones of 17 and 24 mm, respectively), L. donovani promastigote (IC50 = 13 μM), L. pifanoi promastigote (IC50 = 13 μM), L. pifanoi amastigotes (IC50 = 29.53 μM) | Total synthesis and analogues | [316,317,318,319,320,322,323,324,325] |
| Mollamide B (171) | Cyclic hexapep-tide | Tunicate Didemnum mole | IC50: P. falciparum clones (2.0–2.1 µg/mL), IC90: L. donovani (18 and 35 µg/mL, respectively), EC50: HIV-1 in human peripheral blood mononuclear cells (48.7 µM) | Total synthesis of analogues | [326] |
| Peptidolipins B (172) and C (173) | Cyclic lipo- heptapeptide | Marine Nocardia sp. cultivated from ascidian Trididemnum orbiculatum | MSSA, MRSA (MIC > 64 μg/mL) | - | [327] |
| Plitidepsin (174) | Cyclic depsipep-tide | tunicate Aplidium albicans | SARS-CoV-2 in human cell line (IC50 = 0.73, CC50 = 200 nM) and in pneumocyte-like cells (IC50 = 1.62, CC50 = 65.43 nM) | Total synthesis and analogues | [321,328,329] |
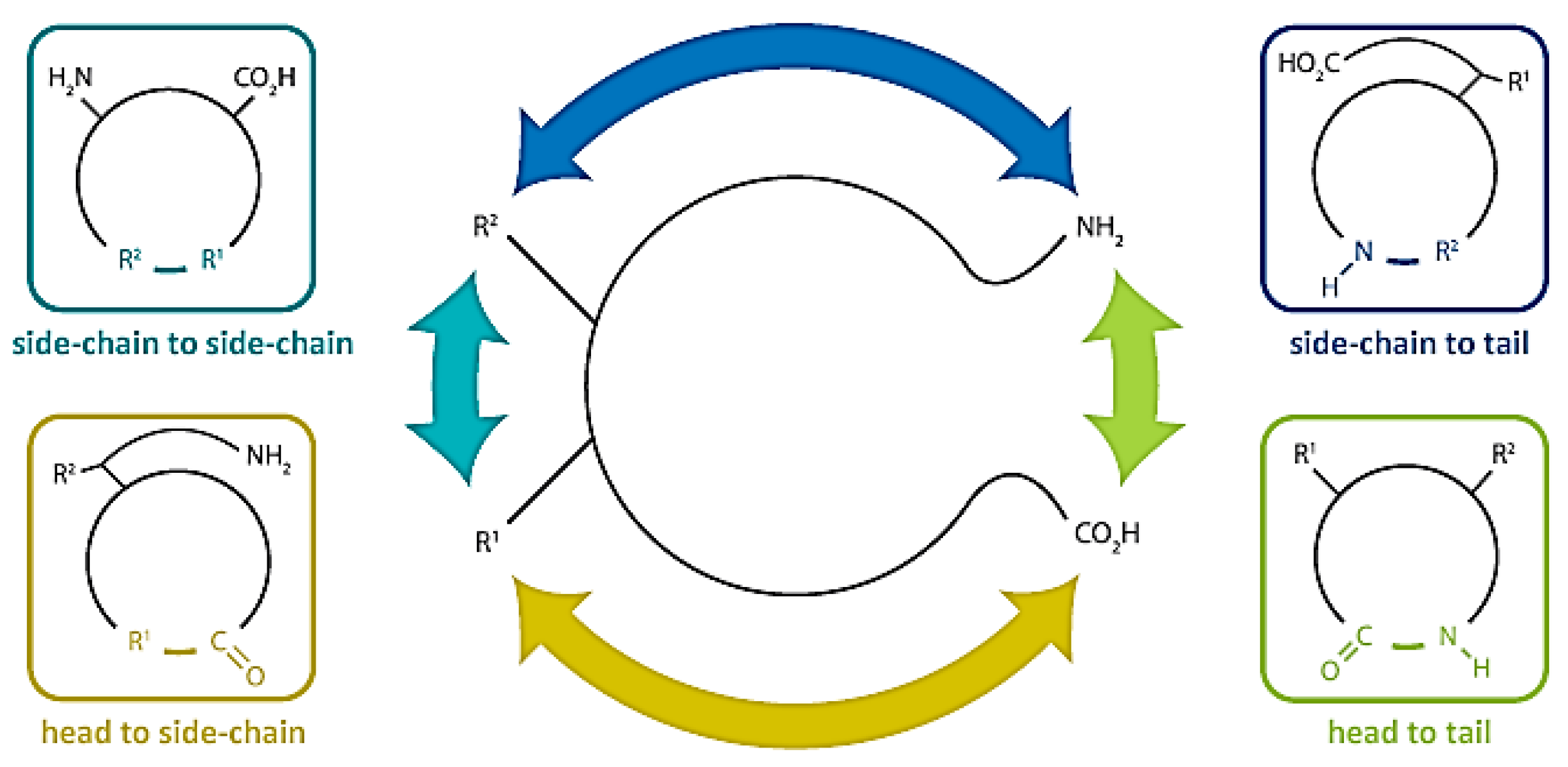
3. Synthetic Methods to Obtain Cyclic Peptides
Total Synthesis of Natural Cyclic Peptides
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Loretz, B.; Oh, Y.-K.; Hudson, S.; Gu, Z.; Lehr, C.-M. Drug delivery for fighting infectious diseases: A global perspective. Drug Deliv. Transl. Res. 2021, 11, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- WHO. Antimicrobial Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 12 April 2021).
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. Available online: https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(21)02724-0/fulltext (accessed on 12 April 2021). [CrossRef]
- Livermore, D.M.; British Society for Antimicrobial Chemotherapy Working Party on The Urgent Need: Regenerating Antibacterial Drug Discovery and Development; Blaser, M.; Carrs, O.; Cassell, G.; Fishman, N.; Guidos, R.; Levy, S.; Powers, J.; Norrby, R.; et al. Discovery research: The scientific challenge of finding new antibiotics. J. Antimicrob. Chemother. 2011, 66, 1941–1944. [Google Scholar] [CrossRef] [PubMed]
- Amyes, S.G. Magic Bullets, Lost Horizons: The Rise and Fall of Antibiotics, 1st ed.; CRC Press: London, UK, 2001; p. 272. [Google Scholar]
- Walsh, C.T.; Wencewicz, T.A. Prospects for new antibiotics: A molecule-centered perspective. J. Antibiot. 2014, 67, 7–22. [Google Scholar] [CrossRef]
- Sun, C.; Hunt, D.K.; Clark, R.B.; Lofland, D.; O’Brien, W.J.; Plamondon, L.; Xiao, X.-Y. Synthesis and antibacterial activity of pentacyclines: A novel class of tetracycline analogs. J. Med. Chem. 2011, 54, 3704–3731. [Google Scholar] [CrossRef]
- Campoy, S.; Adrio, J.L. Antifungals. Biochem. Pharmacol. 2017, 133, 86–96. [Google Scholar] [CrossRef]
- Thompson, G.R.; Cadena, J.; Patterson, T.F. Overview of antifungal agents. Clin. Chest Med. 2009, 30, 203–215. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Diekema, D.J. Epidemiology of invasive candidiasis: A persistent public health problem. Clin. Microbiol. Rev. 2007, 20, 133–163. [Google Scholar] [CrossRef]
- Caston-Osorio, J.; Rivero, A.; Torre-Cisneros, J. Epidemiology of invasive fungal infection. Int. J. Antimicrob. Agents 2008, 32, S103–S109. [Google Scholar] [CrossRef]
- Sobel, J.D.; Nyirjesy, P. Oteseconazole: An advance in treatment of recurrent vulvovaginal candidiasis. Future Microbiol. 2021, 16, 1453–1461. [Google Scholar] [CrossRef]
- De Clercq, E. Recent highlights in the development of new antiviral drugs. Curr. Opin. Microbiol. 2005, 8, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Woolhouse, M.; Scott, F.; Hudson, Z.; Howey, R.; Chase-Topping, M. Human viruses: Discovery and emergence. Philos. Trans. R. Soc. B 2012, 367, 2864–2871. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Li, G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.T.; Leung, K.; Leung, G.M. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: A modelling study. Lancet 2020, 395, 689–697. [Google Scholar] [CrossRef]
- Nash, T.E. Parasitic Diseases that Cause Seizures: Parasitic Diseases that Cause Seizures. Epilepsy Curr. 2014, 14, 29–34. [Google Scholar] [CrossRef]
- Renslo, A.R.; McKerrow, J.H. Drug discovery and development for neglected parasitic diseases. Nat. Chem. Biol. 2006, 2, 701–710. [Google Scholar] [CrossRef]
- Pozio, E. World distribution of Trichinella spp. infections in animals and humans. Vet. Parasitol. 2007, 149, 3–21. [Google Scholar] [CrossRef]
- Garcia, H.H.; Gonzalez, A.E.; Gilman, R.H. Diagnosis, treatment and control of Taenia solium cysticercosis. Curr. Opin. Infect. Dis. 2003, 16, 411–419. [Google Scholar] [CrossRef]
- Pink, R.; Hudson, A.; Mouriès, M.-A.; Bendig, M. Opportunities and challenges in antiparasitic drug discovery. Nat. Rev. Drug Discov. 2005, 4, 727–740. [Google Scholar] [CrossRef]
- Watkins, B.M. Drugs for the control of parasitic diseases: Current status and development. Trends Parasitol. 2003, 19, 477–478. [Google Scholar] [CrossRef]
- Upcroft, J.A.; Dunn, L.A.; Wal, T.; Tabrizi, S.; Delgadillo-Correa, M.G.; Johnson, P.J.; Garland, S.; Siba, P.; Upcroft, P. Metronidazole resistance in Trichomonas vaginalis from highland women in Papua New Guinea. Sex Health 2009, 6, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Anthwal, A.; Rajesh, U.C.; Rawat, M.; Kushwaha, B.; Maikhuri, J.P.; Sharma, V.L.; Gupta, G.; Rawat, D.S. Novel metronidazole–chalcone conjugates with potential to counter drug resistance in Trichomonas vaginalis. Eur. J. Med. Chem. 2014, 79, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, P.; Cooper, B.S.; Coast, J.; Oppong, R.; Thuy, N.D.T.; Phodha, T.; Celhay, O.; Guerin, P.J.; Wertheim, H.; Lubell, Y. Enumerating the economic cost of antimicrobial resistance per antibiotic consumed to inform the evaluation of interventions affecting their use. Antimicrob. Resist. Infect. Control. 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Groseclose, S.L.; Buckeridge, D.L. Public health surveillance systems: Recent advances in their use and evaluation. Annu. Rev. Public Health 2017, 38, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef]
- Laxminarayan, R.; Matsoso, P.; Pant, S.; Brower, C.; Røttingen, J.-A.; Klugman, K.; Davies, S. Access to effective antimicrobials: A worldwide challenge. Lancet 2016, 387, 168–175. [Google Scholar] [CrossRef]
- Martens, E.; Demain, A.L. The antibiotic resistance crisis, with a focus on the United States. J. Antibiot. 2017, 70, 520–526. [Google Scholar] [CrossRef]
- Andersson, D.I.; Hughes, D.; Kubicek-Sutherland, J.Z. Mechanisms and consequences of bacterial resistance to antimicrobial peptides. Drug Resist. Updates 2016, 26, 43–57. [Google Scholar] [CrossRef]
- Anjum, K.; Abbas, S.Q.; Shah, S.A.A.; Akhter, N.; Batool, S.; ul Hassan, S.S. Marine sponges as a drug treasure. Biomol. Ther. 2016, 24, 347. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.; Rodríguez, A.D.; Taglialatela-Scafati, O.; Fusetani, N. Marine pharmacology in 2009–2011: Marine compounds with antibacterial, antidiabetic, antifungal, anti-inflammatory, antiprotozoal, antituberculosis, and antiviral activities; affecting the immune and nervous systems, and other miscellaneous mechanisms of action. Mar. Drugs 2013, 11, 2510–2573. [Google Scholar] [PubMed]
- Cheung, R.C.F.; Ng, T.B.; Wong, J.H. Marine peptides: Bioactivities and applications. Mar. Drugs 2015, 13, 4006–4043. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.; Rodríguez, A.D.; Berlinck, R.G.; Fusetani, N. Marine pharmacology in 2007–8: Marine compounds with antibacterial, anticoagulant, antifungal, anti-inflammatory, antimalarial, antiprotozoal, antituberculosis, and antiviral activities; affecting the immune and nervous system, and other miscellaneous mechanisms of action. Comp. Biochem. Physiol. C 2011, 153, 191–222. [Google Scholar]
- Aneiros, A.; Garateix, A. Bioactive peptides from marine sources: Pharmacological properties and isolation procedures. J. Chromatogr. B 2004, 803, 41–53. [Google Scholar] [CrossRef]
- Donia, M.; Hamann, M.T. Marine natural products and their potential applications as anti-infective agents. Lancet Infect. Dis. 2003, 3, 338–348. [Google Scholar] [CrossRef]
- Koslow, J.A. The silent deep: The discovery, ecology, and conservation of the deep sea. Oceanography 2007, 23, 228. [Google Scholar]
- Russo, P.; Del Bufalo, A.; Fini, M. Deep sea as a source of novel-anticancer drugs: Update on discovery and preclinical/clinical evaluation in a systems medicine perspective. EXCLI J. 2015, 14, 228. [Google Scholar]
- Xu, L.; Meng, W.; Cao, C.; Wang, J.; Shan, W.; Wang, Q. Antibacterial and antifungal compounds from marine fungi. Mar. Drugs 2015, 13, 3479–3513. [Google Scholar] [CrossRef]
- Alves, A.; Sousa, E.; Kijjoa, A.; Pinto, M. Marine-derived compounds with potential use as cosmeceuticals and nutricosmetics. Molecules 2020, 25, 2536. [Google Scholar] [CrossRef]
- Mehbub, M.F.; Lei, J.; Franco, C.; Zhang, W. Marine sponge derived natural products between 2001 and 2010: Trends and opportunities for discovery of bioactives. Mar. Drugs 2014, 12, 4539–4577. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, I.; Kim, S.-K. Immense essence of excellence: Marine microbial bioactive compounds. Mar. Drugs 2010, 8, 2673–2701. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, J.; Hu, G.; Yu, J.; Zhu, X.; Lin, Y.; Chen, S.; Yuan, J. Statistical research on the bioactivity of new marine natural products discovered during the 28 years from 1985 to 2012. Mar. Drugs 2015, 13, 202–221. [Google Scholar] [CrossRef] [PubMed]
- Winder, P.L.; Pomponi, S.A.; Wright, A.E. Natural Products from the Lithistida: A Review of the Literature since 2000. Mar. Drugs 2011, 9, 2643–2682. [Google Scholar] [CrossRef]
- Duray, H.; Hatfill, J.; Pellis, R. Venom peptides as human pharmaceuticals. Sci. Med. 1997, 4, 6–15. [Google Scholar]
- Wang, X.; Gong, X.; Li, P.; Lai, D.; Zhou, L. Structural diversity and biological activities of cyclic depsipeptides from fungi. Molecules 2018, 23, 169. [Google Scholar] [CrossRef]
- May Zin, W.W.; Buttachon, S.; Dethoup, T.; Fernandes, C.; Cravo, S.; Pinto, M.M.; Gales, L.; Pereira, J.A.; Silva, A.; Sekeroglu, N. New cyclotetrapeptides and a new diketopiperzine derivative from the marine sponge-associated fungus Neosartorya glabra KUFA 0702. Mar. Drugs 2016, 14, 136. [Google Scholar] [CrossRef]
- Prompanya, C.; Fernandes, C.; Cravo, S.; Pinto, M.M.; Dethoup, T.; Silva, A.; Kijjoa, A. A new cyclic hexapeptide and a new isocoumarin derivative from the marine sponge-associated fungus Aspergillus similanensis KUFA 0013. Mar. Drugs 2015, 13, 1432–1450. [Google Scholar] [CrossRef]
- Cooper, B.M.; Iegre, J.; O’Donovan, D.H.; Halvarsson, M.Ö.; Spring, D.R. Peptides as a platform for targeted therapeutics for cancer: Peptide–drug conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Zompra, A.A.; Galanis, A.S.; Werbitzky, O.; Albericio, F. Manufacturing peptides as active pharmaceutical ingredients. Future Med. Chem. 2009, 1, 361–377. [Google Scholar] [CrossRef]
- Otvos, L., Jr. Peptide-based drug design: Here and now. Methods Mol. Biol. 2008, 494, 1–8. [Google Scholar] [PubMed]
- Goodwin, D.; Simerska, P.; Toth, I. Peptides as therapeutics with enhanced bioactivity. Curr. Med. Chem. 2012, 19, 4451–4461. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, V.M.; Frank, R.; Boehnke, S.; Schütz, C.L.; Hampel, G.; Iffland, D.S.; Bings, N.H.; Hey-Hawkins, E.; Beck-Sickinger, A.G. Receptor-mediated uptake of boron-rich neuropeptide y analogues for boron neutron capture therapy. ChemMedChem 2015, 10, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M. Design and development of peptides and peptide mimetics as antagonists for therapeutic intervention. Future Med. Chem. 2010, 2, 1813–1822. [Google Scholar] [CrossRef]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical modifications designed to improve peptide stability: Incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef]
- Guharoy, M.; Chakrabarti, P. Secondary structure based analysis and classification of biological interfaces: Identification of binding motifs in protein–protein interactions. Bioinformatics 2007, 23, 1909–1918. [Google Scholar] [CrossRef]
- DeLorbe, J.E.; Clements, J.H.; Whiddon, B.B.; Martin, S.F. Thermodynamic and structural effects of macrocyclic constraints in protein–ligand interactions. ACS Med. Chem. Lett. 2010, 1, 448–452. [Google Scholar] [CrossRef]
- DeLorbe, J.E.; Clements, J.H.; Teresk, M.G.; Benfield, A.P.; Plake, H.R.; Millspaugh, L.E.; Martin, S.F. Thermodynamic and Structural Effects of Conformational Constraints in Protein–Ligand Interactions. Entropic Paradoxy Associated with Ligand Preorganization. J. Am. Chem. Soc. 2009, 131, 16758–16770. [Google Scholar] [CrossRef]
- Tapeinou, A.; Matsoukas, M.T.; Simal, C.; Tselios, T. Review cyclic peptides on a merry-go-round; towards drug design. Pept. Sci. 2015, 104, 453–461. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into protein–ligand interactions: Mechanisms, models, and methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Lee, M.S.; Gardner, B.; Kahn, M.; Nakanishi, H. The three dimensional solution structure of a constrained peptidomimetic in water and in chloroform observation of solvent induced hydrophobic cluster. FEBS Lett. 1995, 359, 113–118. [Google Scholar] [CrossRef]
- Uma, K.; Kishore, R.; Balaram, P. Stereochemical constraints in peptide design: Analysis of the influence of a disulfide bridge and an α-aminoisobutyryl residue on the conformation of a hexapeptide. Biopolymers 1993, 33, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.; Guo, L.; Liu, R.; Marik, J.; Lam, K. Peptide ligands targeting integrin α3β1 in non-small cell lung cancer. Lung Cancer 2006, 52, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Rezai, T.; Yu, B.; Millhauser, G.L.; Jacobson, M.P.; Lokey, R.S. Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers. J. Am. Chem. Soc. 2006, 128, 2510–2511. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.-U.; Kodadek, T. Quantitative comparison of the relative cell permeability of cyclic and linear peptides. Chem. Biol. 2007, 14, 671–677. [Google Scholar] [CrossRef]
- Hussack, G.; Hirama, T.; Ding, W.; MacKenzie, R.; Tanha, J. Engineered single-domain antibodies with high protease resistance and thermal stability. PLoS ONE 2011, 6, e28218. [Google Scholar] [CrossRef]
- Lindgren, M.; Hällbrink, M.; Prochiantz, A.; Langel, Ü. Cell-penetrating peptides. Trends Pharmacol. Sci. 2000, 21, 99–103. [Google Scholar] [CrossRef]
- Cini, E.; Bifulco, G.; Menchi, G.; Rodriquez, M.; Taddei, M. Synthesis of Enantiopure 7-substituted Azepane-2-carboxylic acids as templates for conformationally constrained Peptidomimetics. Eur. J. Org. Chem. 2012, 2012, 2133–2141. [Google Scholar] [CrossRef]
- Grigoryan, G.; Reinke, A.W.; Keating, A.E. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature 2009, 458, 859–864. [Google Scholar] [CrossRef]
- Grauer, A.; König, B. Peptidomimetics—A versatile route to biologically active compounds. Eur. J. Org. Chem. 2009, 2009, 5099–5111. [Google Scholar] [CrossRef]
- Mandell, D.J.; Kortemme, T. Computer-aided design of functional protein interactions. Nat. Chem. Biol. 2009, 5, 797–807. [Google Scholar] [CrossRef]
- Terrett, N. Drugs in middle space. MedChemComm 2013, 4, 474–475. [Google Scholar] [CrossRef]
- Joo, S.H. Cyclic peptides as therapeutic agents and biochemical tools. Biomol. Ther. 2012, 20, 19. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Fenner, A.M. Drug discovery from marine microbes. Microb. Ecol. 2013, 65, 800–806. [Google Scholar] [CrossRef]
- Rocha-Martin, J.; Harrington, C.; Dobson, A.D.; O’Gara, F. Emerging strategies and integrated systems microbiology technologies for biodiscovery of marine bioactive compounds. Mar. Drugs 2014, 12, 3516–3559. [Google Scholar] [CrossRef]
- Kang, H.K.; Seo, C.H.; Park, Y. Marine peptides and their anti-infective activities. Mar. Drugs 2015, 13, 618–654. [Google Scholar] [CrossRef]
- Bionda, N.; Stawikowski, M.; Stawikowska, R.; Cudic, M.; López-Vallejo, F.; Treitl, D.; Medina-Franco, J.; Cudic, P. Effects of cyclic lipodepsipeptide structural modulation on stability, antibacterial activity, and human cell toxicity. ChemMedChem 2012, 7, 871–882. [Google Scholar] [CrossRef]
- Raaijmakers, J.M.; De Bruijn, I.; Nybroe, O.; Ongena, M. Natural functions of lipopeptides from Bacillus and Pseudomonas: More than surfactants and antibiotics. FEMS Microbiol. Rev. 2010, 34, 1037–1062. [Google Scholar] [CrossRef]
- Jensen, K.J.; Brask, J. Carbohydrates in peptide and protein design. Pept. Sci. 2005, 80, 747–761. [Google Scholar] [CrossRef]
- Taevernier, L.; Wynendaele, E.; Gevaert, B.; De Spiegeleer, B. Chemical classification of cyclic depsipeptides. Curr. Protein Pept. Sci. 2017, 18, 425–452. [Google Scholar] [CrossRef]
- Moss, G.; Smith, P.; Tavernier, D. Glossary of class names of organic compounds and reactivity intermediates based on structure (IUPAC Recommendations 1995). Pure Appl. Chem. 1995, 67, 1307–1375. [Google Scholar] [CrossRef]
- Dixon, H.B.F. Nomenclature and symbolism for amino acids and peptides. Pure Appl. Chem. 1984, 56, 595–624. Available online: https://febs.onlinelibrary.wiley.com/doi/10.1111/j.1432-1033.1984.tb07877.x (accessed on 12 April 2021).
- Sieber, S.A.; Marahiel, M.A. Molecular mechanisms underlying nonribosomal peptide synthesis: Approaches to new antibiotics. Chem. Rev. 2005, 105, 715–738. [Google Scholar] [CrossRef]
- Fischbach, M.A.; Walsh, C.T. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: Logic, machinery, and mechanisms. Chem. Rev. 2006, 106, 3468–3496. [Google Scholar] [CrossRef]
- Thompson, F.T.C. Biossíntese de Metabólitos Secundários. In Biotecnologia Marinha; FURG, Ed.; PPG-Mar: Rio Grande, Mexico, 2020; pp. 91–95. [Google Scholar]
- Reimer, J.M.; Haque, A.S.; Tarry, M.J.; Schmeing, T.M. Piecing together nonribosomal peptide synthesis. Curr. Opin. Struct. Biol. 2018, 49, 104–113. [Google Scholar] [CrossRef]
- Gulick, A.M. Structural insight into the necessary conformational changes of modular nonribosomal peptide synthetases. Curr. Opin. Chem. Biol. 2016, 35, 89–96. [Google Scholar] [CrossRef]
- Schwarzer, D.; Finking, R.; Marahiel, M.A. Nonribosomal peptides: From genes to products. Nat. Prod. Rep. 2003, 20, 275–287. [Google Scholar] [CrossRef]
- Marahiel, M.A.; Stachelhaus, T.; Mootz, H.D. Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem. Rev. 1997, 97, 2651–2674. [Google Scholar] [CrossRef]
- Alonzo, D.A.; Schmeing, T.M. Biosynthesis of depsipeptides, or Depsi: The peptides with varied generations. Protein Sci. 2020, 29, 2316–2347. [Google Scholar] [CrossRef]
- Donadio, S.; Monciardini, P.; Sosio, M. Polyketide synthases and nonribosomal peptide synthetases: The emerging view from bacterial genomics. Nat. Prod. Rep. 2007, 24, 1073–1109. [Google Scholar] [CrossRef]
- Horsman, M.E.; Hari, T.P.; Boddy, C.N. Polyketide synthase and non-ribosomal peptide synthetase thioesterase selectivity: Logic gate or a victim of fate? Nat. Prod. Rep. 2016, 33, 183–202. [Google Scholar] [CrossRef]
- Wilson, D.J.; Shi, C.; Teitelbaum, A.M.; Gulick, A.M.; Aldrich, C.C. Characterization of AusA: A dimodular nonribosomal peptide synthetase responsible for the production of aureusimine pyrazinones. Biochemistry 2013, 52, 926–937. [Google Scholar] [CrossRef]
- Manavalan, B.; Murugapiran, S.K.; Lee, G.; Choi, S. Molecular modeling of the reductase domain to elucidate the reaction mechanism of reduction of peptidyl thioester into its corresponding alcohol in non-ribosomal peptide synthetases. BMC Struct. Biol. 2010, 10, 1–14. [Google Scholar] [CrossRef]
- Bloudoff, K.; Schmeing, T.M. Structural and functional aspects of the nonribosomal peptide synthetase condensation domain superfamily: Discovery, dissection and diversity. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1587–1604. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, N.; Cacho, R.A.; Gong, Z.; Liu, Z.; Qin, W.; Tang, C.; Tang, Y.; Zhou, J. Structural basis of nonribosomal peptide macrocyclization in fungi. Nat. Chem. Biol. 2016, 12, 1001–1003. [Google Scholar] [CrossRef]
- Bloudoff, K.; Fage, C.D.; Marahiel, M.A.; Schmeing, T.M. Structural and mutational analysis of the nonribosomal peptide synthetase heterocyclization domain provides insight into catalysis. Proc. Natl. Acad. Sci. USA 2017, 114, 95–100. [Google Scholar] [CrossRef]
- Yuwen, L.; Zhang, F.-L.; Chen, Q.-H.; Lin, S.-J.; Zhao, Y.-L.; Li, Z.-Y. The role of aromatic L-amino acid decarboxylase in bacillamide C biosynthesis by Bacillus atrophaeus C89. Sci. Rep. 2013, 3, 1–10. [Google Scholar] [CrossRef]
- Chang, C.-Y.; Lohman, J.R.; Huang, T.; Michalska, K.; Bigelow, L.; Rudolf, J.D.; Jedrzejczak, R.; Yan, X.; Ma, M.; Babnigg, G. Structural insights into the free-standing condensation enzyme SgcC5 catalyzing ester-bond formation in the biosynthesis of the enediyne antitumor antibiotic C-1027. Biochemistry 2018, 57, 3278–3288. [Google Scholar] [CrossRef]
- Kohli, R.M.; Walsh, C.T.; Burkart, M.D. Biomimetic synthesis and optimization of cyclic peptide antibiotics. Nature 2002, 418, 658–661. [Google Scholar] [CrossRef]
- Albericio, F. Developments in peptide and amide synthesis. Curr. Opin. Chem. Biol. 2004, 8, 211–221. [Google Scholar] [CrossRef]
- Jiang, S.; Li, Z.; Ding, K.; Roller, P.P. Recent progress of synthetic studies to peptide and peptidomimetic cyclization. Curr. Org. Chem. 2008, 12, 1502–1542. [Google Scholar] [CrossRef]
- Lambert, J.N.; Mitchell, J.P.; Roberts, K.D. The synthesis of cyclic peptides. J. Chem. Soc. Perkin Trans. 1 2001, 471–484. [Google Scholar] [CrossRef]
- Sánchez, A.; Vázquez, A. Bioactive peptides: A review. Food Qual. Saf. 2017, 1, 29–46. [Google Scholar] [CrossRef]
- Phyo, Y.Z.; Ribeiro, J.; Fernandes, C.; Kijjoa, A.; Pinto, M.M. Marine natural peptides: Determination of absolute configuration using liquid chromatography methods and evaluation of bioactivities. Molecules 2018, 23, 306. [Google Scholar] [CrossRef]
- Abdalla, M.A.; McGaw, L.J. Natural cyclic peptides as an attractive modality for therapeutics: A mini review. Molecules 2018, 23, 2080. [Google Scholar] [CrossRef]
- Zorzi, A.; Deyle, K.; Heinis, C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [Google Scholar] [CrossRef]
- Cardoso, J.; Nakayama, D.G.; Sousa, E.; Pinto, E. Marine-derived compounds and prospects for their antifungal application. Molecules 2020, 25, 5856. [Google Scholar] [CrossRef]
- Barbosa, F.; Pinto, E.; Kijjoa, A.; Pinto, M.; Sousa, E. Targeting antimicrobial drug resistance with marine natural products. Int. J. Antimicrob. Agents 2020, 56, 106005. [Google Scholar] [CrossRef]
- Anand, T.P.; Chellaram, C.; Kuberan, G.; Archana, H. Bioactive peptides from marine sources-a review. Indian J. Innov. Dev. 2012, 1, 61–64. [Google Scholar]
- Bewley, C.A.; Faulkner, D.J. Lithistid sponges: Star performers or hosts to the stars. Angew. Chem. Int. Ed. 1998, 37, 2162–2178. [Google Scholar] [CrossRef]
- Schmidt, E.; Obraztsova, A.; Davidson, S.; Faulkner, D.; Haygood, M. Identification of the antifungal peptide-containing symbiont of the marine sponge Theonella swinhoei as a novel δ-proteobacterium,“Candidatus Entotheonella palauensis”. Mar. Biol. 2000, 136, 969–977. [Google Scholar] [CrossRef]
- Amelia, T.S.M.; Suaberon, F.A.C.; Vad, J.; Fahmi, A.D.M.; Saludes, J.P.; Bhubalan, K. Recent Advances of Marine Sponge-Associated Microorganisms as a Source of Commercially Viable Natural Products. Mar. Biotechnol. 2022, 1–21. [Google Scholar] [CrossRef]
- Liu, L.; Zheng, Y.-Y.; Shao, C.-L.; Wang, C.-Y. Metabolites from marine invertebrates and their symbiotic microorganisms: Molecular diversity discovery, mining, and application. Mar. Life Sci. Technol. 2019, 1, 60–94. [Google Scholar] [CrossRef]
- Kobayashi, M.; Tanaka, J.-I.; Katori, T.; Matsuura, M.; Yamashita, M.; Kitagawa, I. Marine natural products. XXII: The absolute stereostructure of swinholide A, a potent cytotoxic dimeric macrolide from the Okinawan marine sponge Theonella swinhoei. Chem. Pharm. Bull. 1990, 38, 2409–2418. [Google Scholar] [CrossRef]
- Li, H.-Y.; Matsunaga, S.; Fusetani, N. Halicylindramides A-C, antifungal and cytotoxic depsipeptides from the marine sponge Halichondria cylindrata. J. Med. Chem. 1995, 38, 338–343. [Google Scholar] [CrossRef]
- Zampella, A.; Sepe, V.; Luciano, P.; Bellotta, F.; Monti, M.C.; D’Auria, M.V.; Jepsen, T.; Petek, S.; Adeline, M.-T.; Laprévôte, O. Homophymine A, an anti-HIV cyclodepsipeptide from the sponge Homophymia sp. J. Org. Chem. 2008, 73, 5319–5327. [Google Scholar] [CrossRef]
- Zabriskie, T.M.; Klocke, J.A.; Ireland, C.M.; Marcus, A.H.; Molinski, T.F.; Faulkner, D.J.; Xu, C.; Clardy, J. Jaspamide, a modified peptide from a Jaspis sponge, with insecticidal and antifungal activity. J. Am. Chem. Soc. 1986, 108, 3123–3124. [Google Scholar] [CrossRef]
- Scott, V.; Boehme, R.; Matthews, T. New class of antifungal agents: Jasplakinolide, a cyclodepsipeptide from the marine sponge, Jaspis species. Antimicrob. Agents Chemother. 1988, 32, 1154–1157. [Google Scholar] [CrossRef]
- Fusetani, N.; Matsunaga, S. Bioactive sponge peptides. Chem. Rev. 1993, 93, 1793–1806. [Google Scholar] [CrossRef]
- Plaza, A.; Bifulco, G.; Masullo, M.; Lloyd, J.R.; Keffer, J.L.; Colin, P.L.; Hooper, J.N.; Bell, L.J.; Bewley, C.A. Mutremdamide A and koshikamides C–H, peptide inhibitors of HIV-1 entry from different Theonella species. J. Org. Chem. 2010, 75, 4344–4355. [Google Scholar] [CrossRef]
- Rashid, M.A.; Gustafson, K.R.; Cartner, L.K.; Shigematsu, N.; Pannell, L.K.; Boyd, M.R. Microspinosamide, a New HIV-Inhibitory Cyclic Depsipeptide from the Marine Sponge Sidonops microspinosa. J. Nat. Prod. 2001, 64, 117–121. [Google Scholar] [CrossRef]
- Plaza, A.; Gustchina, E.; Baker, H.L.; Kelly, M.; Bewley, C.A. Mirabamides A–D, depsipeptides from the sponge Siliquariaspongia mirabilis that inhibit HIV-1 fusion. J. Nat. Prod. 2007, 70, 1753–1760. [Google Scholar] [CrossRef]
- Lu, Z.; Van Wagoner, R.M.; Harper, M.K.; Baker, H.L.; Hooper, J.N.; Bewley, C.A.; Ireland, C.M. Mirabamides E–H, HIV-inhibitory depsipeptides from the sponge Stelletta clavosa. J. Nat. Prod. 2011, 74, 185–193. [Google Scholar] [CrossRef]
- Gulakowski, R.J.; McMahon, J.B.; Staley, P.G.; Moran, R.A.; Boyd, M.R. A semiautomated multiparameter approach for anti-HIV drug screening. J. Virol. Methods 1991, 33, 87–100. [Google Scholar] [CrossRef]
- Oku, N.; Gustafson, K.R.; Cartner, L.K.; Wilson, J.A.; Shigematsu, N.; Hess, S.; Pannell, L.K.; Boyd, M.R.; McMahon, J.B. Neamphamide A, a new HIV-inhibitory depsipeptide from the Papua New Guinea marine sponge Neamphius huxleyi. J. Nat. Prod. 2004, 67, 1407–1411. [Google Scholar] [CrossRef]
- Yamano, Y.; Arai, M.; Kobayashi, M. Neamphamide B, new cyclic depsipeptide, as an anti-dormant mycobacterial substance from a Japanese marine sponge of Neamphius sp. Bioorg. Med. Chem. Lett. 2012, 22, 4877–4881. [Google Scholar] [CrossRef]
- Ford, P.W.; Gustafson, K.R.; McKee, T.C.; Shigematsu, N.; Maurizi, L.K.; Pannell, L.K.; Williams, D.E.; Dilip de Silva, E.; Lassota, P.; Allen, T.M. Papuamides A–D, HIV-Inhibitory and Cytotoxic Depsipeptides from the Sponges Theonella mirabilis and Theonella swinhoei Collected in Papua New Guinea. J. Am. Chem. Soc. 1999, 121, 5899–5909. [Google Scholar] [CrossRef]
- Youssef, D.T.; Shaala, L.A.; Mohamed, G.A.; Badr, J.M.; Bamanie, F.H.; Ibrahim, S.R. Theonellamide G, a potent antifungal and cytotoxic bicyclic glycopeptide from the Red Sea marine sponge Theonella swinhoei. Mar. Drugs 2014, 12, 1911–1923. [Google Scholar] [CrossRef]
- Ratnayake, A.S.; Bugni, T.S.; Feng, X.; Harper, M.K.; Skalicky, J.J.; Mohammed, K.A.; Andjelic, C.D.; Barrows, L.R.; Ireland, C.M. Theopapuamide, a cyclic depsipeptide from a Papua New Guinea lithistid sponge Theonella swinhoei. J. Nat. Prod. 2006, 69, 1582–1586. [Google Scholar] [CrossRef]
- Plaza, A.; Bifulco, G.; Keffer, J.L.; Lloyd, J.R.; Baker, H.L.; Bewley, C.A. Celebesides A–C and theopapuamides B–D, depsipeptides from an Indonesian sponge that inhibit HIV-1 entry. J. Org. Chem. 2009, 74, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Bewley, C.A.; He, H.; Williams, D.H.; Faulkner, D.J. Aciculitins A–C: Cytotoxic and antifungal cyclic peptides from the lithistid sponge Aciculites orientalis. J. Am. Chem. Soc. 1996, 118, 4314–4321. [Google Scholar] [CrossRef]
- Ng-Choi, I.; Oliveras, À.; Feliu, L.; Planas, M. Solid-phase synthesis of biaryl cyclic peptides containing a histidine-phenylalanine linkage. Int. J. Pept. Res. Ther. 2020, 26, 695–707. [Google Scholar] [CrossRef]
- Zampella, A.; D’Auria, M.V.; Paloma, L.G.; Casapullo, A.; Minale, L.; Debitus, C.; Henin, Y. Callipeltin A, an anti-HIV cyclic depsipeptide from the New Caledonian Lithistida sponge Callipelta sp. J. Am. Chem. Soc. 1996, 118, 6202–6209. [Google Scholar] [CrossRef]
- Liang, B.; Carroll, P.J.; Joullié, M.M. Progress toward the total synthesis of callipeltin A (I): Asymmetric synthesis of (3S, 4 R)-3,4-dimethylglutamine. Org. Lett. 2000, 2, 4157–4160. [Google Scholar] [CrossRef]
- Hansen, D.B.; Wan, X.; Carroll, P.J.; Joullié, M.M. Stereoselective synthesis of four stereoisomers of β-methoxytyrosine, a component of callipeltin A. J. Org. Chem. 2005, 70, 3120–3126. [Google Scholar] [CrossRef]
- Kikuchi, M.; Konno, H. Total synthesis of callipeltin B and M, peptidyl marine natural products. Org. Lett. 2014, 16, 4324–4327. [Google Scholar] [CrossRef]
- Zampella, A.; D’Auria, M.V. Stereoselective synthesis of (2R,3R,4R)-3-hydroxy-2,4,6-trimethylheptanoic acid and determination of the absolute stereochemistry of the natural product from callipeltin A. Tetrahedron Asymmetry 2002, 13, 1237–1239. [Google Scholar] [CrossRef]
- Daletos, G.; Kalscheuer, R.; Koliwer-Brandl, H.; Hartmann, R.; De Voogd, N.J.; Wray, V.; Lin, W.; Proksch, P. Callyaerins from the marine sponge Callyspongia aerizusa: Cyclic peptides with antitubercular activity. J. Nat. Prod. 2015, 78, 1910–1925. [Google Scholar] [CrossRef]
- Zhang, S.; De Leon Rodriguez, L.M.; Leung, I.K.; Cook, G.M.; Harris, P.W.; Brimble, M.A. Total Synthesis and Conformational Study of Callyaerin A: Anti-Tubercular Cyclic Peptide Bearing a Rare Rigidifying (Z)-2,3-Diaminoacrylamide Moiety. Angew. Chem. Int. Ed. 2018, 57, 3631–3635. [Google Scholar] [CrossRef]
- Clark, D.P.; Carroll, J.; Naylor, S.; Crews, P. An antifungal cyclodepsipeptide, cyclolithistide A, from the sponge Theonella swinhoei. J. Org. Chem. 1998, 63, 8757–8764. [Google Scholar] [CrossRef]
- Grieco, P.A.; Perez-Medrano, A. Total synthesis of the mixed peptide-polypropionate based cyclodepsipeptide (+)-geodiamolide B. Tetrahedron Lett. 1988, 29, 4225–4228. [Google Scholar] [CrossRef]
- Chan, W.R.; Tinto, W.F.; Manchand, P.S.; Todaro, L.J. Stereostructures of geodiamolides A and B, novel cyclodepsipeptides from the marine sponge Geodia sp. J. Org. Chem. 1987, 52, 3091–3093. [Google Scholar] [CrossRef]
- White, J.D.; Amedio, J.C., Jr. Total synthesis of geodiamolide A, a novel cyclodepsipeptide of marine origin. J. Org. Chem. 1989, 54, 736–738. [Google Scholar] [CrossRef]
- Amagata, T.; Morinaka, B.I.; Amagata, A.; Tenney, K.; Valeriote, F.A.; Lobkovsky, E.; Clardy, J.; Crews, P. A chemical study of cyclic depsipeptides produced by a sponge-derived fungus. J. Nat. Prod. 2006, 69, 1560–1565. [Google Scholar] [CrossRef]
- Yeo, S.-H.; Seo, H.-J.; Lim, D.-Y. Synthesis of halicylindramide a mimetics containing lactone isosteres. Bull. Korean Chem. Soc. 2011, 32, 2916–2920. [Google Scholar] [CrossRef]
- Seo, H.; Lim, D. Total synthesis of Halicylindramide A. J. Org. Chem. 2009, 74, 906–909. [Google Scholar] [CrossRef]
- Bellotta, F.; D’Auria, M.V.; Sepe, V.; Zampella, A. Synthetic studies on homophymine A: Stereoselective synthesis of (2R,3R,4R,6R)-3-hydroxy-2,4,6-trimethyloctanoic acid. Tetrahedron 2009, 65, 3659–3663. [Google Scholar] [CrossRef]
- Ohtaka, J.; Hamajima, A.; Nemoto, T.; Hamada, Y. Efficient diastereoselective synthesis of (2R, 3R, 4R)-2-amino-3-hydroxy-4, 5-dimethylhexanoic acid, the lactone linkage unit of homophymine A. Chem. Pharm. Bull. 2013, 61, 245–250. [Google Scholar] [CrossRef]
- Kobayashi, J.I.; Tsuda, M.; Nakamura, T.; Mikami, Y.; Shigemori, H. Hymenamides A and B, new proline-rich cyclic heptapeptides from the Okinawan marine sponge Hymeniacidon sp. Tetrahedron 1993, 49, 2391–2402. [Google Scholar] [CrossRef]
- Shiki, Y.; Onai, M.; Sugiyama, D.; Osada, S.; Fujita, I.; Kodama, H. Synthesis and biological activities of cyclic peptide, hymenamide analogs. In Peptides for Youth; Springer: New York, NY, USA, 2009; pp. 323–324. [Google Scholar]
- Tsuda, M.; Shigemori, H.; Mikami, Y.; Kobayashi, J.i. Hymenamides C-E, new cyclic heptapeptides with two proline residues from the okinawan marine sponge hymeniacidon sp. Tetrahedron 1993, 49, 6785–6796. [Google Scholar] [CrossRef]
- Chu, K.S.; Negrete, G.R.; Konopelski, J.P. Asymmetric total synthesis of (+)-jasplakinolide. J. Org. Chem. 1991, 56, 5196–5202. [Google Scholar] [CrossRef]
- Grieco, P.A.; Hon, Y.S.; Perez-Medrano, A. Convergent, enantiospecific total synthesis of the novel cyclodepsipeptide (+)-jasplakinolide (jaspamide). J. Am. Chem. Soc. 1988, 110, 1630–1631. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Moon, D.K. Enantioselective total synthesis of (+)-jasplakinolide. Org. Lett. 2007, 9, 2425–2427. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.; Mangalindan, G.C.; Bojo, Z.P.; Antemano, R.R.; Rodriguez, N.O.; Concepcion, G.P.; Samson, S.C.; de Guzman, D.; Cruz, L.J.; Tasdemir, D. Microcionamides A and B, bioactive peptides from the Philippine sponge Clathria (Thalysias) abietina. J. Org. Chem. 2004, 69, 4170–4176. [Google Scholar] [CrossRef] [PubMed]
- Bewley, C.A.; Debitus, C.; Faulkner, D.J. Microsclerodermins A and B. Antifungal cyclic peptides from the lithistid sponge Microscleroderma sp. J. Am. Chem. Soc. 1994, 116, 7631–7636. [Google Scholar] [CrossRef]
- Schmidt, E.W.; Faulkner, D.J. Microsclerodermins C-E, antifungal cyclic peptides from the lithistid marine sponges Theonella sp. and Microscleroderma sp. Tetrahedron 1998, 54, 3043–3056. [Google Scholar] [CrossRef]
- Qureshi, A.; Colin, P.L.; Faulkner, D.J. Microsclerodermins F–I, Antitumor and Antifungal Cyclic Peptides from the Lithistid Sponge Microscleroderma sp. Tetrahedron 2000, 56, 3679–3685. [Google Scholar] [CrossRef]
- Zhang, X.; Jacob, M.R.; Rao, R.R.; Wang, Y.-H.; Agarwal, A.K.; Newman, D.J.; Khan, I.A.; Clark, A.M.; Li, X.-C. Antifungal cyclic peptides from the marine sponge Microscleroderma herdmani. Med. Chem. Res. 2012, 2, 7. [Google Scholar]
- Zhu, J.; Ma, D. Total synthesis of microsclerodermin E. Angew. Chem. Int. Ed. 2003, 42, 5348–5351. [Google Scholar] [CrossRef]
- Santhakumar, G.; Payne, R.J. Studies toward the Total Synthesis and Stereochemical Assignment of Microspinosamide. Aust. J. Chem. 2016, 68, 1885–1889. [Google Scholar] [CrossRef]
- Ramamoorthy, G.; Acevedo, C.M.; Alvira, E.; Lipton, M.A. Synthesis and spectroscopic correlation of the diastereoisomers of 2,3-dihydroxy-2,6,8-trimethyldeca-(4Z,6E)-dienoic acid: Implications for the structures of papuamides A–D and mirabamides A–D. Tetrahedron Asymmetry 2008, 19, 2546–2554. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Okada, Y.; Matsunaga, S.; van Soest, R.W.; Fusetani, N. Nagahamide A, an antibacterial depsipeptide from the marine sponge Theonella swinhoei. Org. Lett. 2002, 4, 3039–3042. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, D.K.; Chaudhuri, S.R.; Sahoo, G.; Gurjar, M.K. Stereoselective synthesis of the polyketide chain of nagahamide A. Tetrahedron Asymmetry 2006, 17, 2609–2616. [Google Scholar] [CrossRef]
- D’Auria, M.V.; Paloma, L.G.; Minale, L.; Zampella, A.; Debitus, C.; Perez, J. Neosiphoniamolide A, a novel cyclodepsipeptide, with antifungal activity from the marine sponge Neosiphonia superstes. J. Nat. Prod. 1995, 58, 121–123. [Google Scholar] [CrossRef]
- Makino, K.; Nagata, E.; Hamada, Y. Synthesis of tripeptide hydrolysate from papuamide A: Determination of absolute stereostructure of β-methoxytyrosine. Tetrahedron Lett. 2005, 46, 6827–6830. [Google Scholar] [CrossRef]
- Okamoto, N.; Hara, O.; Makino, K.; Hamada, Y. Diastereoselective synthesis of all stereoisomers of β-methoxytyrosine, a component of papuamides. J. Org. Chem. 2002, 67, 9210–9215. [Google Scholar] [CrossRef]
- Xie, W.; Ding, D.; Zi, W.; Li, G.; Ma, D. Total synthesis and structure assignment of papuamide B, a potent marine cyclodepsipeptide with anti-HIV properties. Angew. Chem. 2008, 120, 2886–2890. [Google Scholar] [CrossRef]
- Makino, K.; Nagata, E.; Hamada, Y. Practical synthesis of (2S,3R)-3hydroxy-3-methylproline, a constituent of papuamides, using a diastereoselective tandem Michael-aldol reaction. Tetrahedron Lett. 2005, 46, 8159–8162. [Google Scholar] [CrossRef]
- Gulavita, N.K.; Gunasekera, S.P.; Pomponi, S.A.; Robinson, E.V. Polydiscamide A: A new bioactive depsipeptide from the marine sponge Discodermia sp. J. Org. Chem. 1992, 57, 1767–1772. [Google Scholar] [CrossRef]
- Santhakumar, G.; Payne, R.J. Total synthesis of polydiscamides B, C, and D via a convergent native chemical ligation–oxidation strategy. Org. Lett. 2014, 16, 4500–4503. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Rashid, M.A.; Cartner, L.K.; Bokesch, H.R.; Wilson, J.A.; McMahon, J.B.; Gustafson, K.R. Stellettapeptins A and B, HIV-inhibitory cyclic depsipeptides from the marine sponge Stelletta sp. Tetrahedron Lett. 2015, 56, 4215–4219. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Singh, S.; Sharma, A.; Chennupati, S.V.; Maharaj, S. First total synthesis and biological screening of a proline-rich cyclopeptide from a Caribbean marine sponge. Mar. Drugs 2016, 14, 228. [Google Scholar] [CrossRef] [PubMed]
- Bewley, C.A.; Faulkner, D.J. Theonegramide, an antifungal glycopeptide from the Philippine lithistid sponge Theonella swinhoei. J. Org. Chem. 1994, 59, 4849–4852. [Google Scholar] [CrossRef]
- Tohdo, K.; Hamada, Y.; Shioiri, T. Theonellamide F synthetic studies. Stereoselective synthesis of (3S, 4S, 5E, 7E)-3-amino-8-(4-bromophenyl)-4-hydroxy-6-methyl-5, 7-octadienoic acid (aboa). Tetrahedron Lett. 1992, 33, 2031–2034. [Google Scholar] [CrossRef]
- Tsuda, M.; Shimbo, K.; Kubota, T.; Mikami, Y.; Kobayashi, J.i. Two theonellapeptolide congeners from marine sponge Theonella sp. Tetrahedron 1999, 55, 10305–10314. [Google Scholar] [CrossRef]
- Kuranaga, T.; Enomoto, A.; Tan, H.; Fujita, K.; Wakimoto, T. Total synthesis of theonellapeptolide Id. Org. Lett. 2017, 19, 1366–1369. [Google Scholar] [CrossRef]
- Stincone, P.; Brandelli, A. Marine bacteria as source of antimicrobial compounds. Crit. Rev. Biotechnol. 2020, 40, 306–319. [Google Scholar] [CrossRef]
- Bürstner, N.; Roggo, S.; Ostermann, N.; Blank, J.; Delmas, C.; Freuler, F.; Gerhartz, B.; Hinniger, A.; Hoepfner, D.; Liechty, B. Gift from nature: Cyclomarin A kills mycobacteria and malaria parasites by distinct modes of action. ChemBioChem 2015, 16, 2433–2436. [Google Scholar] [CrossRef]
- Intaraudom, C.; Rachtawee, P.; Suvannakad, R.; Pittayakhajonwut, P. Antimalarial and antituberculosis substances from Streptomyces sp. BCC26924. Tetrahedron 2011, 67, 7593–7597. [Google Scholar] [CrossRef]
- Morgan, K.D.; Andersen, R.J.; Ryan, K.S. Piperazic acid-containing natural products: Structures and biosynthesis. Nat. Prod. Rep. 2019, 36, 1628–1653. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.B.; Seo, J. Antimicrobial peptides under clinical investigation. Pept. Sci. 2019, 111, e24122. [Google Scholar] [CrossRef]
- Li, W.; Leet, J.E.; Ax, H.A.; Gustavson, D.R.; Brown, D.M.; Turner, L.; Brown, K.; Clark, J.; Yang, H.; Fung-Tomc, J. Nocathiacins, new thiazolyl peptide antibiotics from Nocardia sp. I. Taxonomy, fermentation and biological activities. J. Antibiot. 2003, 56, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Pucci, M.J.; Bronson, J.J.; Barrett, J.F.; DenBleyker, K.L.; Discotto, L.F.; Fung-Tomc, J.C.; Ueda, Y. Antimicrobial evaluation of nocathiacins, a thiazole peptide class of antibiotics. Antimicrob. Agents Chemother. 2004, 48, 3697–3701. [Google Scholar] [CrossRef] [PubMed]
- Naidu, B.N.; Sorenson, M.E.; Zhang, Y.; Kim, O.K.; Matiskella, J.D.; Wichtowski, J.A.; Connolly, T.P.; Li, W.; Lam, K.S.; Bronson, J.J. Nocathiacin I analogues: Synthesis, in vitro and in vivo biological activity of novel semi-synthetic thiazolyl peptide antibiotics. Bioorg. Med. Chem. Lett. 2004, 14, 5573–5577. [Google Scholar] [CrossRef]
- Chiba, H.; Agematu, H.; Kaneto, R.; Terasawa, T.; Sakai, K.; Dobashi, K.; Yoshioka, T. Rhodopeptins (Mer-N1033), novel cyclic tetrapeptides with antifungal activity from Rhodococcus sp. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. J. Antibiot. 1999, 52, 695–699. [Google Scholar] [CrossRef][Green Version]
- Nakayama, K.; Kawato, H.C.; Inagaki, H.; Nakajima, R.; Kitamura, A.; Someya, K.; Ohta, T. Synthesis and antifungal activity of rhodopeptin analogues. 2. Modification of the west amino acid moiety. Org. Lett. 2000, 2, 977–980. [Google Scholar] [CrossRef]
- Zhou, B.; Achanta, P.S.; Shetye, G.; Chen, S.-N.; Lee, H.; Jin, Y.-Y.; Cheng, J.; Lee, M.-J.; Suh, J.-W.; Cho, S. Rufomycins or Ilamycins: Naming Clarifications and Definitive Structural Assignments. J. Nat. Prod. 2021, 84, 2644–2663. [Google Scholar] [CrossRef]
- Kazmaier, U.; Junk, L. Recent Developments on the Synthesis and Bioactivity of Ilamycins/Rufomycins and Cyclomarins, Marine Cyclopeptides That Demonstrate Anti-Malaria and Anti-Tuberculosis Activity. Mar. Drugs 2021, 19, 446. [Google Scholar] [CrossRef]
- Nakayama, Y.; Ozawa, H.; Tahara, K.; Umezawa, H. Studies on ilamycin. J. Antibiot. Ser. A 1962, 15, 49–50. [Google Scholar]
- Takita, T.; Ohi, K.; Okami, Y.; Maeda, K.; Umezawa, H. New antibiotics, ilamycins. J. Antibiot. Ser A 1962, 15, 46–48. [Google Scholar]
- Shibata, M.; Higashide, E.; Yamamoto, H.; Nakazawa, K.; Iwasaki, H.; Ueyanagi, J.; Miyake, A. Studies on Streptomycetes: Part I. Streptomyces atratus nov. sp., Producing New Antituberculous Antibiotics Rufomycin A and B Part II. Rufomycin A and B, New Antituberculous Antibiotics. Agric. Biol. Chem. 1962, 26, 228–237. [Google Scholar] [CrossRef]
- Renner, M.K.; Shen, Y.-C.; Cheng, X.-C.; Jensen, P.R.; Frankmoelle, W.; Kauffman, C.A.; Fenical, W.; Lobkovsky, E.; Clardy, J. Cyclomarins A–C, new antiinflammatory cyclic peptides produced by a marine bacterium (Streptomyces sp.). J. Am. Chem. Soc. 1999, 121, 11273–11276. [Google Scholar] [CrossRef]
- Kumamoto, T.; Koshino, H.; Watanabe, D. M10709, a new cyclic peptide antibiotic from clinically isolated Streptomyces sp. Heterocycles 2010, 80, 281–288. [Google Scholar]
- Zhou, B.; Shetye, G.; Yu, Y.; Santarsiero, B.D.; Klein, L.L.; Abad-Zapatero, C.; Wolf, N.M.; Cheng, J.; Jin, Y.; Lee, H. Antimycobacterial rufomycin analogues from Streptomyces atratus strain MJM3502. J. Nat. Prod. 2020, 83, 657–667. [Google Scholar] [CrossRef]
- Choules, M.P.; Wolf, N.M.; Lee, H.; Anderson, J.R.; Grzelak, E.M.; Wang, Y.; Ma, R.; Gao, W.; McAlpine, J.B.; Jin, Y.-Y. Rufomycin targets ClpC1 proteolysis in Mycobacterium tuberculosis and M. abscessus. Antimicrob. Agents Chemother. 2019, 63, e02204–e02218. [Google Scholar] [CrossRef]
- Wolf, N.; Lee, H.; Nam, J.; Hong, J.; Duc, N.; Ho, N.; Lee, H.; Suh, J.; Pauli, G.; Franzblau, S. Structures of CIpC1-NTD with potent anti-TB cyclic peptides Rufomycin and Ecumicin: Implications for the mechanism of action and design of therapeutic agents. Acta Cryst. A 2019, 75, A59. [Google Scholar] [CrossRef]
- Tanabe, K.; Lamping, E.; Adachi, K.; Takano, Y.; Kawabata, K.; Shizuri, Y.; Niimi, M.; Uehara, Y. Inhibition of fungal ABC transporters by unnarmicin A and unnarmicin C, novel cyclic peptides from marine bacterium. Biochem. Biophys. Res. Commun. 2007, 364, 990–995. [Google Scholar] [CrossRef]
- Pimentel-Elardo, S.M.; Kozytska, S.; Bugni, T.S.; Ireland, C.M.; Moll, H.; Hentschel, U. Anti-parasitic compounds from Streptomyces sp. strains isolated from Mediterranean sponges. Mar. Drugs 2010, 8, 373–380. [Google Scholar] [CrossRef]
- Lim, Y.-H.; Chang, J.-H.; Kim, J.-H.; Suh, J.-W.; Jung, J.-K.; Lee, C.-H. Structure Elucidation of a Potent Anti-MRSA Antibiotic, AM3, Produced by Streptomyces sp. Appl. Biol. Chem. 1995, 38, 516–521. [Google Scholar]
- Pettit, G.R.; Knight, J.C.; Herald, D.L.; Pettit, R.K.; Hogan, F.; Mukku, V.J.; Hamblin, J.S.; Dodson, M.J.; Chapuis, J.-C. Antineoplastic agents. 570. Isolation and structure elucidation of bacillistatins 1 and 2 from a marine Bacillus silvestris. J. Nat. Prod. 2009, 72, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Hu, S.; Knight, J.C.; Chapuis, J.-C. Antineoplastic agents. 571. Total synthesis of bacillistatin 2. J. Nat. Prod. 2009, 72, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Pesic, A.; Baumann, H.I.; Kleinschmidt, K.; Ensle, P.; Wiese, J.; Süssmuth, R.D.; Imhoff, J.F. Champacyclin, a new cyclic octapeptide from Streptomyces strain C42 isolated from the Baltic Sea. Mar. Drugs 2013, 11, 4834–4857. [Google Scholar] [CrossRef] [PubMed]
- Barbie, P.; Kazmaier, U. Total synthesis of cyclomarins A, C and D, marine cyclic peptides with interesting anti-tuberculosis and anti-malaria activities. Org. Biomol. Chem. 2016, 14, 6036–6054. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, Q.; Liu, X.; Chen, Y.; Zhang, Y.; Sun, A.; Zhang, W.; Zhang, J.; Ju, J. Cyclic hexapeptides from the deep South China Sea-derived Streptomyces scopuliridis SCSIO ZJ46 active against pathogenic Gram-positive bacteria. J. Nat. Prod. 2014, 77, 1937–1941. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.X.; Liu, C.; Liu, N.; Wu, Y.; Zhao, Q.J.; Hu, H.G.; Li, X.; Zou, Y. Total synthesis and antibacterial study of cyclohexapeptides desotamide B, wollamide B and their analogs. Chem. Biodivers. 2018, 15, e1700414. [Google Scholar] [CrossRef]
- Tsutsumi, L.S.; Tan, G.T.; Sun, D. Solid-phase synthesis of cyclic hexapeptides wollamides A, B and desotamide B. Tetrahedron Lett. 2017, 58, 2675–2680. [Google Scholar] [CrossRef]
- Sun, P.; Maloney, K.N.; Nam, S.-J.; Haste, N.M.; Raju, R.; Aalbersberg, W.; Jensen, P.R.; Nizet, V.; Hensler, M.E.; Fenical, W. Fijimycins A–C, three antibacterial etamycin-class depsipeptides from a marine-derived Streptomyces sp. Biorg. Med. Chem. 2011, 19, 6557–6562. [Google Scholar] [CrossRef]
- Yang, L.; Tan, R.-X.; Wang, Q.; Huang, W.-Y.; Yin, Y.-X. Antifungal cyclopeptides from Halobacillus litoralis YS3106 of marine origin. Tetrahedron Lett. 2002, 43, 6545–6548. [Google Scholar] [CrossRef]
- Dahiya, R.; Pathak, D. First total synthesis and biological evaluation of halolitoralin A. J. Serb. Chem. Soc. 2007, 72, 101–107. [Google Scholar] [CrossRef]
- Martín, J.; Sousa, D.S.; Crespo, G.; Palomo, S.; González, I.; Tormo, J.R.; De la Cruz, M.; Anderson, M.; Hill, R.T.; Vicente, F. Kocurin, the true structure of PM181104, an anti-methicillin-resistant Staphylococcus aureus (MRSA) thiazolyl peptide from the marine-derived bacterium Kocuria palustris. Mar. Drugs 2013, 11, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Gerard, J.M.; Haden, P.; Kelly, M.T.; Andersen, R.J. Loloatins A–D, cyclic decapeptide antibiotics produced in culture by a tropical marine bacterium. J. Nat. Prod. 1999, 62, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Scherkenbeck, J.; Chen, H.; Haynes, R.K. Solid-Phase Syntheses of Loloatins A–C. Eur. J. Org. Chem. 2002, 2002, 2350–2355. [Google Scholar] [CrossRef]
- Jiang, L.; Huang, P.; Ren, B.; Song, Z.; Zhu, G.; He, W.; Zhang, J.; Oyeleye, A.; Dai, H.; Zhang, L. Antibacterial polyene-polyol macrolides and cyclic peptides from the marine-derived Streptomyces sp. MS110128. Appl. Microbiol. Biotechnol. 2021, 105, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Elbatrawi, Y.M.; Kang, C.W.; Del Valle, J.R. Total Synthesis of L-156,373 and an oxoPiz Analogue via a Submonomer Approach. Org. Lett. 2018, 20, 2707–2710. [Google Scholar] [CrossRef]
- Zhou, X.; Huang, H.; Chen, Y.; Tan, J.; Song, Y.; Zou, J.; Tian, X.; Hua, Y.; Ju, J. Marthiapeptide A, an anti-infective and cytotoxic polythiazole cyclopeptide from a 60 L scale fermentation of the deep sea-derived Marinactinospora thermotolerans SCSIO 00652. J. Nat. Prod. 2012, 75, 2251–2255. [Google Scholar] [CrossRef]
- Zhang, Y.; Islam, M.A.; McAlpine, S.R. Synthesis of the natural product marthiapeptide A. Org. Lett. 2015, 17, 5149–5151. [Google Scholar] [CrossRef]
- Raju, R.; Khalil, Z.G.; Piggott, A.M.; Blumenthal, A.; Gardiner, D.L.; Skinner-Adams, T.S.; Capon, R.J. Mollemycin A: An antimalarial and antibacterial glyco-hexadepsipeptide-polyketide from an Australian marine-derived Streptomyces sp.(CMB-M0244). Org. Lett. 2014, 16, 1716–1719. [Google Scholar] [CrossRef]
- Enck, S.; Tremmel, P.; Eckhardt, S.; Marsch, M.; Geyer, A. Stereoselective synthesis of highly functionalized thiopeptide thiazole fragments from uronic acid/cysteine condensation products: Access to the core dipeptide of the thiazomycins and nocathiacins. Tetrahedron 2012, 68, 7166–7178. [Google Scholar] [CrossRef]
- Hrnciar, P.; Ueda, Y.; Huang, S.; Leet, J.E.; Bronson, J.J. Synthesis of novel nocathiacin-class antibiotics. Condensation of glycolaldehyde with primary amides and tandem reductive amination of amadori-rearranged 2-oxoethyl intermediates. J. Org. Chem. 2002, 67, 8789–8793. [Google Scholar] [CrossRef]
- Naidu, B.N.; Li, W.; Sorenson, M.E.; Connolly, T.P.; Wichtowski, J.A.; Zhang, Y.; Kim, O.K.; Matiskella, J.D.; Lam, K.S.; Bronson, J.J. Organic reactions in frozen water: Michael addition of amines and thiols to the dehydroalanine side chain of nocathiacins. Tetrahedron Lett. 2004, 45, 1059–1063. [Google Scholar] [CrossRef]
- Naidu, B.N.; Sorenson, M.E.; Matiskella, J.D.; Li, W.; Sausker, J.B.; Zhang, Y.; Connolly, T.P.; Lam, K.S.; Bronson, J.J.; Pucci, M.J. Synthesis and antibacterial activity of nocathiacin I analogues. Bioorg. Med. Chem. Lett. 2006, 16, 3545–3549. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Farthing, A.K.; Dropinski, J.F.; Meinke, P.T.; McCallum, C.; Hickey, E.; Liu, K. Synthesis and antibacterial activity of novel water-soluble nocathiacin analogs. Bioorg. Med. Chem. Lett. 2013, 23, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Um, S.; Choi, T.J.; Kim, H.; Kim, B.Y.; Kim, S.-H.; Lee, S.K.; Oh, K.-B.; Shin, J.; Oh, D.-C. Ohmyungsamycins A and B: Cytotoxic and antimicrobial cyclic peptides produced by Streptomyces sp. from a volcanic island. J. Org. Chem. 2013, 78, 12321–12329. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.; Jang, J.; Sim, J.; Son, W.S.; Ahn, H.C.; Kim, T.S.; Shin, Y.H.; Lim, C.; Lee, S.; An, H. Conformation-Enabled Total Syntheses of Ohmyungsamycins A and B and Structural Revision of Ohmyungsamycin B. Angew. Chem. 2018, 130, 3123–3127. [Google Scholar] [CrossRef]
- Kunze, B.; Böhlendorf, B.; Reichenbach, H.; Höfle, G. Pedein A and B: Production, isolation, structure elucidation and biological properties of new antifungal cyclopeptides from Chondromyces pediculatus (Myxobacteria). J. Antibiot. 2008, 61, 18–26. [Google Scholar] [CrossRef]
- Nakayama, K.; Kawato, H.C.; Inagaki, H.; Ohta, T. Novel peptidomimetics of the antifungal cyclic peptide rhodopeptin: Design of mimetics utilizing scaffolding methodology. Org. Lett. 2001, 3, 3447–3450. [Google Scholar] [CrossRef]
- Moore, B.S.; Trischman, J.A.; Seng, D.; Kho, D.; Jensen, P.R.; Fenical, W. Salinamides, antiinflammatory depsipeptides from a marine streptomycete. J. Org. Chem. 1999, 64, 1145–1150. [Google Scholar] [CrossRef]
- Tan, L.; Ma, D. Total Synthesis of Salinamide A: A Potent Anti-Inflammatory Bicyclic Depsipeptide. Angew. Chem. Int. Ed. 2008, 47, 3614–3617. [Google Scholar] [CrossRef]
- Hassan, H.M.; Degen, D.; Jang, K.H.; Ebright, R.H.; Fenical, W. Salinamide F, new depsipeptide antibiotic and inhibitor of bacterial RNA polymerase from a marine-derived Streptomyces sp. J. Antibiot. 2015, 68, 206–209. [Google Scholar] [CrossRef]
- Höltzel, A.; Jack, R.W.; Nicholson, G.J.; Jung, G.; Gebhardt, K.; Fiedler, H.-P.; Süssmuth, R.D. Streptocidins AD, Novel Cyclic Decapeptide Antibiotics Produced by Streptomyces sp. Tü 6071 II. Structure Elucidation. J. Antibiot. 2001, 54, 434–440. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Qin, C.; Zhong, X.; Ng, N.L.; Bu, X.; Chan, W.S.; Guo, Z. Facile solid-phase synthesis of cyclic decapeptide antibiotic streptocidins A–D. Tetrahedron Lett. 2004, 45, 217–220. [Google Scholar] [CrossRef]
- Schmidt, E.W.; Bewley, C.A.; Faulkner, D.J. Theopalauamide, a bicyclic glycopeptide from filamentous bacterial symbionts of the lithistid sponge Theonella swinhoei from Palau and Mozambique. J. Org. Chem. 1998, 63, 1254–1258. [Google Scholar] [CrossRef]
- Romero, F.; Espliego, F.; Baz, J.P.; De Quesada, T.G.; Grávalos, D.; De La Calle, F.; Fernández-Puentes, J.L. Thiocoraline, a new depsipeptide with antitumor activity produced by a marine Micromonospora I. Taxonomy, fermentation, isolation, and biological activities. J. Antibiot. 1997, 50, 734–737. [Google Scholar] [CrossRef]
- Boger, D.L.; Ichikawa, S. Total syntheses of thiocoraline and BE-22179: Establishment of relative and absolute stereochemistry. J. Am. Chem. Soc. 2000, 122, 2956–2957. [Google Scholar] [CrossRef]
- Tulla-Puche, J.; Auriemma, S.; Falciani, C.; Albericio, F. Orthogonal chemistry for the synthesis of thiocoraline–triostin hybrids. Exploring their structure–activity relationship. J. Med. Chem. 2013, 56, 5587–5600. [Google Scholar] [CrossRef]
- Engelhardt, K.; Degnes, K.F.; Kemmler, M.; Bredholt, H.; Fjærvik, E.; Klinkenberg, G.; Sletta, H.; Ellingsen, T.E.; Zotchev, S.B. Production of a new thiopeptide antibiotic, TP-1161, by a marine Nocardiopsis species. Appl. Environ. Microbiol. 2010, 76, 4969–4976. [Google Scholar] [CrossRef]
- Kishimoto, S.; Tsunematsu, Y.; Nishimura, S.; Hayashi, Y.; Hattori, A.; Kakeya, H. Tumescenamide C, an antimicrobial cyclic lipodepsipeptide from Streptomyces sp. Tetrahedron 2012, 68, 5572–5578. [Google Scholar] [CrossRef]
- Takahashi, N.; Kaneko, K.; Kakeya, H. Total Synthesis and Antimicrobial Activity of Tumescenamide C and Its Derivatives. J. Org. Chem. 2020, 85, 4530–4535. [Google Scholar] [CrossRef]
- Gisin, B.; Merrifield, R.; Tosteson, D. Solid-phase synthesis of the cyclododecadepsipeptide valinomycin. J. Am. Chem. Soc. 1969, 91, 2691–2695. [Google Scholar] [CrossRef]
- Kuisle, O.; Quiñoá, E.; Riguera, R. A general methodology for automated solid-phase synthesis of depsides and depsipeptides. Preparation of a valinomycin analogue. J. Org. Chem. 1999, 64, 8063–8075. [Google Scholar] [CrossRef] [PubMed]
- Annese, C.; Fanizza, I.; Calvano, C.D.; D’Accolti, L.; Fusco, C.; Curci, R.; Williard, P.G. Selective synthesis of hydroxy analogues of valinomycin using dioxiranes. Org. Lett. 2011, 13, 5096–5099. [Google Scholar] [CrossRef] [PubMed]
- Uzair, B.; Tabassum, S.; Rasheed, M.; Rehman, S.F. Exploring marine cyanobacteria for lead compounds of pharmaceutical importance. Sci. World J. 2012, 2012, 179782. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.; Vasconcelos, V. Cyanobactins from cyanobacteria: Current genetic and chemical state of knowledge. Mar. Drugs 2015, 13, 6910–6946. [Google Scholar] [CrossRef]
- Mi, Y.; Zhang, J.; He, S.; Yan, X. New peptides isolated from marine cyanobacteria, an overview over the past decade. Mar. Drugs 2017, 15, 132. [Google Scholar] [CrossRef]
- Almaliti, J.; Malloy, K.L.; Glukhov, E.; Spadafora, C.; Gutiérrez, M.; Gerwick, W.H. Dudawalamides A–D, antiparasitic cyclic depsipeptides from the marine cyanobacterium Moorea producens. J. Nat. Prod. 2017, 80, 1827–1836. [Google Scholar] [CrossRef]
- Ogawa, H.; Iwasaki, A.; Sumimoto, S.; Kanamori, Y.; Ohno, O.; Iwatsuki, M.; Ishiyama, A.; Hokari, R.; Otoguro, K.; Ōmura, S. Janadolide, a Cyclic Polyketide–Peptide Hybrid Possessing a tert-Butyl Group from an Okeania sp. Marine Cyanobacterium. J. Nat. Prod. 2016, 79, 1862–1866. [Google Scholar] [CrossRef]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Tan, L.T. Lagunamides A and B: Cytotoxic and antimalarial cyclodepsipeptides from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2010, 73, 1810–1814. [Google Scholar] [CrossRef]
- Montaser, R.; Paul, V.J.; Luesch, H. Pitipeptolides C–F, antimycobacterial cyclodepsipeptides from the marine cyanobacterium Lyngbya majuscula from Guam. Phytochemistry 2011, 72, 2068–2074. [Google Scholar] [CrossRef]
- Luesch, H.; Pangilinan, R.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Pitipeptolides A and B, new cyclodepsipeptides from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2001, 64, 304–307. [Google Scholar] [CrossRef]
- Müller, D.; Krick, A.; Kehraus, S.; Mehner, C.; Hart, M.; Küpper, F.C.; Saxena, K.; Prinz, H.; Schwalbe, H.; Janning, P. Brunsvicamides A–C: Sponge-Related Cyanobacterial Peptides with Mycobacterium tuberculosis Protein Tyrosine Phosphatase Inhibitory Activity. J. Med. Chem. 2006, 49, 4871–4878. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.; Renner, S.; Waldmann, H.; Arndt, H.D. Synthesis and Structure-Activity Correlation of a Brunsvicamide-Inspired Cyclopeptide Collection. ChemBioChem 2009, 10, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Junk, L.; Kazmaier, U. Total synthesis and configurational revision of mozamide A, a hydroxy-brunsvicamide. J. Org. Chem. 2019, 84, 2489–2500. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.; Arndt, H.-D.; Waldmann, H. Solid-support based total synthesis and stereochemical correction of brunsvicamide A. Org. Lett. 2008, 10, 3199–3202. [Google Scholar] [CrossRef] [PubMed]
- Marquez, B.L.; Watts, K.S.; Yokochi, A.; Roberts, M.A.; Verdier-Pinard, P.; Jimenez, J.I.; Hamel, E.; Scheuer, P.J.; Gerwick, W.H. Structure and absolute stereochemistry of hectochlorin, a potent stimulator of actin assembly. J. Nat. Prod. 2002, 65, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Cetusic, J.R.; Green, F.R.; Graupner, P.R.; Oliver, M.P. Total synthesis of hectochlorin. Org. Lett. 2002, 4, 1307–1310. [Google Scholar] [CrossRef]
- Ojima, D.; Mine, H.; Iwasaki, A.; Suenaga, K. Total synthesis of janadolide. Tetrahedron Lett. 2018, 59, 1360–1362. [Google Scholar] [CrossRef]
- Gorges, J.; Kazmaier, U. Matteson Homologation-Based Total Synthesis of Lagunamide A. Org. Lett. 2018, 20, 2033–2036. [Google Scholar] [CrossRef]
- Dai, L.; Chen, B.; Lei, H.; Wang, Z.; Liu, Y.; Xu, Z.; Ye, T. Total synthesis and stereochemical revision of lagunamide A. Chem. Commun. 2012, 48, 8697–8699. [Google Scholar] [CrossRef]
- Huang, W.; Ren, R.-G.; Dong, H.-Q.; Wei, B.-G.; Lin, G.-Q. Diverse synthesis of marine cyclic depsipeptide lagunamide A and its analogues. J. Org. Chem. 2013, 78, 10747–10762. [Google Scholar] [CrossRef]
- Liu, H.-M.; Chang, C.-Y.; Lai, Y.-C.; Yang, M.-D.; Chang, C.-Y. An efficient synthesis of the C27–C45 fragment of lagunamide A, a cyclodepsipeptide with potent cytotoxic and antimalarial properties. Tetrahedron Asymmetry 2014, 25, 187–192. [Google Scholar] [CrossRef]
- Pal, S.; Chakraborty, T.K. Toward the total synthesis of a lagunamide B analogue. Tetrahedron Lett. 2014, 55, 3469–3472. [Google Scholar] [CrossRef]
- MacMillan, J.B.; Ernst-Russell, M.A.; De Ropp, J.S.; Molinski, T.F. Lobocyclamides A-C, Lipopeptides from a cryptic cyanobacterial mat containing Lyngbya confervoides. J. Org. Chem. 2002, 67, 8210–8215. [Google Scholar] [CrossRef] [PubMed]
- Frankmölle, W.P.; Larsen, L.K.; Caplan, F.R.; Patterson, G.M.; Knübel, G.; Levine, I.A.; Moore, R.E. Antifungal cyclic peptides from the terrestrial blue-green alga Anabaena laxa I. Isolation and biological properties. J. Antibiot. 1992, 45, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Milligan, K.E.; Marquez, B.L.; Williamson, R.T.; Gerwick, W.H. Lyngbyabellin B, a toxic and antifungal secondary metabolite from the marine cyanobacterium Lyngbya m ajuscula. J. Nat. Prod. 2000, 63, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Pirovani, R.V.; Brito, G.A.; Barcelos, R.C.; Pilli, R.A. Enantioselective total synthesis of (+)-lyngbyabellin M. Mar. Drugs 2015, 13, 3309–3324. [Google Scholar] [CrossRef]
- Yokokawa, F.; Sameshima, H.; Katagiri, D.; Aoyama, T.; Shioiri, T. Total syntheses of lyngbyabellins A and B, potent cytotoxic lipopeptides from the marine cyanobacterium Lyngbya majuscula. Tetrahedron 2002, 58, 9445–9458. [Google Scholar] [CrossRef]
- Yokokawa, F.; Sameshima, H.; Shioiri, T. Total synthesis of lyngbyabellin A, a potent cytotoxic metabolite from the marine cyanobacterium Lyngbya majuscula. Tetrahedron Lett. 2001, 42, 4171–4174. [Google Scholar] [CrossRef]
- Zainuddin, E.N.; Jansen, R.; Nimtz, M.; Wray, V.; Preisitsch, M.; Lalk, M.; Mundt, S. Lyngbyazothrins A–D, Antimicrobial Cyclic Undecapeptides from the Cultured Cyanobacterium Lyngbya sp. J. Nat. Prod. 2009, 72, 1373–1378. [Google Scholar] [CrossRef]
- Wang, X.J.; Zhang, J.T.; Liu, D.W.; Tang, L.J.; Feng, J.M.; Chen, S.P. Study on Synthesis of 2, 2-dimethyl-3-hydroxy-7-octynoic acid. In Advanced Materials Research; Trans Tech Publications Ltd.: Zurich, Switzerland, 2013; pp. 1103–1107. [Google Scholar]
- Koeller, K.M.; Wong, C.-H. Enzymes for chemical synthesis. Nature 2001, 409, 232–240. [Google Scholar] [CrossRef]
- Linington, R.G.; Edwards, D.J.; Shuman, C.F.; McPhail, K.L.; Matainaho, T.; Gerwick, W.H. Symplocamide A, a potent cytotoxin and chymotrypsin inhibitor from the marine cyanobacterium Symploca sp. J. Nat. Prod. 2008, 71, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Stolze, S.C.; Meltzer, M.; Ehrmann, M.; Kaiser, M. Solid phase total synthesis of the 3-amino-6-hydroxy-2-piperidone (Ahp) cyclodepsipeptide and protease inhibitor Symplocamide A. Chem. Commun. 2010, 46, 8857–8859. [Google Scholar] [CrossRef] [PubMed]
- Jaki, B.; Zerbe, O.; Heilmann, J.; Sticher, O. Two novel cyclic peptides with antifungal activity from the cyanobacterium Tolypothrix byssoidea (EAWAG 195). J. Nat. Prod. 2001, 64, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Linington, R.G.; González, J.; Urena, L.-D.; Romero, L.I.; Ortega-Barría, E.; Gerwick, W.H. Venturamides A and B: Antimalarial constituents of the panamanian marine Cyanobacterium Oscillatoria sp. J. Nat. Prod. 2007, 70, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, P.; Zhang, Y.; Zhang, X.; Liu, J.; Du, Y. One-Pot Enantiomeric Synthesis of Thiazole-Containing Amino Acids: Total Synthesis of Venturamides A and B. J. Org. Chem. 2018, 83, 3897–3905. [Google Scholar] [CrossRef]
- Jin, L.; Quan, C.; Hou, X.; Fan, S. Potential pharmacological resources: Natural bioactive compounds from marine-derived fungi. Mar. Drugs 2016, 14, 76. [Google Scholar] [CrossRef]
- Nakajima, S. The origin of cephalosporins. Yakushigaku Zasshi 2003, 37, 119–127. [Google Scholar]
- Ohyama, T.; Iwadate-Kurihara, Y.; Ishikawa, T.; Miyakoshi, S.; Hamano, K.; Inukai, M. Arborcandins A, B, C, D, E and F, Novel 1, 3-β-Glucan Synthase Inhibitors Physico-chemical Properties and Structure Elucidation. J. Antibiot. 2003, 56, 1024–1032. [Google Scholar] [CrossRef]
- Ohyama, T.; Kurihara, Y.; Ono, Y.; Ishikawa, T.; Miyakoshi, S.; Hamano, K.; Araei, M.; Suzuki, T.; Igari, H.; Suzuki, Y. Arborcandins A, B, C, D, E and F, novel 1, 3-β-glucan synthase inhibitors: Production and biological activity. J. Antibiot. 2000, 53, 1108–1116. [Google Scholar] [CrossRef]
- Balkovec, J.M. Section review: Anti-infectives: Lipopeptide antifungal agents. Expert Opin. Investig. Drugs 1994, 3, 65–82. [Google Scholar] [CrossRef]
- Takesako, K.; Ikai, K.; Haruna, F.; Endo, M.; Shimanaka, K.; Sono, E.; Nakamura, T.; Kato, I.; Yamaguchi, H. Aureobasidins, new antifungal antibiotics taxonomy, fermentation, isolation, and properties. J. Antibiot. 1991, 44, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Herath, K.; Harris, G.; Jayasuriya, H.; Zink, D.; Smith, S.; Vicente, F.; Bills, G.; Collado, J.; González, A.; Jiang, B. Isolation, structure and biological activity of phomafungin, a cyclic lipodepsipeptide from a widespread tropical Phoma sp. Biorg. Med. Chem. 2009, 17, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Shao, C.-L.; Fu, X.-M.; Kong, C.-J.; She, Z.-G.; Wang, C.-Y. Lumazine peptides penilumamides B–D and the cyclic pentapeptide asperpeptide A from a gorgonian-derived Aspergillus sp. fungus. J. Nat. Prod. 2014, 77, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Bao, J.; Zhang, X.-Y.; Tu, Z.-C.; Shi, Y.-M.; Qi, S.-H. Asperterrestide A, a cytotoxic cyclic tetrapeptide from the marine-derived fungus Aspergillus terreus SCSGAF0162. J. Nat. Prod. 2013, 76, 1182–1186. [Google Scholar] [CrossRef]
- Ohsawa, K.; Sugai, M.; Zhang, L.; Masuda, Y.; Yoshida, M.; Doi, T. Total synthesis and structural revision of cyclotetrapeptide asperterrestide A. J. Org. Chem. 2019, 84, 6765–6779. [Google Scholar] [CrossRef] [PubMed]
- Maharani, R.; Puspitasari, D.; Huspa, D.; Hidayat, A.; Sumiarsa, D.; Hidayat, I. Synthesis of t-Butyl (2R)-Hydroxyisovalerate, A Precursor of Aureobasidin B. In Proceedings of the IOP Conference Series: Materials Science and Engineering, Purwokerto, Indonesia, 15–16 September 2016; IOP Publishing Ltd.: Bristol, UK, 2017; p. 012044. [Google Scholar]
- Kurome, T.; Inami, K.; Inoue, T.; Ikai, K.; Takesako, K.; Kato, I.; Shiba, T. Total synthesis of an antifungal cyclic depsipeptide aureobasidin A. Tetrahedron 1996, 52, 4327–4346. [Google Scholar] [CrossRef]
- Isaka, M.; Srisanoh, U.; Lartpornmatulee, N.; Boonruangprapa, T. ES-242 derivatives and cycloheptapeptides from Cordyceps sp. strains BCC 16173 and BCC 16176. J. Nat. Prod. 2007, 70, 1601–1604. [Google Scholar] [CrossRef]
- Dahiya, R.; Gautam, H. Solution phase synthesis and bioevaluation of cordyheptapeptide B. Bull. Pharm. Res. 2011, 1, 1–10. [Google Scholar]
- Puentes, A.R.; Neves Filho, R.A.; Rivera, D.G.; Wessjohann, L.A. Total synthesis of cordyheptapeptide A. Synlett 2017, 28, 1971–1974. [Google Scholar]
- Li, C.; Wang, J.; Luo, C.; Ding, W.; Cox, D.G. A new cyclopeptide with antifungal activity from the co-culture broth of two marine mangrove fungi. Nat. Prod. Res. 2014, 28, 616–621. [Google Scholar] [CrossRef]
- Du, F.-Y.; Zhang, P.; Li, X.-M.; Li, C.-S.; Cui, C.-M.; Wang, B.-G. Cyclohexadepsipeptides of the isaridin class from the marine-derived fungus Beauveria felina EN-135. J. Nat. Prod. 2014, 77, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.-C.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Induced production of emericellamides A and B from the marine-derived fungus Emericella sp. in competing co-culture. J. Nat. Prod. 2007, 70, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liang, S.; Tan, W.; Xu, Z.; Ye, T. Total synthesis of emericellamides A and B. Tetrahedron 2009, 65, 2695–2702. [Google Scholar] [CrossRef]
- Pradhan, T.K.; Reddy, K.M.; Ghosh, S. Total synthesis of emericellamides A and B. Tetrahedron Asymmetry 2013, 24, 1042–1051. [Google Scholar] [CrossRef]
- Ren, R.-G.; Ma, J.-Y.; Mao, Z.-Y.; Liu, Y.-W.; Wei, B.-G. Asymmetric synthesis of emericellamide B. Chin. Chem. Lett. 2015, 26, 1209–1215. [Google Scholar] [CrossRef]
- Jenkins, K.M.; Renner, M.K.; Jensen, P.R.; Fenical, W. Exumolides A and B: Antimicroalgal cyclic depsipeptides produced by a marine fungus of the genus Scytalidium. Tetrahedron Lett. 1998, 39, 2463–2466. [Google Scholar] [CrossRef]
- Rahmadani, A.; Masruhim, M.A.; Rijai, L.; Hidayat, A.T.; Supratman, U.; Maharani, R. Total synthesis of cyclohexadepsipeptides exumolides A and B. Tetrahedron 2021, 83, 131987. [Google Scholar] [CrossRef]
- Sato, T.; Ishiyama, D.; Honda, R.; Senda, H.; Konno, H.; Tokumasu, S.; Kanazawa, S. Glomosporin, a novel antifungal cyclic depsipeptide from Glomospora sp. J. Antibiot. 2000, 53, 597–602. [Google Scholar] [CrossRef]
- Lee, K.K.; Gloer, J.B.; Scott, J.A.; Malloch, D. Petriellin A: A novel antifungal depsipeptide from the coprophilous fungus Petriella sordida. J. Org. Chem. 1995, 60, 5384–5385. [Google Scholar] [CrossRef]
- Sleebs, M.M.; Scanlon, D.; Karas, J.; Maharani, R.; Hughes, A.B. Total synthesis of the antifungal depsipeptide petriellin A. J. Org. Chem. 2011, 76, 6686–6693. [Google Scholar] [CrossRef]
- Zheng, J.; Zhu, H.; Hong, K.; Wang, Y.; Liu, P.; Wang, X.; Peng, X.; Zhu, W. Novel cyclic hexapeptides from marine-derived fungus, Aspergillus sclerotiorum PT06-1. Org. Lett. 2009, 11, 5262–5265. [Google Scholar] [CrossRef] [PubMed]
- Diamandas, M.; Moreira, R.; Taylor, S.D. Solid-Phase Total Synthesis of Dehydrotryptophan-Bearing Cyclic Peptides Tunicyclin B, Sclerotide A, CDA3a, and CDA4a Using a Protected β-Hydroxytryptophan Building Block. Org. Lett. 2021, 23, 3048–3052. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xu, Z.; Wang, Y.; Hong, K.; Liu, P.; Zhu, W. Cyclic tripeptides from the halotolerant fungus Aspergillus sclerotiorum PT06-1. J. Nat. Prod. 2010, 73, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Casertano, M.; Menna, M.; Imperatore, C. The ascidian-derived metabolites with antimicrobial properties. Antibiotics 2020, 9, 510. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fu, C.; Wang, G. Microbial diversity associated with ascidians: A review of research methods and application. Symbiosis 2017, 71, 19–26. [Google Scholar] [CrossRef]
- Derby, C.D. Escape by inking and secreting: Marine molluscs avoid predators through a rich array of chemicals and mechanisms. Biol. Bull. 2007, 213, 274–289. [Google Scholar] [CrossRef]
- Calin-Jageman, R.J.; Fischer, T.M. Behavioral adaptation of the aplysia siphon-withdrawal response is accompanied by sensory adaptation. Behav. Neurosci. 2007, 121, 200. [Google Scholar] [CrossRef]
- Love-Chezem, T.; Aggio, J.F.; Derby, C.D. Defense through sensory inactivation: Sea hare ink reduces sensory and motor responses of spiny lobsters to food odors. J. Exp. Biol. 2013, 216, 1364–1372. [Google Scholar] [CrossRef]
- Berriman, J.; Kay, M.; Reed, D.; Rassweiler, A.; Goldstein, D.; Wright, W. Shifts in attack behavior of an important kelp forest predator within marine reserves. Mar. Ecol. Prog. Ser. 2015, 522, 193–201. [Google Scholar] [CrossRef]
- Desriac, F.; Jégou, C.; Balnois, E.; Brillet, B.; Chevalier, P.L.; Fleury, Y. Antimicrobial peptides from marine proteobacteria. Mar. Drugs 2013, 11, 3632–3660. [Google Scholar] [CrossRef]
- Hamann, M.T.; Otto, C.S.; Scheuer, P.J.; Dunbar, D.C. Kahalalides: Bioactive peptides from a marine mollusk Elysia rufescens and its algal diet Bryopsis sp. J. Org. Chem. 1996, 61, 6594–6600. [Google Scholar] [CrossRef] [PubMed]
- Shilabin, A.G.; Kasanah, N.; Wedge, D.E.; Hamann, M.T. Lysosome and HER3 (ErbB3) selective anticancer agent kahalalide F: Semisynthetic modifications and antifungal lead-exploration studies. J. Med. Chem. 2007, 50, 4340–4350. [Google Scholar] [CrossRef] [PubMed]
- Ashour, M.; Edrada, R.; Ebel, R.; Wray, V.; Wätjen, W.; Padmakumar, K.; Müller, W.E.; Lin, W.H.; Proksch, P. Kahalalide derivatives from the Indian sacoglossan mollusk Elysia grandifolia. J. Nat. Prod. 2006, 69, 1547–1553. [Google Scholar] [CrossRef] [PubMed]
- Hamann, M.T.; Scheuer, P.J. Kahalalide F: A bioactive depsipeptide from the sacoglossan mollusk Elysia rufescens and the green alga Bryopsis sp. J. Am. Chem. Soc. 1993, 115, 5825–5826. [Google Scholar] [CrossRef]
- Cruz, L.J.; Luque-Ortega, J.R.; Rivas, L.; Albericio, F. Kahalalide F, an antitumor depsipeptide in clinical trials, and its analogues as effective antileishmanial agents. Mol. Pharm. 2009, 6, 813–824. [Google Scholar] [CrossRef]
- White, K.M.; Rosales, R.; Yildiz, S.; Kehrer, T.; Miorin, L.; Moreno, E.; Jangra, S.; Uccellini, M.B.; Rathnasinghe, R.; Coughlan, L. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 2021, 371, 926–931. [Google Scholar] [CrossRef]
- López-Macià, À.; Jiménez, J.C.; Royo, M.; Giralt, E.; Albericio, F. Synthesis and structure determination of Kahalalide F1, 2. J. Am. Chem. Soc. 2001, 123, 11398–11401. [Google Scholar] [CrossRef]
- Lopez-Macia, A.; Jiménez, J.C.; Royo, M.; Giralt, E.; Albericio, F. Kahalalide B. Synthesis of a natural cyclodepsipeptide. Tetrahedron Lett. 2000, 41, 9765–9769. [Google Scholar] [CrossRef]
- Bourel-Bonnet, L.; Rao, K.V.; Hamann, M.T.; Ganesan, A. Solid-phase total synthesis of kahalalide A and related analogues. J. Med. Chem. 2005, 48, 1330–1335. [Google Scholar] [CrossRef]
- Izzo, I.; Acosta, G.A.; Tulla-Puche, J.; Cupido, T.; Martin-Lopez, M.J.; Cuevas, C.; Albericio, F. Solid-Phase Synthesis of Aza-Kahalalide F Analogues: (2R, 3R)-2-Amino-3-azidobutanoic acid as precursor of the Aza-Threonine. Eur. J. Org. Chem. 2010, 2010, 2536–2543. [Google Scholar] [CrossRef]
- McKeever, B.; Pattenden, G. Total synthesis of mollamide, a reverse prenyl substituted cytotoxic cyclic peptide from Didemnum molle. Tetrahedron Lett. 1999, 40, 9317–9320. [Google Scholar] [CrossRef]
- Wyche, T.P.; Hou, Y.; Vazquez-Rivera, E.; Braun, D.; Bugni, T.S. Peptidolipins B–F, antibacterial lipopeptides from an ascidian-derived Nocardia sp. J. Nat. Prod. 2012, 75, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Borjan, B.; Steiner, N.; Karbon, S.; Kern, J.; Francesch, A.; Hermann, M.; Willenbacher, W.; Gunsilius, E.; Untergasser, G. The Aplidin analogs PM01215 and PM02781 inhibit angiogenesis in vitro and in vivo. BMC Cancer 2015, 15, 1–16. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rodriguez, I.; Polanco, C.; Cuevas, F.; Mandez, P.; Cuevas, C.; Gallego, P.; Munt, S.; Manzanares, I. Synthetic Methods for Aplidine and New Antitumoral Derivatives, Methods of Making and Using Them. U.S. Patent 7,678,765, 16 March 2010. [Google Scholar]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509. [Google Scholar] [CrossRef]
- Amblard, M.; Fehrentz, J.-A.; Martinez, J.; Subra, G. Methods and protocols of modern solid phase peptide synthesis. Mol. Biotechnol. 2006, 33, 239–254. [Google Scholar] [CrossRef]
- Foster, A.D.; Ingram, J.D.; Leitch, E.K.; Lennard, K.R.; Osher, E.L.; Tavassoli, A. Methods for the creation of cyclic peptide libraries for use in lead discovery. J. Biomol. Screen. 2015, 20, 563–576. [Google Scholar] [CrossRef]
- Dougherty, P.G.; Qian, Z.; Pei, D. Macrocycles as protein–protein interaction inhibitors. Biochem. J. 2017, 474, 1109–1125. [Google Scholar] [CrossRef]
- Schmidt, U.; Langner, J. Cyclotetrapeptides and cyclopentapeptides: Occurrence and synthesis. J. Pept. Res. 1997, 49, 67–73. [Google Scholar] [CrossRef]
- Kemp, D.S.; Rebek, J., Jr. Peptide racemization mechanism. Kinetic isotope effect as a means of distinguishing enolization from oxazolone formation. J. Am. Chem. Soc. 1970, 92, 5792–5793. [Google Scholar] [CrossRef]
- Wen, S.; Packham, G.; Ganesan, A. Macrolactamization versus macrolactonization: Total synthesis of FK228, the depsipeptide histone deacetylase inhibitor. J. Org. Chem. 2008, 73, 9353–9361. [Google Scholar] [CrossRef]
- Chinchilla, R.; Dodsworth, D.J.; Nájera, C.; Soriano, J.M. New polymer-supported allyloxycarbonyl (Alloc) and propargyloxycarbonyl (Proc) amino-protecting reagents. Synlett 2003, 2003, 0809–0812. [Google Scholar]
- Albericio, F. Orthogonal protecting groups for N-α-amino and C-terminal carboxyl functions in solid-phase peptide synthesis. Pept. Sci. 2000, 55, 123–139. [Google Scholar] [CrossRef]
- Venkataraman, K.; Wagle, D. Cyanuric chloride: A useful reagent for converting carboxylic acids into chlorides, esters, amides and peptides. Tetrahedron Lett. 1979, 20, 3037–3040. [Google Scholar] [CrossRef]
- Kamiński, Z.J. 2-Chloro-4, 6-dimethoxy-1, 3, 5-triazine. A new coupling reagent for peptide synthesis. Synthesis 1987, 1987, 917–920. [Google Scholar] [CrossRef]
- Kamiński, Z.; Paneth, P.; Rudziński, J. A study on the activation of carboxylic acids by means of 2-chloro-4,6-dimethoxy-1,3,5-triazine and 2-chloro-4,6-diphenoxy-1,3,5-triazine. J. Org. Chem. 1998, 63, 4248–4255. [Google Scholar] [CrossRef]
- Falchi, A.; Giacomelli, G.; Porcheddu, A.; Taddei, M. 4-(4,6-Dimethoxy [1,3,5] triazin-2-yl)-4-methyl-morpholinium chloride (DMTMM): A valuable alternative to PyBOP for solid phase peptide synthesis. Synlett 2000, 2000, 275–277. [Google Scholar] [CrossRef]
- Sarantakis, D.; Teichman, J.; Lien, E.; Fenichel, R. A novel cyclic undecapeptide, WY-40,770, with prolonged growth hormone release inhibiting activity. Biochem. Biophys. Res. Commun. 1976, 73, 336–342. [Google Scholar] [CrossRef]
- Sheehan, J.C.; Ledis, S.L. Total synthesis of a monocyclic peptide lactone antibiotic, etamycin. J. Am. Chem. Soc. 1973, 95, 875–879. [Google Scholar] [CrossRef]
- Sheehan, J.C.; Hlavka, J.J. The use of water-soluble and basic carbodiimides in peptide synthesis. J. Org. Chem. 1956, 21, 439–441. [Google Scholar] [CrossRef]
- Kunz, H. Synthesen mit 2-Phosphonioethoxycarbonyl-Schutzgruppen: Peptidsynthese in Wasser. Angew. Chem. 1978, 90, 63–64. [Google Scholar] [CrossRef]
- Carpino, L.A. 1-Hydroxy-7-azabenzotriazole. An efficient peptide coupling additive. J. Am. Chem. Soc. 1993, 115, 4397–4398. [Google Scholar] [CrossRef]
- Carpino, L.A.; El-Faham, A.; Minor, C.A.; Albericio, F. Advantageous applications of azabenzotriazole (triazolopyridine)-based coupling reagents to solid-phase peptide synthesis. J. Chem. Soc. Chem. Commun. 1994, 201–203. [Google Scholar] [CrossRef]
- Carpino, L.A.; El-Faham, A.; Albericio, F. Racemization studies during solid-phase peptide synthesis using azabenzotriazole-based coupling reagents. Tetrahedron Lett. 1994, 35, 2279–2282. [Google Scholar] [CrossRef]
- Carpino, L.A.; Imazumi, H.; El-Faham, A.; Ferrer, F.J.; Zhang, C.; Lee, Y.; Foxman, B.M.; Henklein, P.; Hanay, C.; Mügge, C. The uronium/guanidinium peptide coupling reagents: Finally the true uronium salts. Angew. Chem. Int. Ed. 2002, 41, 441–445. [Google Scholar] [CrossRef]
- Reszka, P.; Methling, K.; Lalk, M.; Xiao, Z.; Weisz, K.; Bednarski, P.J. Control of aspartate epimerization during the coupling of caspase specific tetrapeptides with aromatic amines by using N-[[(dimethylamino)-1H-1, 2, 3-triazolo [4, 5-b]-pyridin-1-yl] methylene]-N-methylmethanaminium hexafluorophosphate N-oxide (HATU) as a coupling reagent. Tetrahedron Asymmetry 2008, 19, 49–59. [Google Scholar]
- El-Faham, A.; Funosas, R.S.; Prohens, R.; Albericio, F. COMU: A safer and more effective replacement for benzotriazole-based uronium coupling reagents. Chemistry 2009, 15, 9404–9416. [Google Scholar] [CrossRef]
- Kumar, A.; Jad, Y.E.; de la Torre, B.G.; El-Faham, A.; Albericio, F. Re-evaluating the stability of COMU in different solvents. J. Pept. Sci. 2017, 23, 763–768. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Alvarez, M.; Albericio, F. Amino acid-protecting groups. Chem. Rev. 2009, 109, 2455–2504. [Google Scholar] [CrossRef]
- Jing, X.; Jin, K. A gold mine for drug discovery: Strategies to develop cyclic peptides into therapies. Med. Res. Rev. 2020, 40, 753–810. [Google Scholar] [CrossRef]
- Suarez-Jimenez, G.-M.; Burgos-Hernandez, A.; Ezquerra-Brauer, J.-M. Bioactive peptides and depsipeptides with anticancer potential: Sources from marine animals. Mar. Drugs 2012, 10, 963–986. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Shioiri, T. Recent progress of the synthetic studies of biologically active marine cyclic peptides and depsipeptides. Chem. Rev. 2005, 105, 4441–4482. [Google Scholar] [CrossRef] [PubMed]
- Somogyi, L.; Haberhauer, G.; Rebek, J., Jr. Improved synthesis of functionalized molecular platforms related to marine cyclopeptides. Tetrahedron 2001, 57, 1699–1708. [Google Scholar] [CrossRef]
- Mancini, I.; Defant, A.; Guella, G. Recent synthesis of marine natural products with antibacterial activities. Anti Infect. Agents Med. Chem. 2007, 6, 17–48. [Google Scholar] [CrossRef]
- Macedo, M.W.F.S.; da Cunha, N.B.; Carneiro, J.A.; da Costa, R.A.; de Alencar, S.A.; Cardoso, M.H.; Franco, O.L.; Dias, S.C. Marine organisms as a rich source of biologically active peptides. Front. Mar. Sci. 2021, 8, 667764. [Google Scholar] [CrossRef]
- Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. A rapid esterification by means of mixed anhydride and its application to large-ring lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989. [Google Scholar] [CrossRef]
- Londregan, A.T.; Farley, K.A.; Limberakis, C.; Mullins, P.B.; Piotrowski, D.W. A new and useful method for the macrocyclization of linear peptides. Org. Lett. 2012, 14, 2890–2893. [Google Scholar] [CrossRef]
- Umehara, M.; Negishi, T.; Tashiro, T.; Nakao, Y.; Kimura, J. Structure-related cytotoxic activity of derivatives from kulokekahilide-2, a cyclodepsipeptide in Hawaiian marine mollusk. Bioorg. Med. Chem. Lett. 2012, 22, 7422–7425. [Google Scholar] [CrossRef]
- Parenty, A.; Moreau, X.; Campagne, J.-M. Macrolactonizations in the total synthesis of natural products. Chem. Rev. 2006, 106, 911–939. [Google Scholar] [CrossRef]
- Davies, J.S. The cyclization of peptides and depsipeptides. J. Pept. Sci. 2003, 9, 471–501. [Google Scholar] [CrossRef]
- Coin, I.; Beerbaum, M.; Schmieder, P.; Bienert, M.; Beyermann, M. Solid-phase synthesis of a cyclodepsipeptide: Cotransin. Org. Lett. 2008, 10, 3857–3860. [Google Scholar] [CrossRef] [PubMed]
- Maharani, R.; Brownlee, R.T.; Hughes, A.B.; Abbott, B.M. A total synthesis of a highly N-methylated cyclodepsipeptide [2S, 3S-Hmp]-aureobasidin L using solid-phase methods. Tetrahedron 2014, 70, 2351–2358. [Google Scholar] [CrossRef]
- Shin, D.-S.; Lee, Y.-S. Synthesis of Pentafluorophenyl Esters of Nitroveratryloxycarbonyl-Protected Amino Acids. Synlett 2009, 2009, 3307–3310. [Google Scholar]
- Thomson, S.A.; Josey, J.A.; Cadilla, R.; Gaul, M.D.; Hassman, C.F.; Luzzio, M.J.; Pipe, A.J.; Reed, K.L.; Ricca, D.J.; Wiethe, R.W. Fmoc mediated synthesis of peptide nucleic acids. Tetrahedron 1995, 51, 6179–6194. [Google Scholar] [CrossRef]
- Ferrazzano, L.; Catani, M.; Cavazzini, A.; Martelli, G.; Corbisiero, D.; Cantelmi, P.; Fantoni, T.; Mattellone, A.; De Luca, C.; Felletti, S. Sustainability in peptide chemistry: Current synthesis and purification technologies and future challenges. Green Chem. 2022, 24, 975–1020. [Google Scholar] [CrossRef]
- Al Musaimi, O.; Beatriz, G.; Albericio, F. Greening Fmoc/t Bu solid-phase peptide synthesis. Green Chem. 2020, 22, 996–1018. [Google Scholar] [CrossRef]
- Palladino, P.; Stetsenko, D.A. New TFA-free cleavage and final deprotection in Fmoc solid-phase peptide synthesis: Dilute HCl in fluoro alcohol. Org. Lett. 2012, 14, 6346–6349. [Google Scholar] [CrossRef]
- Albericio, F.; El-Faham, A. Choosing the right coupling reagent for peptides: A twenty-five-year journey. Org. Process Res. Dev. 2018, 22, 760–772. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Novel proton acceptor immonium-type coupling reagents: Application in solution and solid-phase peptide synthesis. Org. Lett. 2007, 9, 4475–4477. [Google Scholar] [CrossRef] [PubMed]
- McKnelly, K.J.; Sokol, W.; Nowick, J.S. Anaphylaxis induced by peptide coupling agents: Lessons learned from repeated exposure to HATU, HBTU, and HCTU. J. Org. Chem. 2019, 85, 1764–1768. [Google Scholar] [CrossRef]
- Marder, O.; Shvo, Y.; Albiricio, F. HCTU and TCTU. New coupling reagents: Development and industrial aspects. Chim. Oggi 2002, 20, 37–41. [Google Scholar] [CrossRef]
- Subiros-Funosas, R.; Moreno, J.A.; Bayo-Puxan, N.; Abu-Rabeah, K.; Ewenson, A.; Atias, D.; Marks, R.S.; Albericio, F. PyClocK, the phosphonium salt derived from 6-Cl-HOBt. Chim. Oggi 2008, 26, 10–12. [Google Scholar]
- Albericio, F.; Cases, M.; Alsina, J.; Triolo, S.A.; Carpino, L.A.; Kates, S.A. On the use of PyAOP, a phosphonium salt derived from HOAt, in solid-phase peptide synthesis. Tetrahedron Lett. 1997, 38, 4853–4856. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. COMU: A third generation of uronium-type coupling reagents. J. Pept. Sci. 2010, 16, 6–9. [Google Scholar] [CrossRef]
- Paul, V.J.; Puglisi, M.P. Chemical mediation of interactions among marine organisms. Nat. Prod. Rep. 2004, 21, 189–209. [Google Scholar] [CrossRef]
- Paul, V.J.; Ritson-Williams, R. Marine chemical ecology. Nat. Prod. Rep. 2008, 25, 662–695. [Google Scholar] [CrossRef]




















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, R.; Pinto, E.; Fernandes, C.; Sousa, E. Marine Cyclic Peptides: Antimicrobial Activity and Synthetic Strategies. Mar. Drugs 2022, 20, 397. https://doi.org/10.3390/md20060397
Ribeiro R, Pinto E, Fernandes C, Sousa E. Marine Cyclic Peptides: Antimicrobial Activity and Synthetic Strategies. Marine Drugs. 2022; 20(6):397. https://doi.org/10.3390/md20060397
Chicago/Turabian StyleRibeiro, Ricardo, Eugénia Pinto, Carla Fernandes, and Emília Sousa. 2022. "Marine Cyclic Peptides: Antimicrobial Activity and Synthetic Strategies" Marine Drugs 20, no. 6: 397. https://doi.org/10.3390/md20060397
APA StyleRibeiro, R., Pinto, E., Fernandes, C., & Sousa, E. (2022). Marine Cyclic Peptides: Antimicrobial Activity and Synthetic Strategies. Marine Drugs, 20(6), 397. https://doi.org/10.3390/md20060397