
Quantitation Overcoming Matrix Effects of Lipophilic Toxins in Mytilus galloprovincialis by Liquid Chromatography-Full Scan High Resolution Mass Spectrometry Analysis (LC-HR-MS)

 ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results
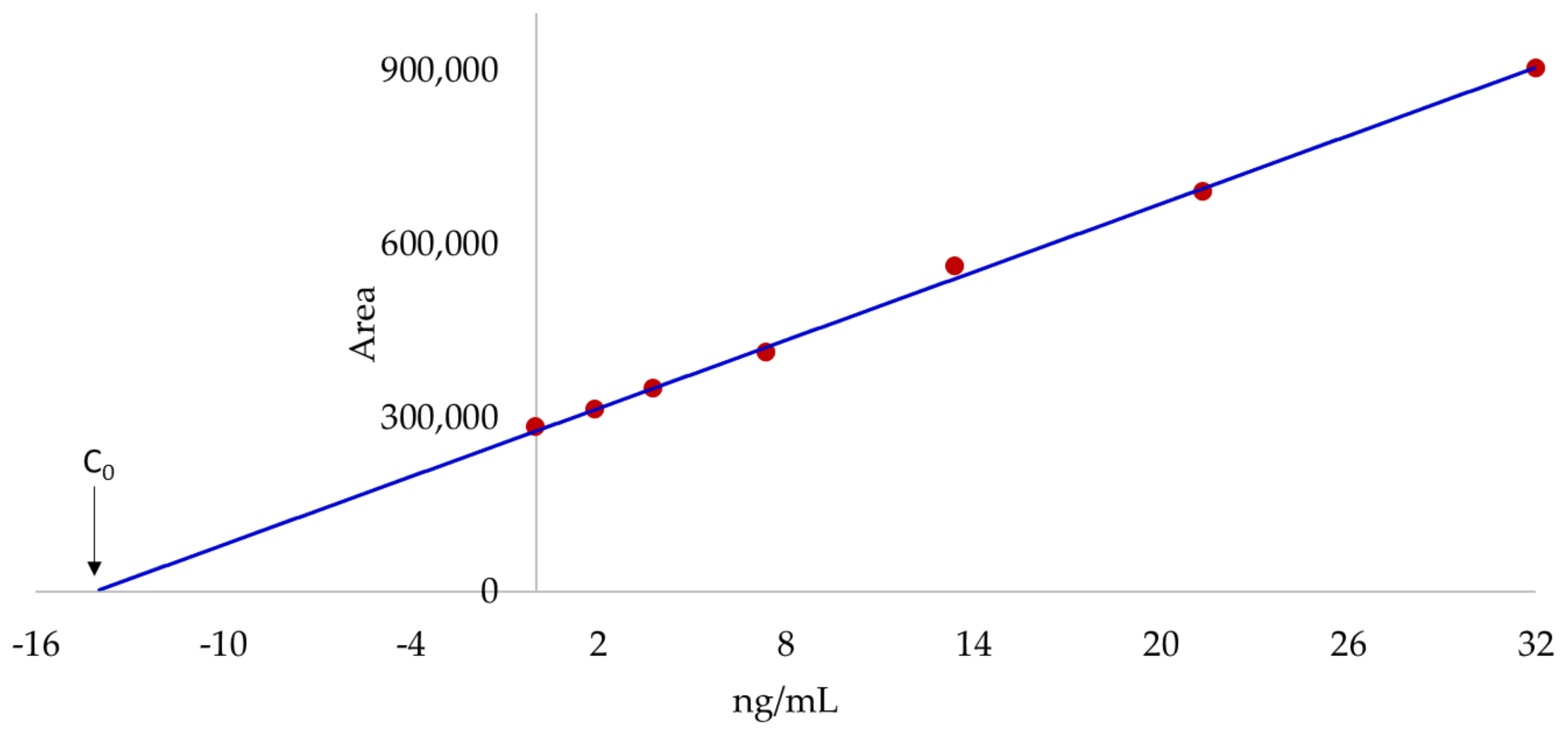
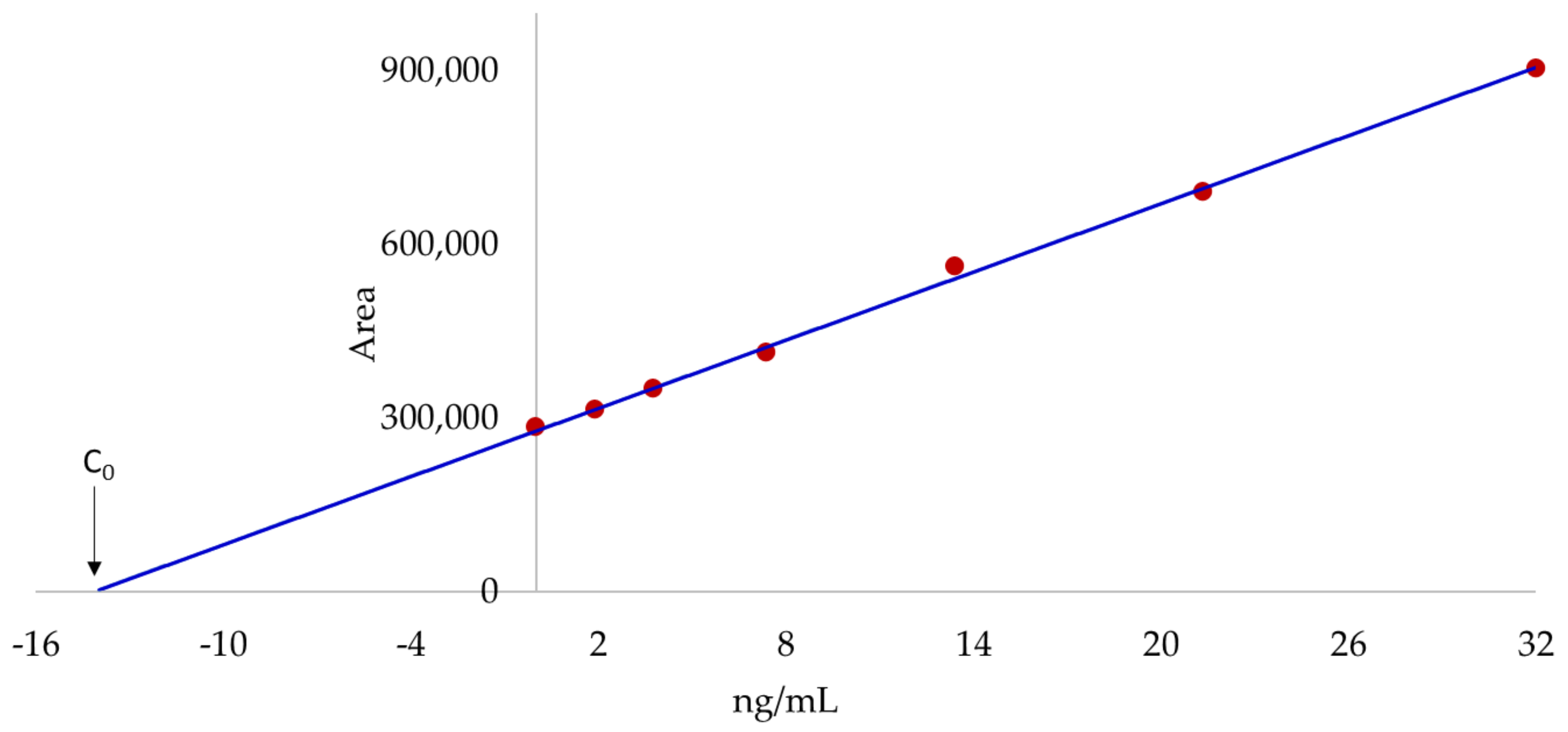
2.1. Accurate Mass-Extracted Ion Chromatogram (AM-XIC) Based Quantitation
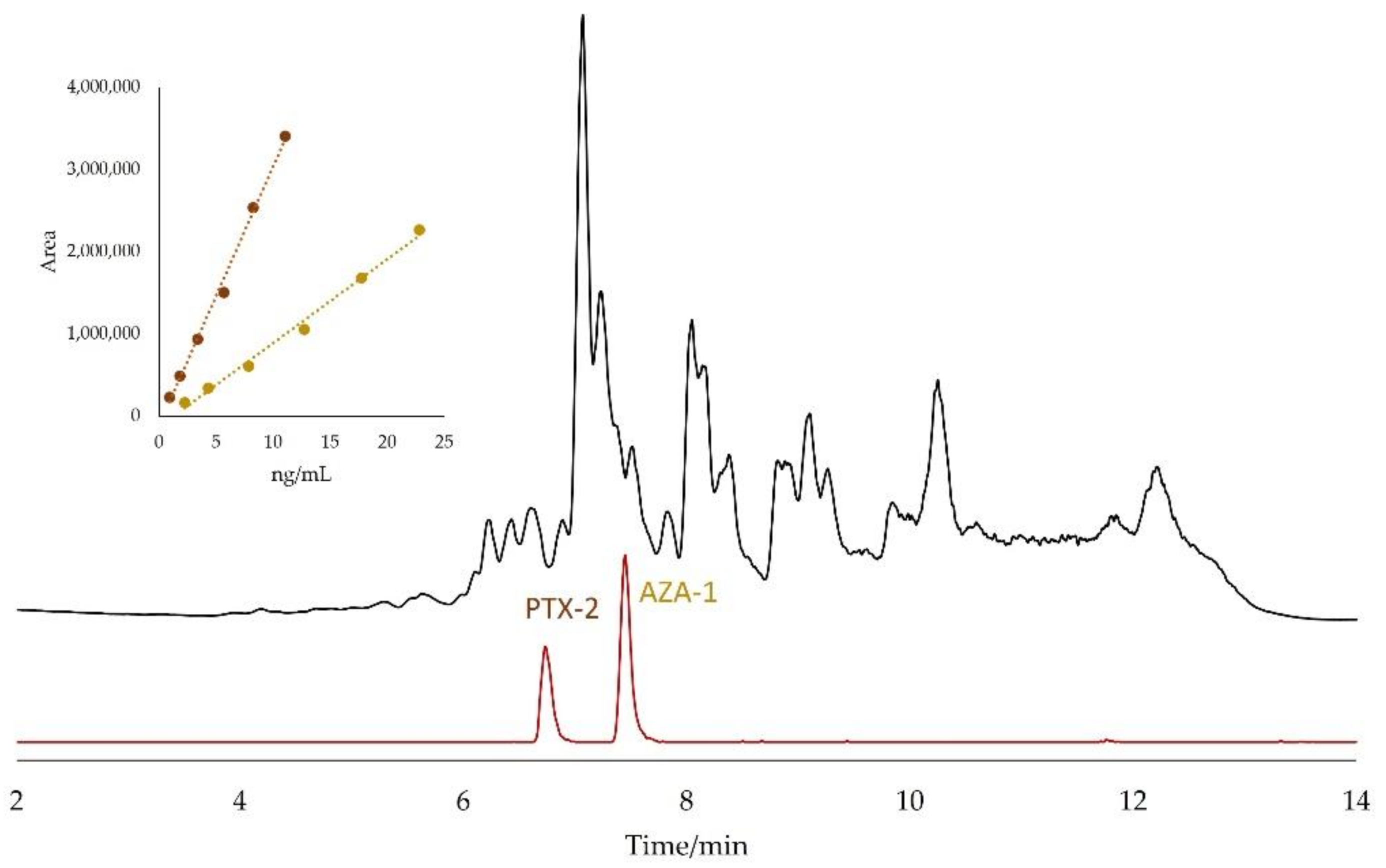
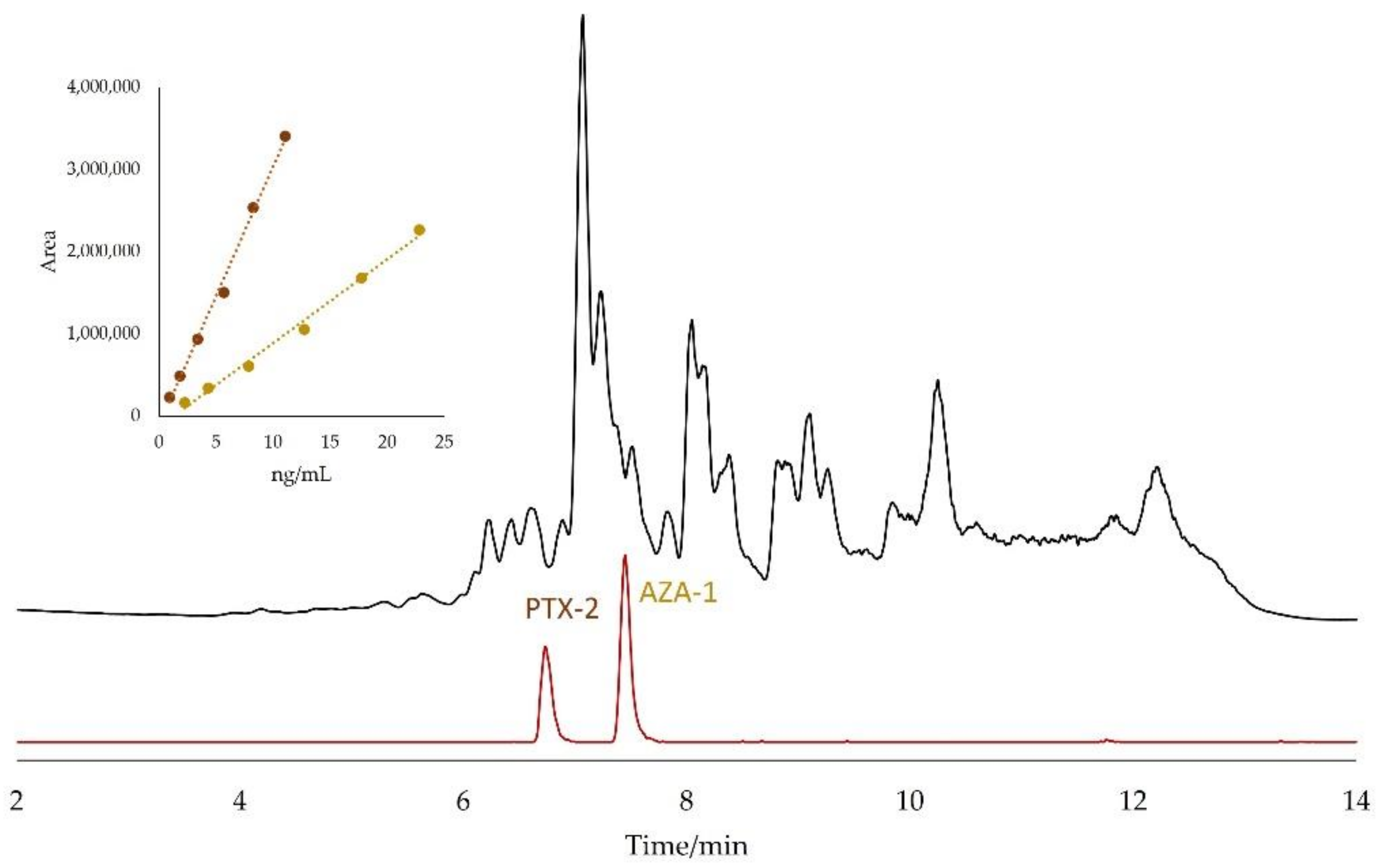
2.2. Matrix Effects

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample and Toxin | Non-Hydrolyzed Extract | % RSD (in-Batch) | Hydrolyzed Extract | % RSD (in-Batch) |
|---|---|---|---|---|
| Fresh mussels | ||||
| OA | 45.2 | 2.4 | 41.1 | 1.9 |
| DTX-1 | 41.9 | 3.9 | 20.5 | 4.2 |
| DTX-2 | 43.8 | 3.3 | 37.5 | 2.5 |
| YTX | 108.6 | 7.6 | 100.1 | 10.1 |
| AZA-1 | 80.6 | 4.5 | 76.6 | 8.2 |
| PTX-2 | 96.7 | 5.5 | 98.1 | 8.8 |
| Reference sample of mussels | ||||
| OA | 51.5 | 0.9 | 33.7 | 5.9 |
| DTX-1 | 35.5 | 0.7 | 25.6 | 4.3 |
| DTX-2 | 50.5 | 4.6 | 26.4 | 8.0 |
| YTX | 57.6 | 9.5 | 76.0 | 4.0 |
| AZA-1 | 71.0 | 1.5 | 71.2 | 4.4 |
| PTX-2 | 65.5 | 0.8 | 77.2 | 3.3 |
| Toxin | Non-Hydrolyzed Extract | % RSD (between Samples) | Hydrolyzed Extract | % RSD (between Samples) |
|---|---|---|---|---|
| OA | 37 a | 33 | 33 a | 23 |
| DTX-1 | 41 a | 21 | 18 b | 24 |
| DTX-2 | 40 a | 24 | 37 a | 27 |
| YTX | 108 a | 17 | 96 a | 17 |
| AZA-1 | 88 a | 21 | 84 a | 17 |
| PTX-2 | 93 a | 23 | 95 a | 7 |
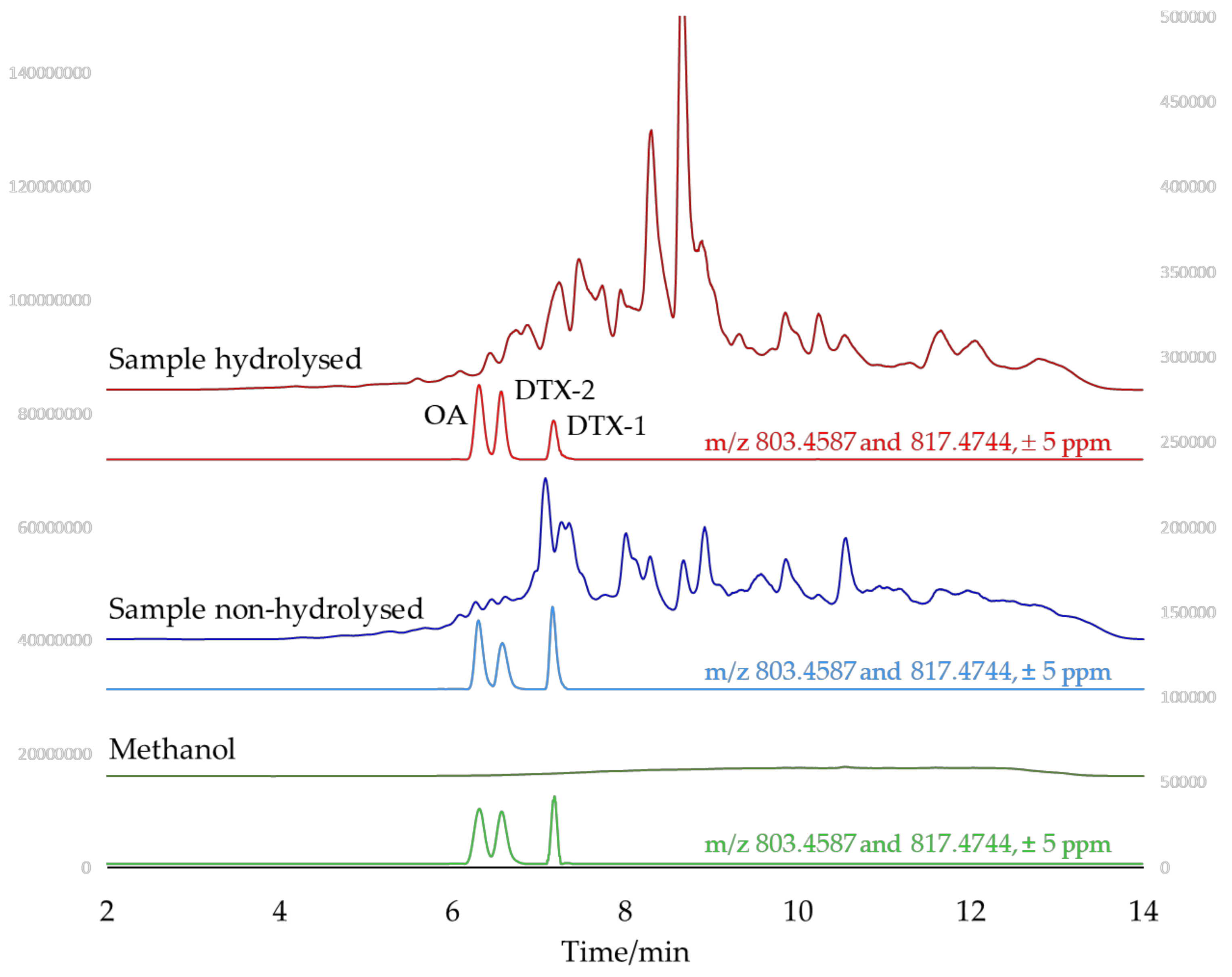
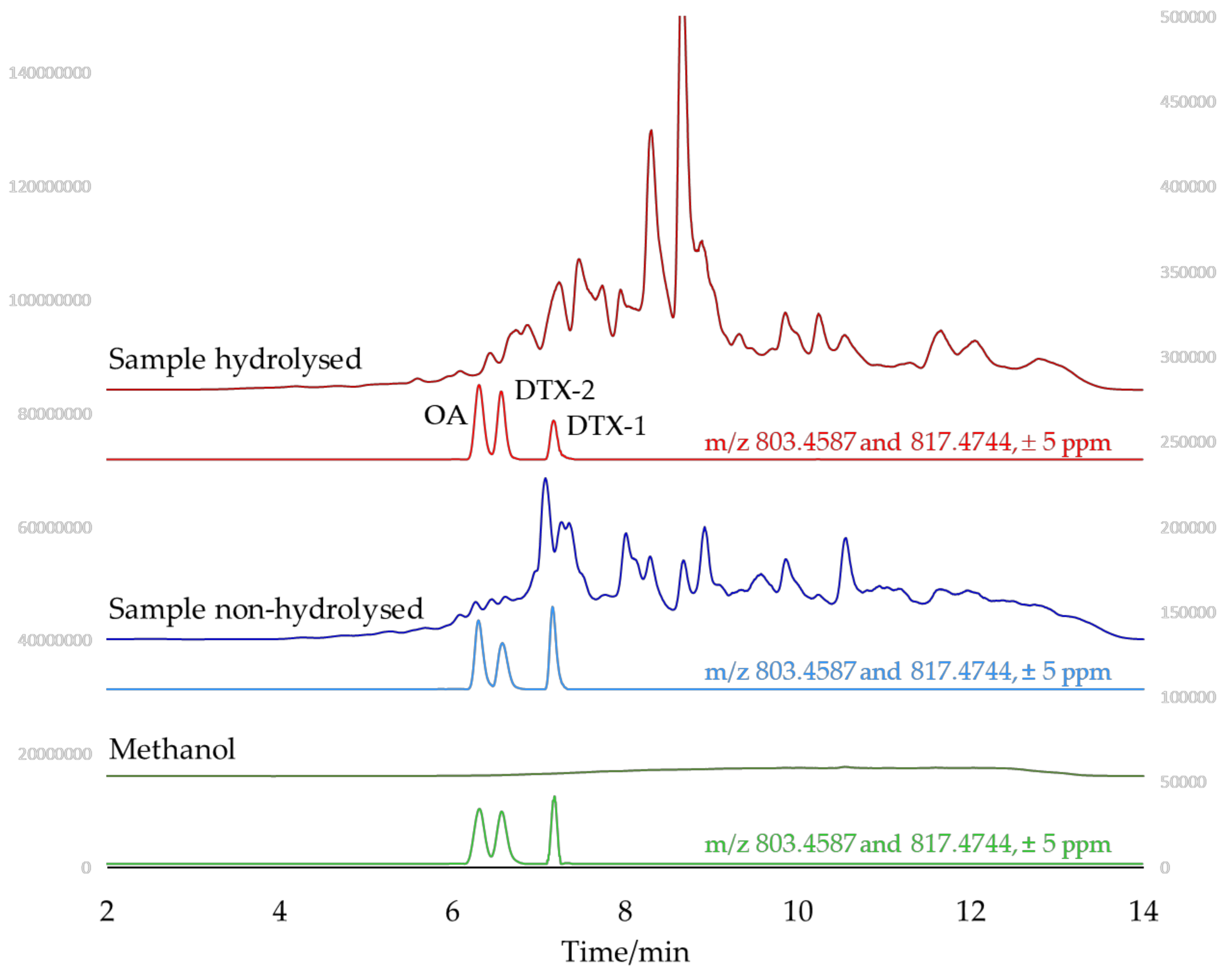
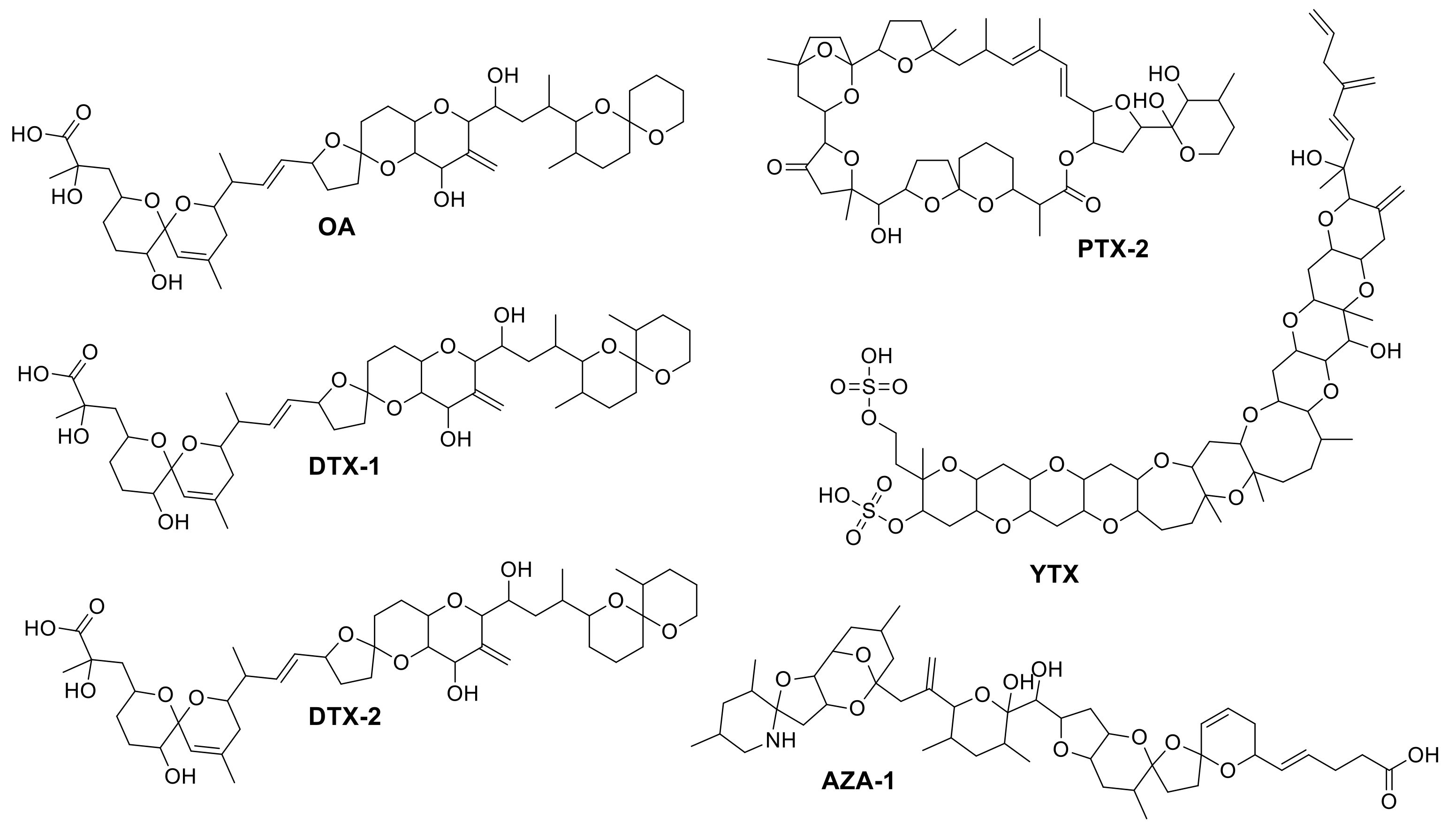
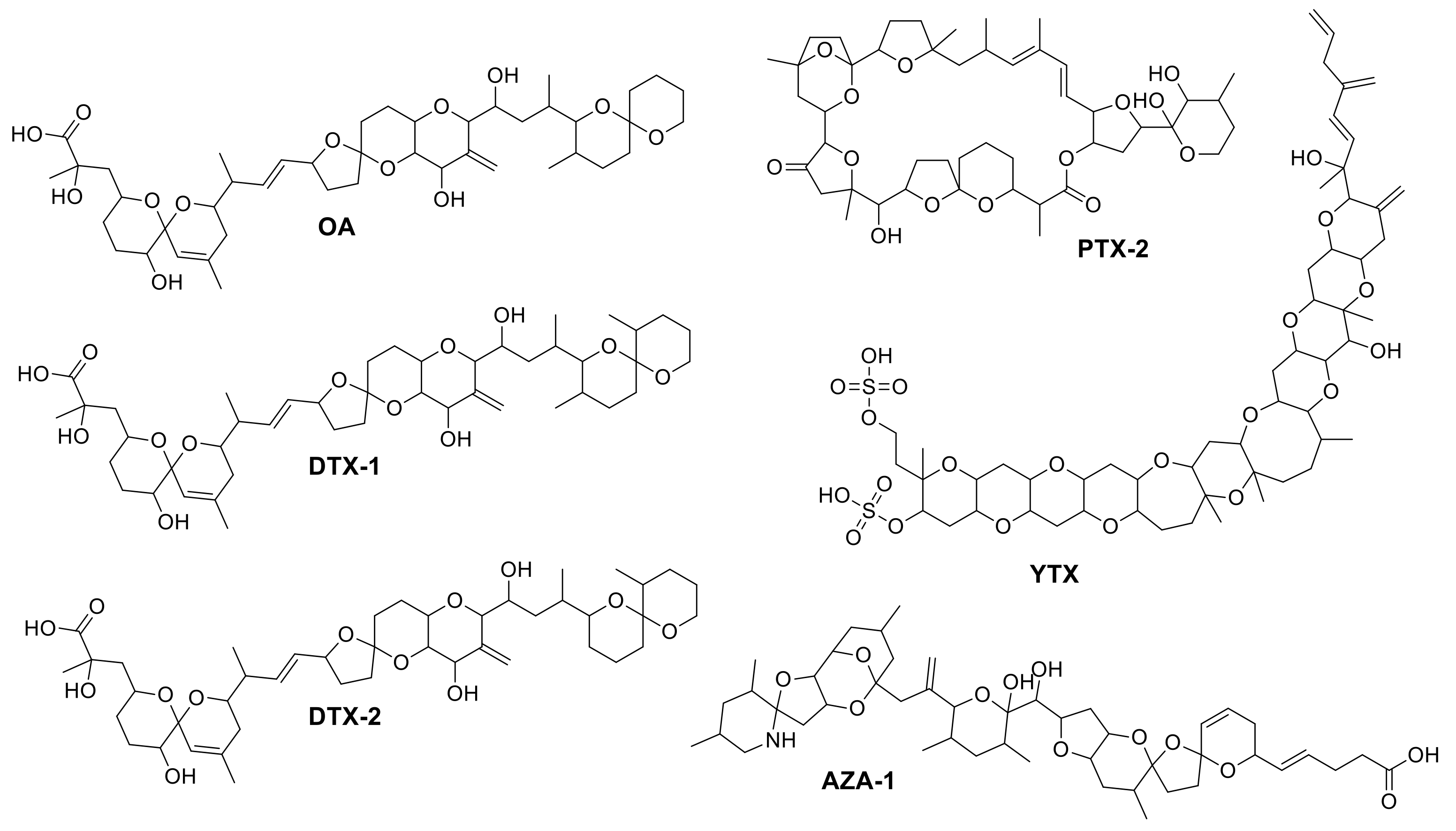
2.3. Identification of Matrix Compounds
2.4. Quantitation by the Standard Addition Method
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Mussel Samples
4.3. Extraction and Addition of Standards
4.4. LC-MS Conditions
4.5. Quantitation and Profile Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Farabegoli, F.; Blanco, L.; Rodriguez, L.; Vieites, J.; Cabado, A. Phycotoxins in Marine Shellfish: Origin, Occurrence and Effects on Humans. Mar. Drugs 2018, 16, 188. [Google Scholar] [CrossRef] [Green Version]
- Visciano, P.; Schirone, M.; Berti, M.; Milandri, A.; Tofalo, R.; Suzzi, G. Marine Biotoxins: Occurrence, Toxicity, Regulatory Limits and Reference Methods. Front. Microbiol. 2016, 7, 1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, K.; Jensen, L. Distribution of diarrhetic shellfish poisoning toxins in consignments of blue mussel. Food Addit. Contam. 2004, 21, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Klijnstra, M.; Faassen, E.; Gerssen, A. A Generic LC-HRMS Screening Method for Marine and Freshwater Phycotoxins in Fish, Shellfish, Water, and Supplements. Toxins 2021, 13, 823. [Google Scholar] [CrossRef]
- Stobo, L.; Lacaze, J.; Scott, A.; Gallacher, S.; Smith, E.; Quilliam, M. Liquid chromatography with mass spectrometry-Detection of lipophilic shellfish toxins. J. AOAC Int. 2005, 88, 1371–1382. [Google Scholar] [CrossRef] [Green Version]
- Regulation (EC) No 853/2004 of the European parliament and of the council of 29 April 2004 laying down specific hygiene rules for food of animal origin. Off. J. Eur. Union 2004, 139, 55–205.
- Commission Delegated Regulation (EU) 2021/1374 of 12 April 2021 amending Annex III to Regulation (EC) No 853/2004 of the European Parliament and of the Council on specific hygiene requirements for food of animal origin. Off. J. Eur. Union 2021, 297, 1–15.
- EURLMB. EU-Harmonised Standard Operating Procedure for Determination of Lipophilic Marine Biotoxins in Molluscs by LC-MS/MS, Version 5. 2015. Available online: https://www.aesan.gob.es/CRLMB/docs/docs/metodos_analiticos_de_desarrollo/EU-Harmonised-SOP-LIPO-LCMSMS_Version5.pdf (accessed on 29 January 2022).
- Rochat, B.; Kottelat, E.; McMullen, J. The future key role of LC-high-resolution-MS analyses in clinical laboratories: A focus on quantification. Bioanalysis 2012, 4, 2939–2958. [Google Scholar] [CrossRef] [PubMed]
- Trufelli, H.; Palma, P.; Famiglini, G.; Cappiello, A. An Overview of Matrix Effects in Liquid Chromatography-Mass Spectrometry. Mass Spectrom. Rev. 2011, 30, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.; Gigliobianco, M.; Magnoni, F.; Censi, R.; Di Martino, P. Compensate for or Minimize Matrix Effects? Strategies for Overcoming Matrix Effects in Liquid Chromatography-Mass Spectrometry Technique: A Tutorial Review. Molecules 2020, 25, 3047. [Google Scholar] [CrossRef] [PubMed]
- Fux, E.; Rode, D.; Bire, R.; Hess, P. Approaches to the evaluation of matrix effects in the liquid chromatography-mass spectrometry (LC-MS) analysis of three regulated lipophilic toxin groups in mussel matrix (Mytilus edulis). Food Addit. Contam. Part A-Chem. Anal. Control. Expo. Risk Assess. 2008, 25, 1024–1032. [Google Scholar] [CrossRef]
- Gerssen, A.; McElhinney, M.; Mulder, P.; Bire, R.; Hess, P.; de Boer, J. Solid phase extraction for removal of matrix effects in lipophilic marine toxin analysis by liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2009, 394, 1213–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Chen, H.; Ji, Y.; Li, T.; Li, A. Evaluation of different strategies to minimize the matrix effects on LC-MS/MS analysis of multiple lipophilic shellfish toxins in both acidic and alkaline chromatographic conditions. Toxicon 2020, 188, 16–26. [Google Scholar] [CrossRef] [PubMed]
- These, A.; Scholz, J.; Preiss-Weigert, A. Sensitive method for the determination of lipophilic marine biotoxins in extracts of mussels and processed shellfish by high-performance liquid chromatography-tandem mass spectrometry based on enrichment by solid-phase extraction. J. Chromatogr. A 2009, 1216, 4529–4538. [Google Scholar] [CrossRef] [PubMed]
- Miles, C.; Kilcoyne, J.; McCarron, P.; Giddings, S.; Waaler, T.; Rundberget, T.; Samdal, I.; Lovberg, K. Selective Extraction and Purification of Azaspiracids from Blue Mussels (Mytilus edulis) Using Boric Acid Gel. J. Agric. Food Chem. 2018, 66, 2962–2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.; Zhou, Y.; Statler, K.; Thomas, J.; Cox, F.; Hall, S.; Barr, J. Quantification of Saxitoxin and Neosaxitoxin in Human Urine Utilizing Isotope Dilution Tandem Mass Spectrometry. J. Anal. Toxicol. 2009, 33, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fux, E.; McMillan, D.; Bire, R.; Hess, P. Development of an ultra-performance liquid chromatography-mass spectrometry method for the detection of lipophilic marine toxins. J. Chromatogr. A 2007, 1157, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Tsukada, K. Matrix effect and correction by standard addition in quantitative liquid chromatographic-mass spectrometric analysis of diarrhetic shellfish poisoning toxins. J. Chromatogr. A 2002, 943, 39–46. [Google Scholar] [CrossRef]
- Rodriguez, I.; Alfonso, A.; Gonzalez-Jartin, J.; Vieytes, M.; Botana, L. A single run UPLC-MS/MS method for detection of all EU-regulated marine toxins. Talanta 2018, 189, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Bruce, G.; Gill, P. Estimates of precision in a standard additions analysis. J. Chem. Educ. 1999, 76, 805–807. [Google Scholar] [CrossRef]
- Zendong, Z.; McCarron, P.; Herrenknecht, C.; Sibat, M.; Amzil, Z.; Cole, R.; Hess, P. High resolution mass spectrometry for quantitative analysis and untargeted screening of algal toxins in mussels and passive samplers. J. Chromatogr. A 2015, 1416, 10–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zendong, Z.; Bertrand, S.; Herrenknecht, C.; Abadie, E.; Jauzein, C.; Lemee, R.; Gouriou, J.; Amzil, Z.; Hess, P. Passive Sampling and High Resolution Mass Spectrometry for Chemical Profiling of French Coastal Areas with a Focus on Marine Biotoxins. Environ. Sci. Technol. 2016, 50, 8522–8529. [Google Scholar] [CrossRef]
- Siriwardane, D.; Wang, C.; Jiang, W.; Mudalige, T. Quantification of phospholipid degradation products in liposomal pharmaceutical formulations by ultra performance liquid chromatography-mass spectrometry (UPLC-MS). Int. J. Pharm. 2020, 578, 119077. [Google Scholar] [CrossRef] [PubMed]
- Khoury, S.; El Banna, N.; Tfaili, S.; Chaminade, P. A study of inter-species ion suppression in electrospray ionization-mass spectrometry of some phospholipid classes. Anal. Bioanal. Chem. 2016, 408, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
| Toxin | Sample | |||
|---|---|---|---|---|
| Pasteurized | Mincha | Medium | Large | |
| OA | 5.4 ± 1.1 | 11.6 ± 1.9 | 11.8 ± 1.4 | 14.1 ± 0.9 |
| DTX-2 | <LOQ | 2.3 ± 0.7 | <LOQ | 3.0 ± 0.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, C.Q.V.; Afonso, I.I.; Lage, S.; Costa, P.R.; Canário, A.V.M.; Da Silva, J.P. Quantitation Overcoming Matrix Effects of Lipophilic Toxins in Mytilus galloprovincialis by Liquid Chromatography-Full Scan High Resolution Mass Spectrometry Analysis (LC-HR-MS). Mar. Drugs 2022, 20, 143. https://doi.org/10.3390/md20020143
Costa CQV, Afonso II, Lage S, Costa PR, Canário AVM, Da Silva JP. Quantitation Overcoming Matrix Effects of Lipophilic Toxins in Mytilus galloprovincialis by Liquid Chromatography-Full Scan High Resolution Mass Spectrometry Analysis (LC-HR-MS). Marine Drugs. 2022; 20(2):143. https://doi.org/10.3390/md20020143
Chicago/Turabian StyleCosta, Camila Q. V., Inês I. Afonso, Sandra Lage, Pedro Reis Costa, Adelino V. M. Canário, and José P. Da Silva. 2022. "Quantitation Overcoming Matrix Effects of Lipophilic Toxins in Mytilus galloprovincialis by Liquid Chromatography-Full Scan High Resolution Mass Spectrometry Analysis (LC-HR-MS)" Marine Drugs 20, no. 2: 143. https://doi.org/10.3390/md20020143
APA StyleCosta, C. Q. V., Afonso, I. I., Lage, S., Costa, P. R., Canário, A. V. M., & Da Silva, J. P. (2022). Quantitation Overcoming Matrix Effects of Lipophilic Toxins in Mytilus galloprovincialis by Liquid Chromatography-Full Scan High Resolution Mass Spectrometry Analysis (LC-HR-MS). Marine Drugs, 20(2), 143. https://doi.org/10.3390/md20020143