1. Introduction
Natural products play an increasingly crucial role in drug discovery, owing to their great chemical and bioactive diversity [
1]. As is well known, fungi are a rich source of novel natural products [
2], and, in the last two decades, the fungal sources of new metabolites have been broadened from terrestrial strains to marine habitats [
3]. The ocean turned out to be an attractive environment, since the search for new biomedicals from marine microorganisms resulted in the isolation of approximately 10,000 metabolites, many of which were endowed with pharmacodynamic properties [
4].
Mangrove is a special marine ecosystem that occurs in tropical and subtropical intertidal estuarine zones, and is characterized by high salinity and rich organic matter [
5], which makes it an extremely diverse microbial resource. Mangrove-associated fungi, as the second-largest ecological group of marine fungi [
6], have proven to be a rich source of natural products, with unique chemical structures and diverse pharmacological activities. Up to now, a large number of metabolites produced by mangrove fungi have been reported, including alkaloids, macrolides, polyketides, quinones, terpenes, and so on [
6], which displayed diverse bioactivity, such as antibacterial, insecticidal, antioxidant, and cytotoxic, etc. These findings indicate that mangrove-derived fungi may possess great potential to produce novel and active secondary metabolites. As part of our ongoing work searching for natural products from mangrove-derived fungi, one fungus,
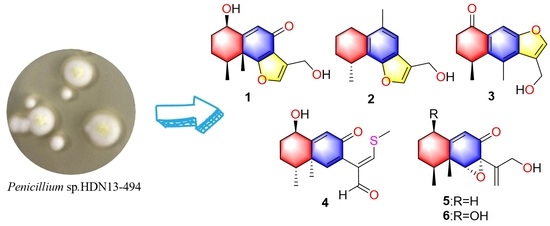
Penicillium sp. HDN13-494, from the root soil sample of a mangrove plant in Wenchang, Hainan, was selected for further chemical studies. Herein, the details of isolation, structural elucidation, and bioactivities of its metabolites are reported.
2. Results and Discussion
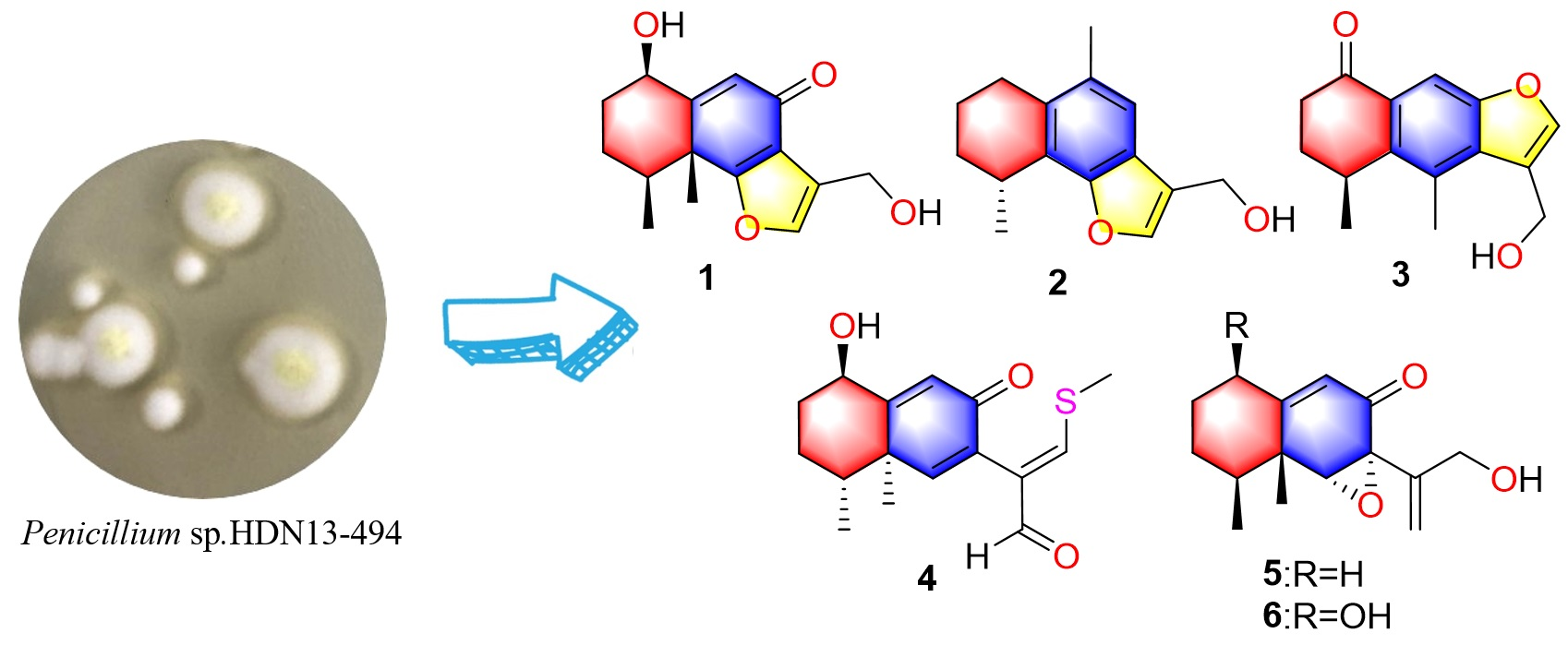
Chemical studies of
Penicillium sp. HDN13-494 led to the isolation of five new sesquiterpenoids (
1–
5) and one known compound (
6) (
Figure 1). Compound
4 represents the first example of eremophilane sesquiterpenoids containing sulfur elements. Citreobenzofuran D (
1) and E (
2) possess a rare cyclic system with 6,12-epoxy-eremophilane-type, which has only been reported twice in natural products [
7,
8].
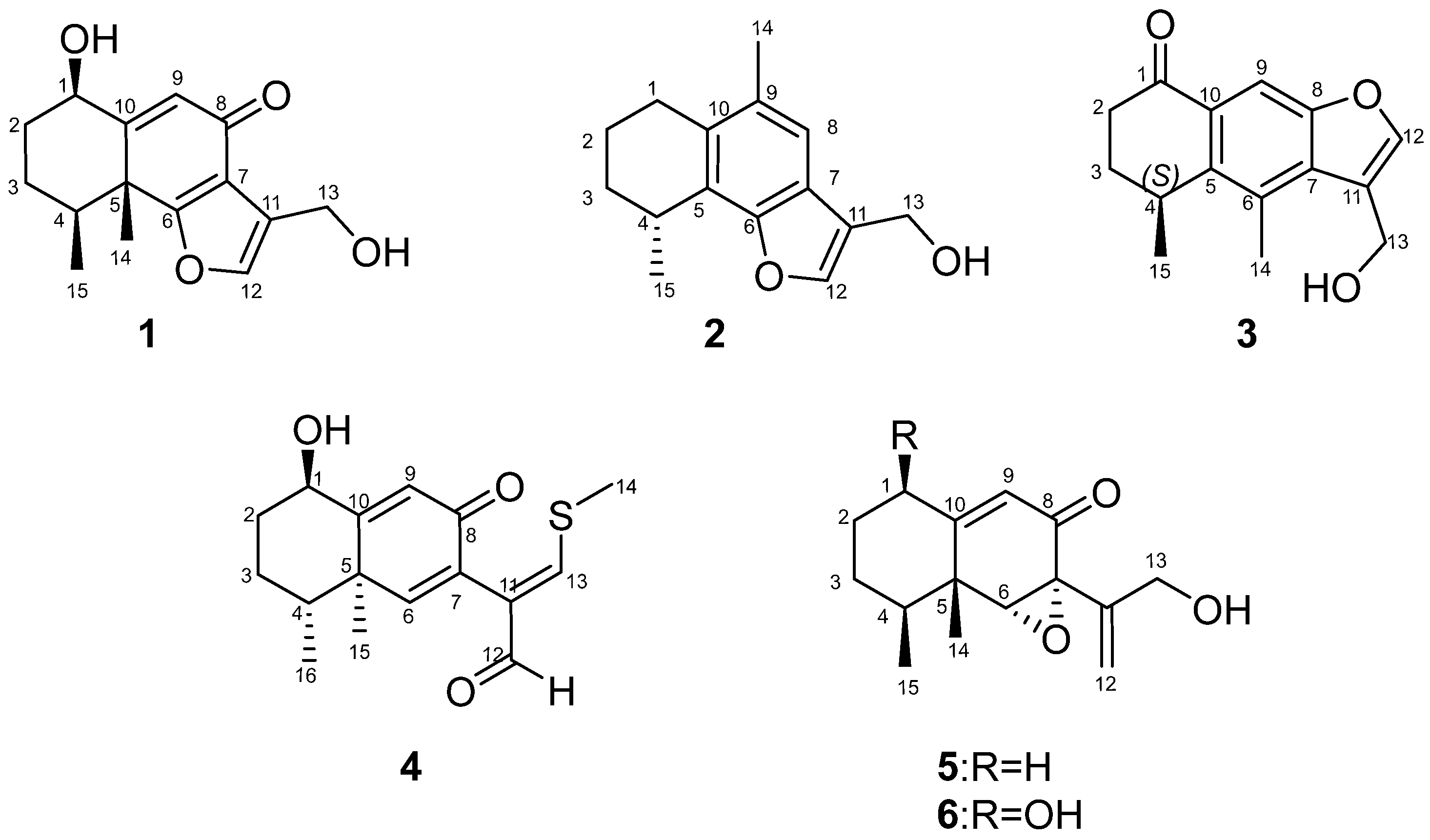
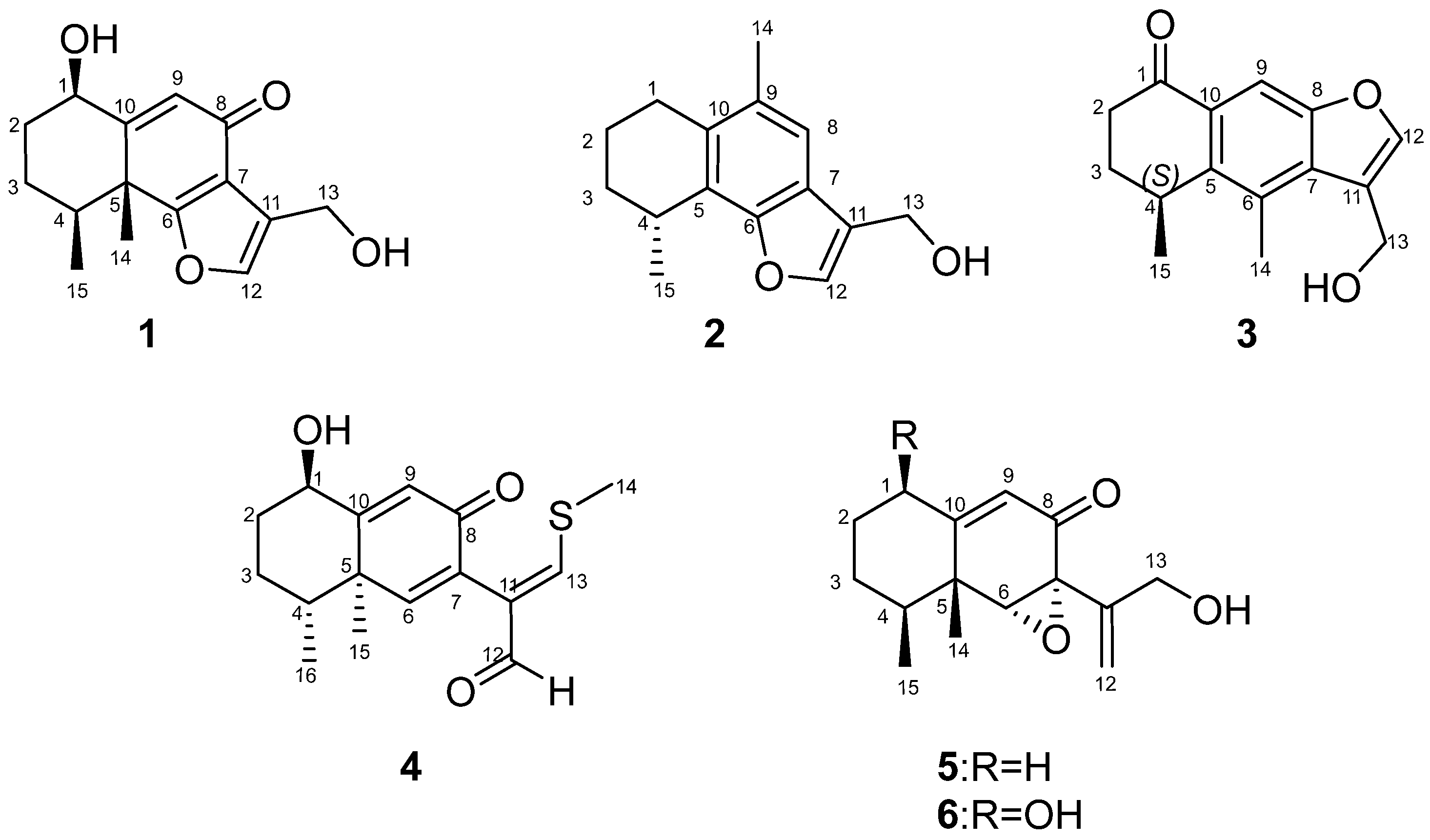
Citreobenzofuran D (
1) was obtained as a brown triclinic crystal with the molecular formula C
15H
18O
4, determined by HRESIMS data (
m/
z 261.1130 [M − H]
−) (
Figure S8). The 1D NMR data (
Table 1 and
Table 2) indicated that
1 shared the same skeleton with isoligularonic acid [
7], except for the appearance of two hydroxyl groups at C-1 (
δC 72.2) and C-13 (
δC 55.2) (
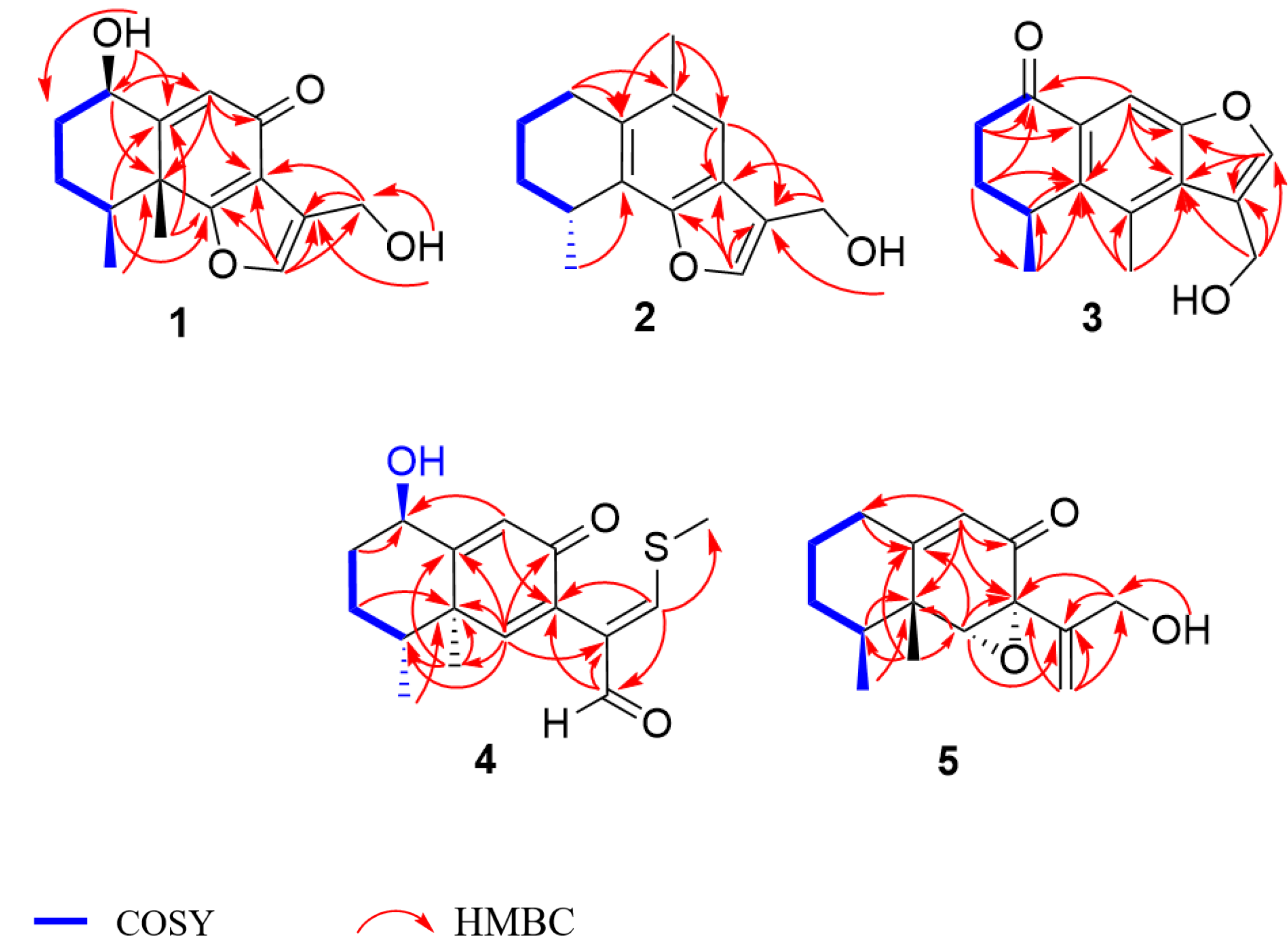
Table 1), which was supported by HMBC correlation from OH-1 to C-1, C-2, C-10, and from OH-13 to C-11 and C-13 (
Figure 2). In addition, the deshielded singlet at C-9 (
δC 125.5) and C-10 (
δC 163.5) indicated the occurrence of alkenylation between them (
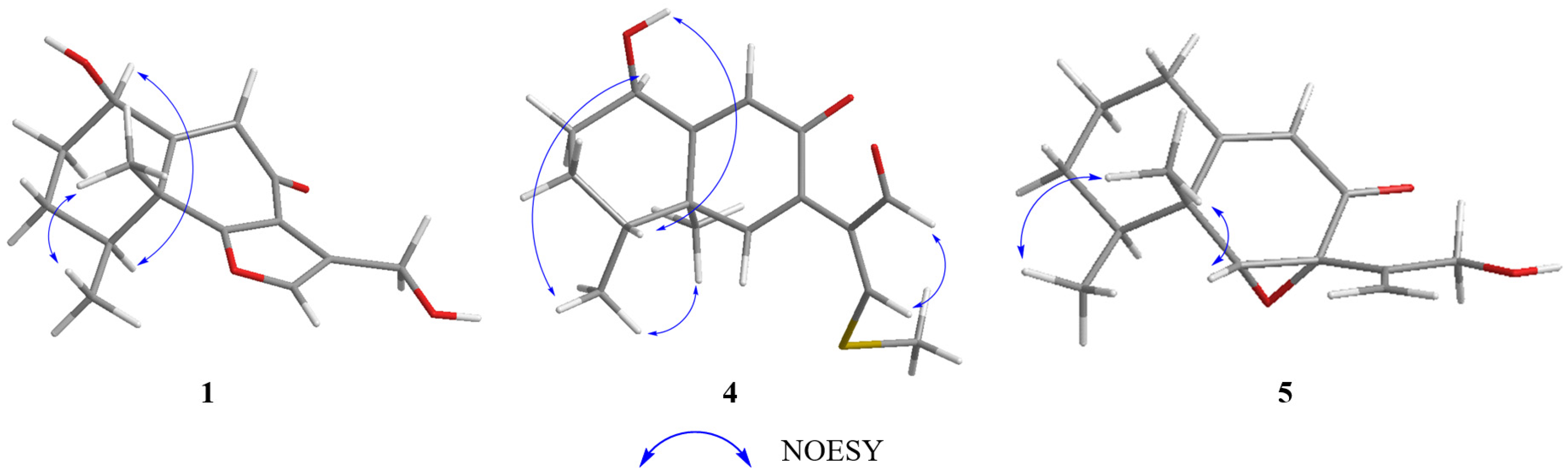
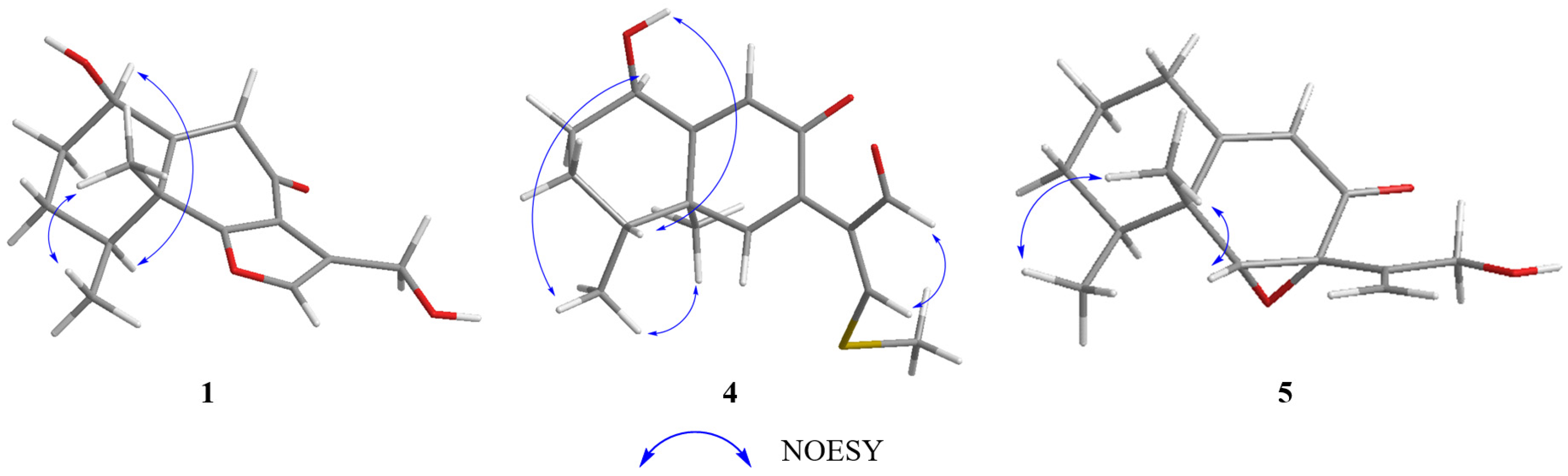
Table 1). The relative configuration of
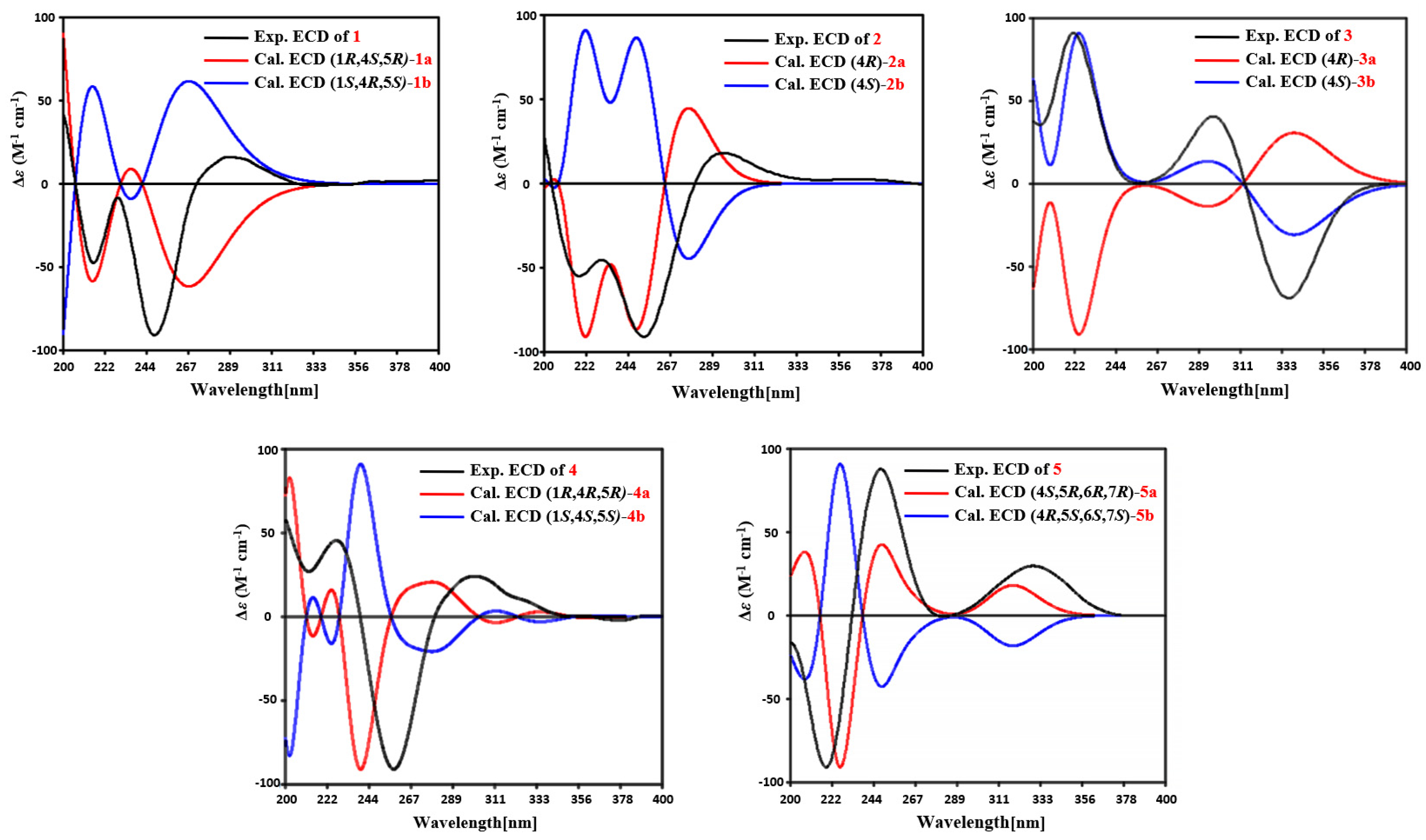
1 was elucidated as 1
R*, 4
S*, 5
R* on the basis of NOESY correlations of H-1 with H-4, and H-14 with H-15 (
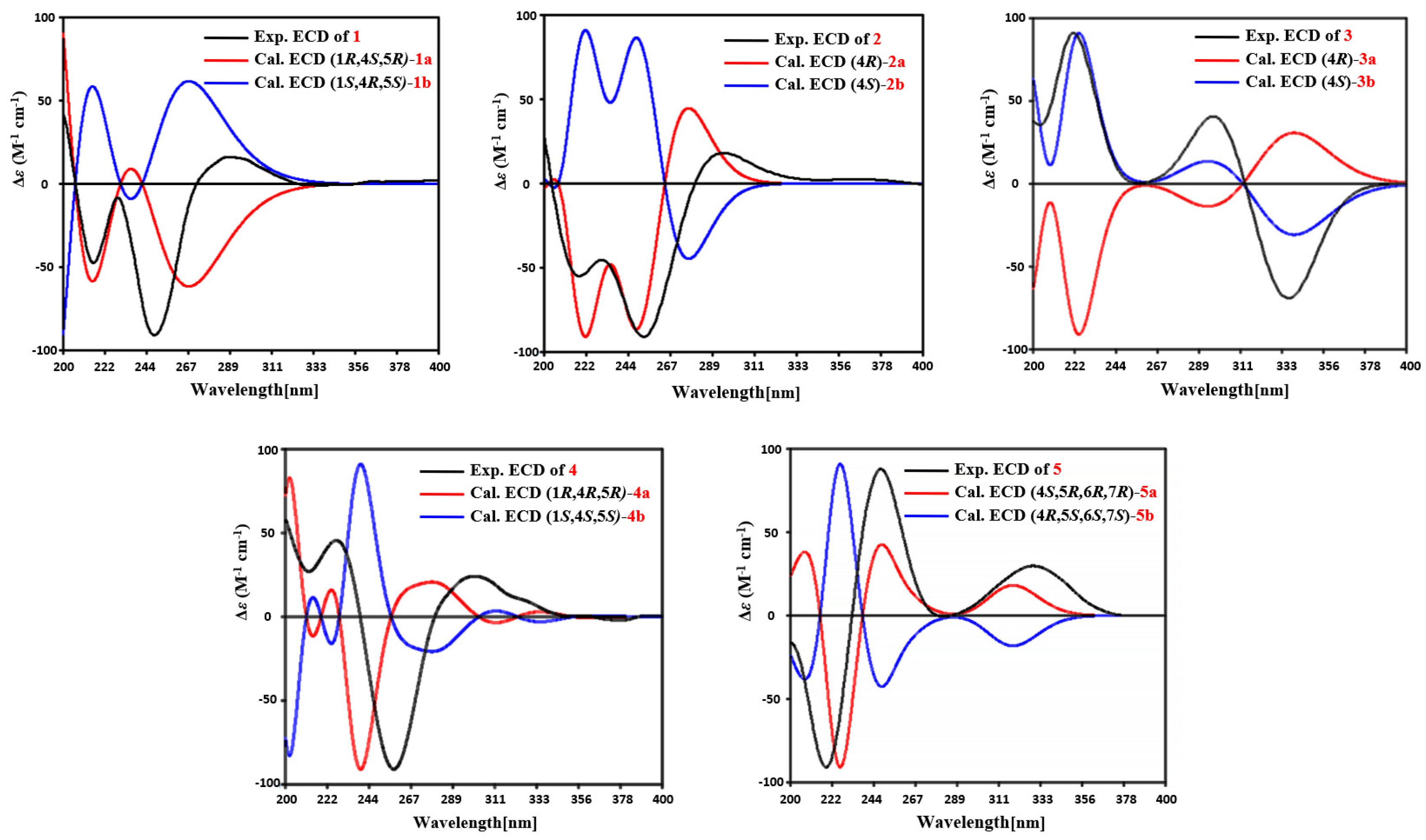
Figure 3). To determine the absolute configuration of
1, the ECD calculations of the optimized conformations of (1
R, 4
S, 5
R)-
1a and (1
S, 4
R, 5
S)-
1b were obtained at the B3LYP/6-31+G(d) level. The overall pattern of the experimental ECD spectrum was in reasonable agreement with the calculated ECD spectrum of
1a (
Figure 4), which indicated the 1
R, 4
S, and 5
R absolute configuration of
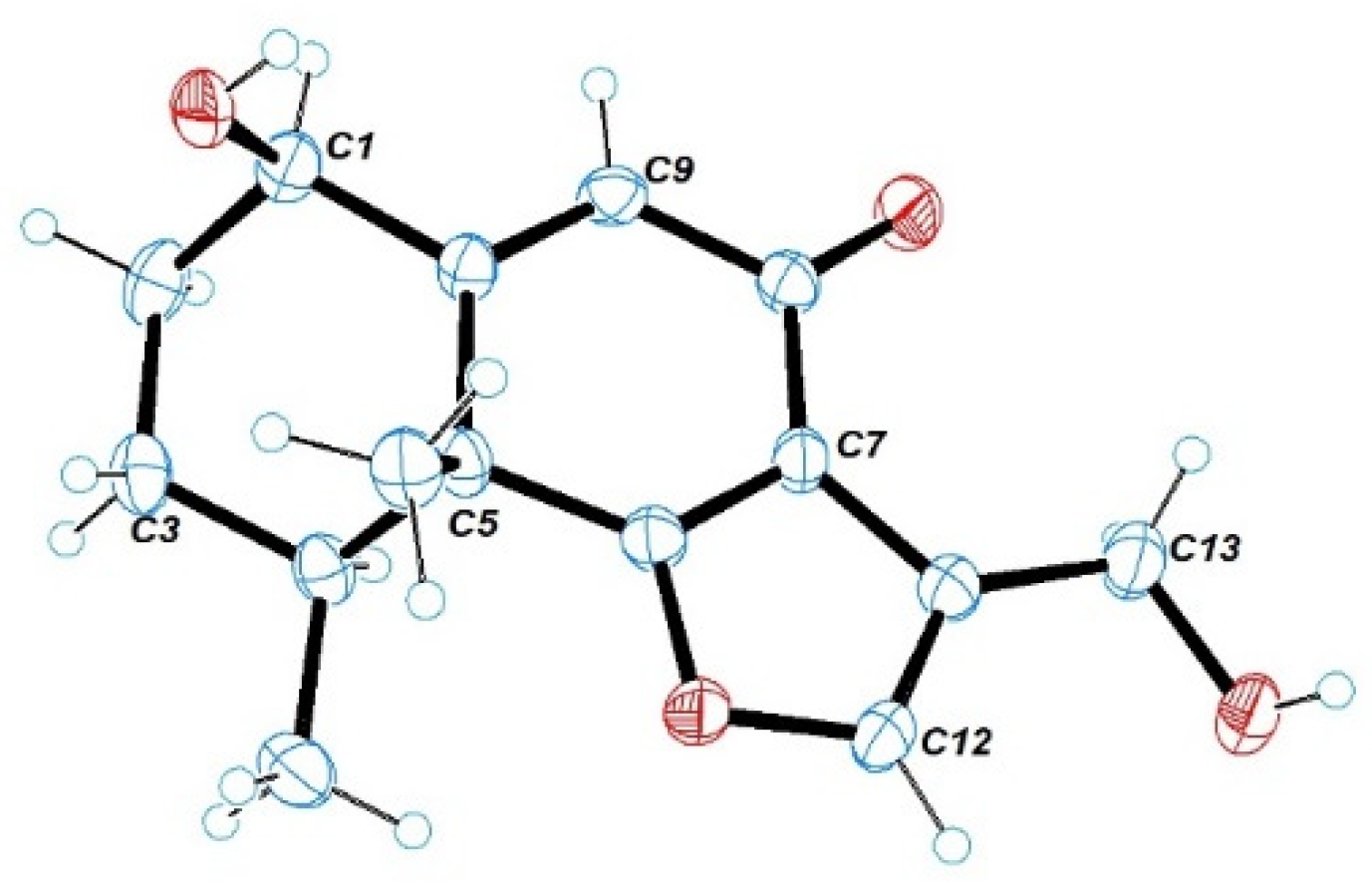
1. Single-crystal X-ray diffraction analysis by Cu K
a radiation further confirmed the absolute configuration of
1 (
Figure 5).
Citreobenzofuran E (
2) was obtained as a green amorphous powder, with the molecular formula C
15H
18O
2, based on the HRESIMS spectrum (
m/
z 229.1227 [M − H]
−) (
Figure S15), indicating seven degrees of unsaturation. Detailed analysis of the 1D NMR data of
2 (
Table 1 and
Table 2) revealed highly structural similarities to the known compound citreobenzofuran B [
8]. The difference was the disappearance of the hydroxyl group at C-3 (
δC 29.6), supported by the shielded singlet at
δC 29.6 (
Table 1), and the HSQC correlation between H
2-3 and C-3 (
Figure S12). The absolute configuration of
2 was assigned as 4
R by comparing the experimental ECD with the calculated ECD (
Figure 4).
Citreobenzofuran F (
3) was obtained as a colorless oil, with the molecular formula C
15H
16O
3, determined by the HRESIMS data (
m/
z 243.1032 [M − H]
−) (
Figure S22). The 1D NMR data (
Table 1 and
Table 2) revealed partial structural similarity to the known compound 3-formyl-4,5-dimethyl-8-oxo-5H-6,7-dihydronaphtho[2,3-
b] furan [
9]. The difference was the replacement of carbonyl at C-13 with a hydroxyl group, which was supported by the chemical shift of C-13 (
δC 54.9) (
Table 1), and the HSQC correlation between H
2-13 and C-13 (
Figure S19). The absolute configuration of
3 was assigned as 4
S by comparing the experimental ECD with the calculated ECD (
Figure 4).
Phomenone A (
4) was obtained as a colorless powder. The HRESIMS spectrum suggested that the molecular formula is C
16H
20O
3S (
m/
z 293.1205 [M + H]
+) (
Figure S30). The
1H and
13C NMR spectra of
4 were similar to those of paraconiothin G [
10], except for the oxidation of the hydroxyl group at C-12 (
δC 188.4) to an aldehyde group, and the appearance of the methylthio group at C-13 (
δC 158.0) (
Table 1). These changes were confirmed by the HMBC correlations from H-13 (
δH 8.01) to C-14 (
δC 17.5) and C-12(
δC 188.4), and from H-12 (
δH 9.28) to C-7(
δC 130.8) and C-11(
δC 134.9) (
Figure 2). The relative configuration of
4 was assigned, by NOESY correlations to between 1-OH and H-4, H-1 and H-15, H-15 and H-16, and H-12 and H-13 (
Figure 3). The agreement between the calculated ECD spectrum of (1
R, 4
R, 5
R)
-4a and the experimental ECD spectrum suggested that the absolute configuration of
4 is 1
R, 4
R, 5
R (
Figure 4).
Phomenone B (
5) was obtained as a colorless oil, with the molecular formula C
15H
20O
3, determined by HRESIMS analysis (
m/
z 247.1343 [M − H]
−) (
Figure S38). According to the 1D NMR data, compound
5 (
Table 1 and
Table 2) was similar to a known mycotoxic sesquiterpenoid, xylarenones A [
11], except for the disappearance of the hydroxyl group at C-1, which was supported by the shielded singlets at
δC 32.6 (
Table 1). The H-6, 14-CH
3, and 15-CH
3 were suggested to be located on the same face of the octahydronaphthalene ring, based on the NOESY correlation of H-6 (
δH 3.36) with H-15 (
δH 1.03), and H-14 (
δH 1.16) with H-15 (
Figure 3). Thus, the relative configuration was determined as 4
S*, 5
R*, 6
R*, 7
R*. The overall pattern of the experimental ECD spectrum was in reasonable agreement with the calculated ECD spectrum of (4
S, 5
R, 6
R, 7
R)-
5a (
Figure 4), which indicated the 4
S, 5
R, 6
R and 7
R absolute configuration of
5.
The known compound was identified as xylarenones A (
6) [
11] by comparison of the spectroscopic data with the literature values.
Compounds
1–
6 were tested for their antimicrobial (
Table 3) and antitumor activity against K562, MDA-MB-231, L-02, H69AR, and ASPC-1. Compound
5 showed promising antimicrobial activity against
B. subtilis, with an MIC value of 6.25 μM, while the other compounds exerted no activity for all the tested strains. Moreover, none of the compounds displayed obvious anticancer activity.
3. Materials and Methods
3.1. General Experimental Procedures
Specific rotations were obtained on a JASCO P-1020 digital polarimeter. UV spectra were recorded on a HITACHI 5430. IR spectra were measured on a Bruker Tensor-27 spectrophotometer in KBr discs. NMR spectra were recorded on a JEOLJN M-ECP 600 spectrometer (JEOL, Tokyo, Japan) or an Agilent 500 MHz DD2 spectrometer using tetramethylsilane as an internal standard. HRMS were obtained on a Thermo Scientific LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) or Micromass Q-TOF ULTIMA GLOBAL GAA076 LC mass spectrometer (Waters Corporation, Milford, MA, USA). Semipreparative HPLC was performed on an ODS column (YMC-Pack ODS-A, 10 × 250 mm, 5 μm, 3 mL/min).
3.2. Materials and Culture Conditions
The fungal strain, Penicillium sp. HDN13-494, was isolated from the root soil sample of a mangrove plant in Wenchang. The strain was identified by internal transcribed spacer (ITS) sequence (GenBank No. OM283301). The strain was incubated in media potato dextrose agar (PDA, 20% potato, 2% dextrose, and 2% agar) at 28 °C for 5 days for sporulation. For compound production, the strains were cultured in PDB (potato dextrose broth) at 28 °C, 180 rpm, for 9 days. The strain was deposited at the Marine Medicinal Bioresources Center, Ocean University of China, Qingdao, China.
3.3. Fermentation
The strain was grown on PDA plates for 5 days at 28 °C. The spores of Penicillium sp. HDN13-494 were inoculated into 500 mL Erlenmeyer flasks containing 150 mL of PDB medium, pH = 7.0 (in seawater collected from Huiquan Bay, Yellow Sea), and cultured at 28 °C for 9 days on a rotary shaker at 180 rpm. A total of 10 L of broth was extracted with EtOAc (4 × 10 L) to generate the extract (30 g).
3.4. Extraction and Purification
All the fermentation broth (10 L) was filtered through cheese cloth to separate the supernatant from the mycelia. The supernatant was extracted with EtOAc (4 × 10 L), and the mycelia was macerated and extracted with methanol (3 × 5 L). All extracts were evaporated under reduced pressure to give a crude gum. The extract was chromatographed over ODS, eluting with a gradient of increasing MeOH/H2O (30–100%) to afford twelve fractions (Fr.1 to Fr.12). Fr.4 was further separated by RP-HPLC, first using 72% MeOH/H2O to yield 4 (3 mg). By using 60% MeCN/H2O separated on an RP-HPLC column, compounds 2 (1.4 mg) and 5 (1.4 mg) were obtained from Fr.5, compound 3 (26.4 mg) was obtained from Fr.6 using 55% MeOH/H2O, while 1 (1.4 mg) and 6 (2.3 mg) were yielded using 58% MeOH/H2O from Fr.7.
Citreobenzofuran D (
1): brown triclinic crystal (MeOH); m.p. 204–205 °C;
−42.2 (
c 0.1, MeOH); UV (MeOH)
λmax (log
ε): 231(1.7), 295(0.6) nm; ECD (
c 0.1 mM, MeOH)
λmax (Δ
ε) 216 (−1.00), 249 (−1.90), 288 (0.34) nm; IR (KBr) ν
max 3259, 2935, 1655, 1208, 1030 cm
−1;
1H and
13C NMR data see
Table 1 and
Table 2; HRESIMS
m/
z 261.1130 [M − H]
− (calcd. for C
15H
17O
4, 261.1132).
Citreobenzofuran E (
2): green amorphous powder;
−14.1 (
c 0.1, MeOH); UV (MeOH)
λmax (log
ε): 214 (1.6), 279 (0.2) nm; ECD (
c 0.1 mM, MeOH)
λmax (Δ
ε) 216 (−0.53), 252 (−0.78), 292 (0.15) nm; IR (KBr) ν
max 3421, 2928, 1684, 1210 cm
−1;
1H and
13C NMR data see
Table 1 and
Table 2; HRESIMS
m/
z 229.1227 [M − H]
− (calcd. for C
15H
17O
2, 229.1234).
Citreobenzofuran F (
3): colorless oil;
−24.0 (
c 0.1, MeOH); UV (MeOH)
λmax (log
ε): 223(1.5), 295(1.0) nm; ECD (
c 0.1 mM, MeOH)
λmax (Δ
ε) 211 (0.47), 298 (0.13), 338 (6.91) nm; IR (KBr) ν
max 3409, 2927, 1668, 1675, 1176 cm
−1;
1H and
13C NMR data see
Table 1 and
Table 2; HRESIMS
m/
z 243.1032 [M − H]− (calcd. for C
15H
15O
3, 243.1027).
Phomenone A (
4): colorless powder;
−4.3 (
c 0.1, MeOH); UV (MeOH)
λmax (log
ε): 201(1.6), 221(1.5), 298 (1.3) nm; ECD (
c 0.1 mM, MeOH)
λmax (Δ
ε) 211 (0.47), 298 (0.13), 338 (6.91) nm; IR (KBr) ν
max 3420, 2930, 1675, 1208 cm
−1;
1H and
13C NMR data see
Table 1 and
Table 2; HRESIMS
m/
z 293.1205 [M + H]
+ (calcd. for C
16H
21O
3S, 293.1206).
Phomenone B (
5): colorless oil;
+336.24 (
c 0.1, MeOH); UV (MeOH)
λmax (log
ε): 202(0.5), 248(1.8) nm; ECD (
c 0.1 mM, MeOH)
λmax (Δ
ε) 216 (−6.80), 248 (7.00), 329 (2.37) nm; IR (KBr) ν
max 3749, 2935, 1671 cm
−1;
1H and
13C NMR data see
Table 1 and
Table 2; HRESIMS
m/
z 247.1343 [M − H]
− (calcd. for C
15H
19O
3, 247.1340).
3.5. X-ray Crystal Structure Analysis
Compound 1 was obtained as brown triclinic crystals from MeOH with the molecular formula C31H40O9. The suitable crystal was selected and analyzed on a CCD area detector diffractometer (Bruker Smart Apex II) using Cu Ka radiation. The crystallographic data for 1 (CCDC 2128584) was deposited in the Cambridge Crystallographic Data Centre.
Crystal data for citreobenzofuran D (1): C31H40O9 (M = 556.63), triclinic, space group P1, a = 7.6499(4) Å, b = 9.7072(7) Å, c = 11.2321(7) Å, α = 103.630(6)°, β = 90.229(5)°, γ = 104.053(5)°, V = 721.01(8) Å3, Z = 1, T = 293(2) K, μ (Cu Kα) = 0.770 mm−1, Dcalc = 1.282 g/cm3, 4156 reflections measured (4.417° ≤ θ ≤ 67.244°), 2898 unique (Rint = 0.0232), which were used in all calculations. The final R1 was 0.0351 (I > 2σ(I)) and ωR2 was 0.0945, flack = −0.1(2).
3.6. Computational Section
Conformational searches were performed by employing the systematic procedure implemented in Spartan′14, using the MMFF (Merck molecular force field). All MMFF minima were reoptimized with DFT calculations at the B3LYP/6-31+G(d) level using the Gaussian09 program. The geometry was optimized starting from various initial conformations, with vibrational frequency calculations confirming the presence of minima. Time-dependent DFT calculations were performed on lowest-energy conformations (>5% population) for each configuration using 20 excited states and using a polarizable continuum model for MeOH. ECD spectra were generated using the program SpecDis by applying a Gaussian band shape with a 0.30 eV width and 25 blue shifts to facilitate comparison to the experimental data.
3.7. Cytotoxicity Assay
Cytotoxic activities of compounds
1–
6 were evaluated against the K562 (using the MTT method), MDA-MB-231, L-02, H69AR and ASPC-1 (using the SRB method) cell lines. Adriamycin (ADM) was used as a positive control. The detailed methodologies for biological testing have been described in previous reports [
12,
13].
3.8. Antimicrobial Activity
Antibacterial activities of compounds
1–
6 were evaluated against
B. subtilis, Proteus vulgaris, Acinetobacter baumannii, Candida. albicans, Escherichia. coli, and MRSA (Methicillin-resistant
Staphylococcus aureus) by using the agar dilution method as previously reported [
14]. Ciprofloxacin was used as a positive control.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}