
The Tetrahydrofuran Motif in Polyketide Marine Drugs



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Polyketide Marine Drugs Containing Tetrahydrofuran Rings
2.1. Macrolides
2.1.1. Amphidinolides
Amphidinolides C and F
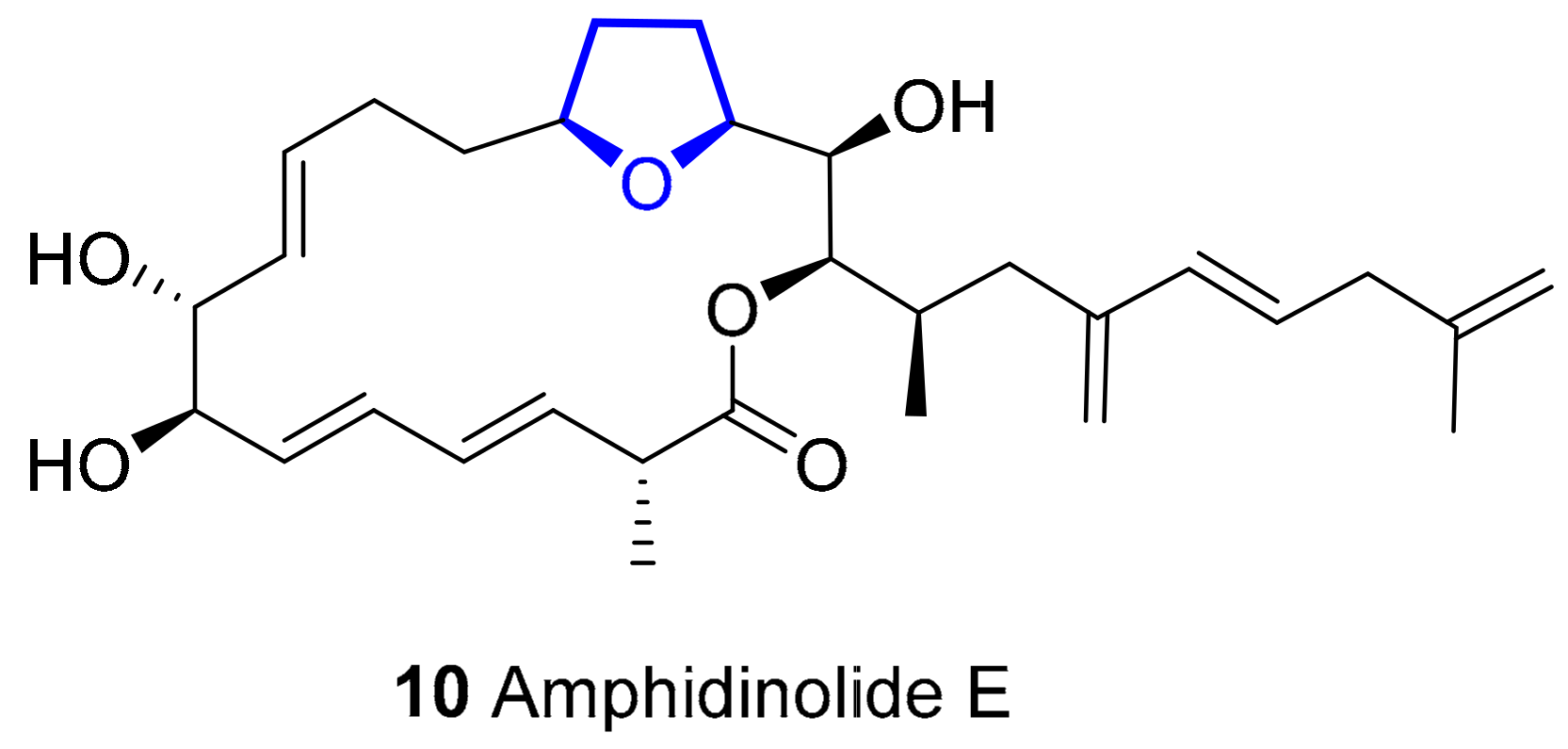
Amphidinolide E
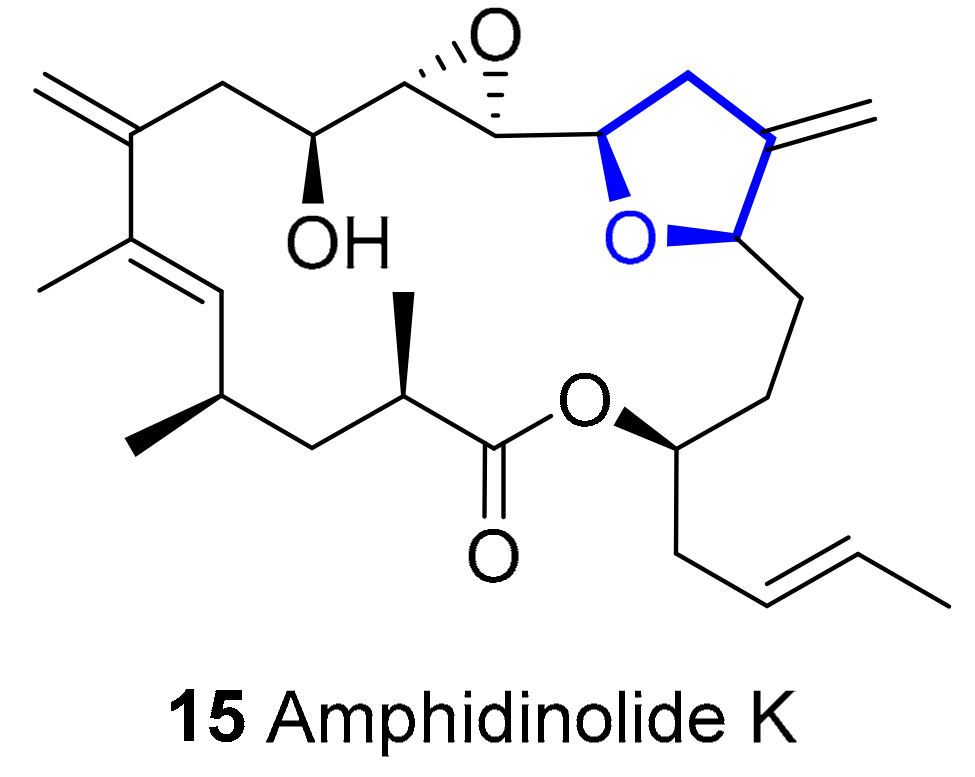
Amphidinolide K
Amphidinolide N/caribenolide I
Amphidinolides T
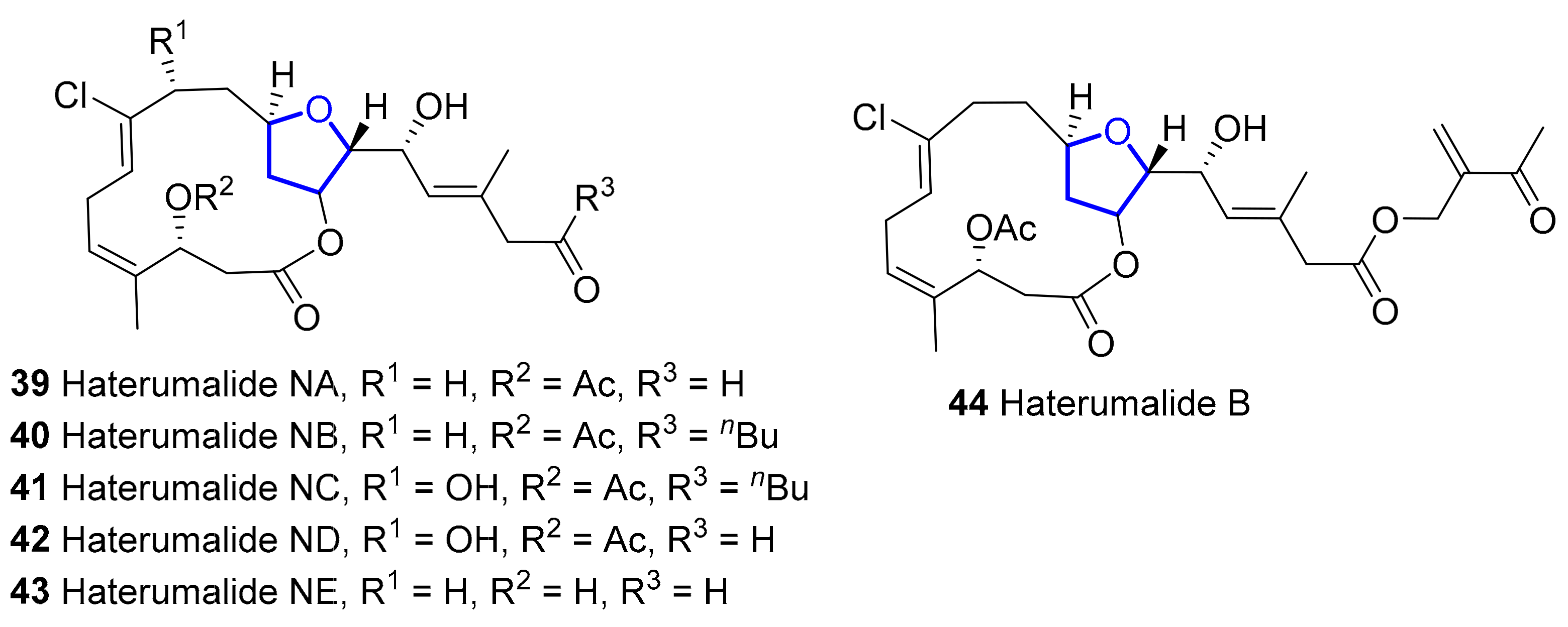
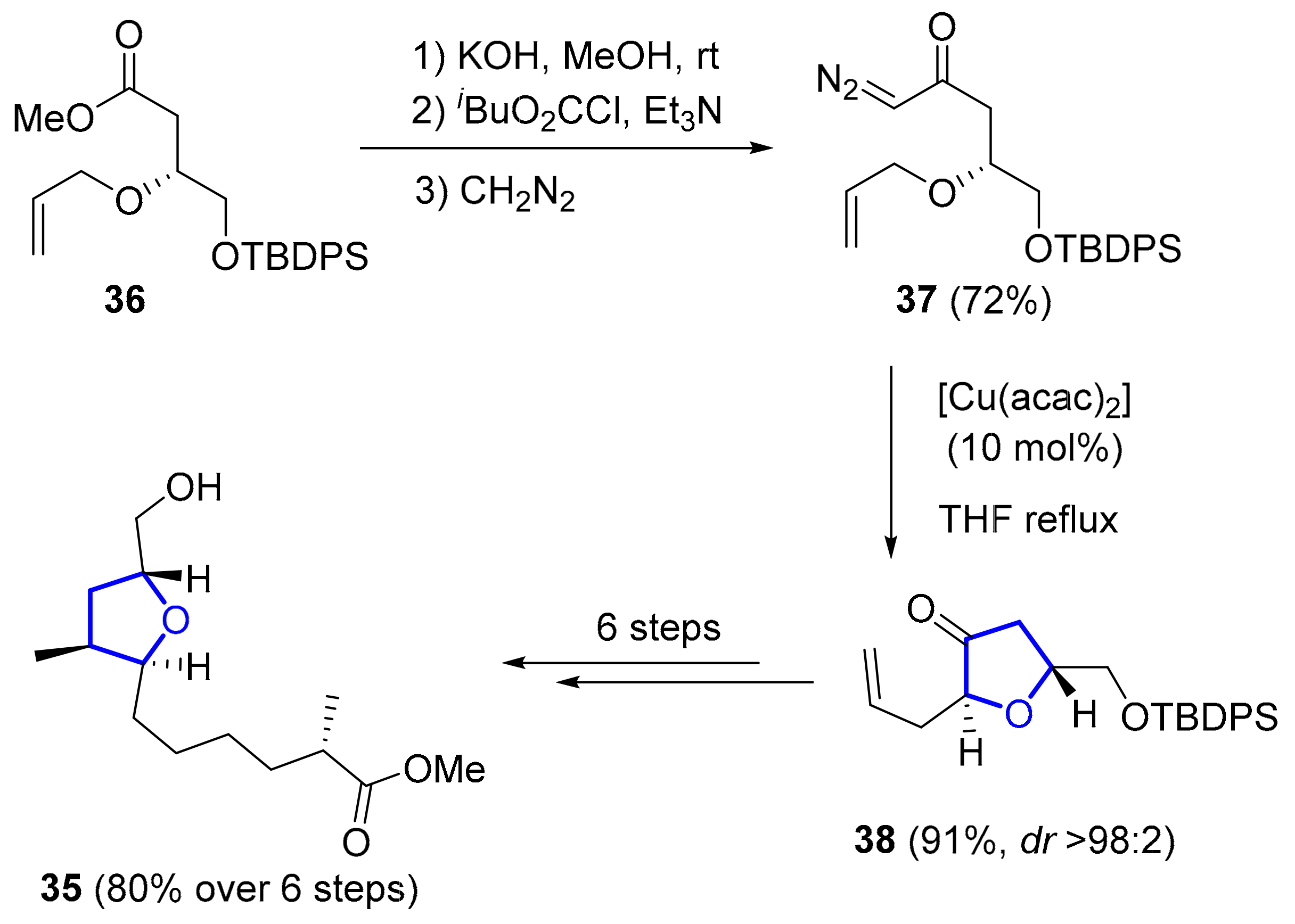
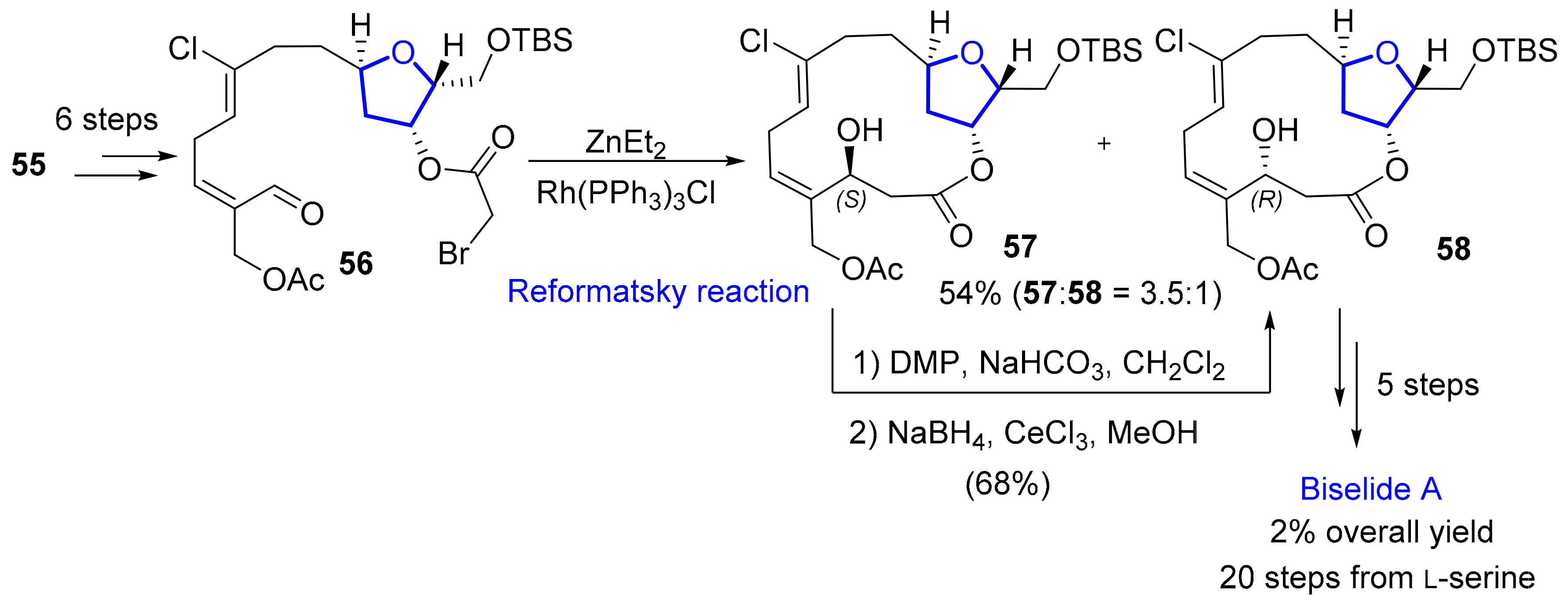
2.1.2. Haterumalides and Biselides
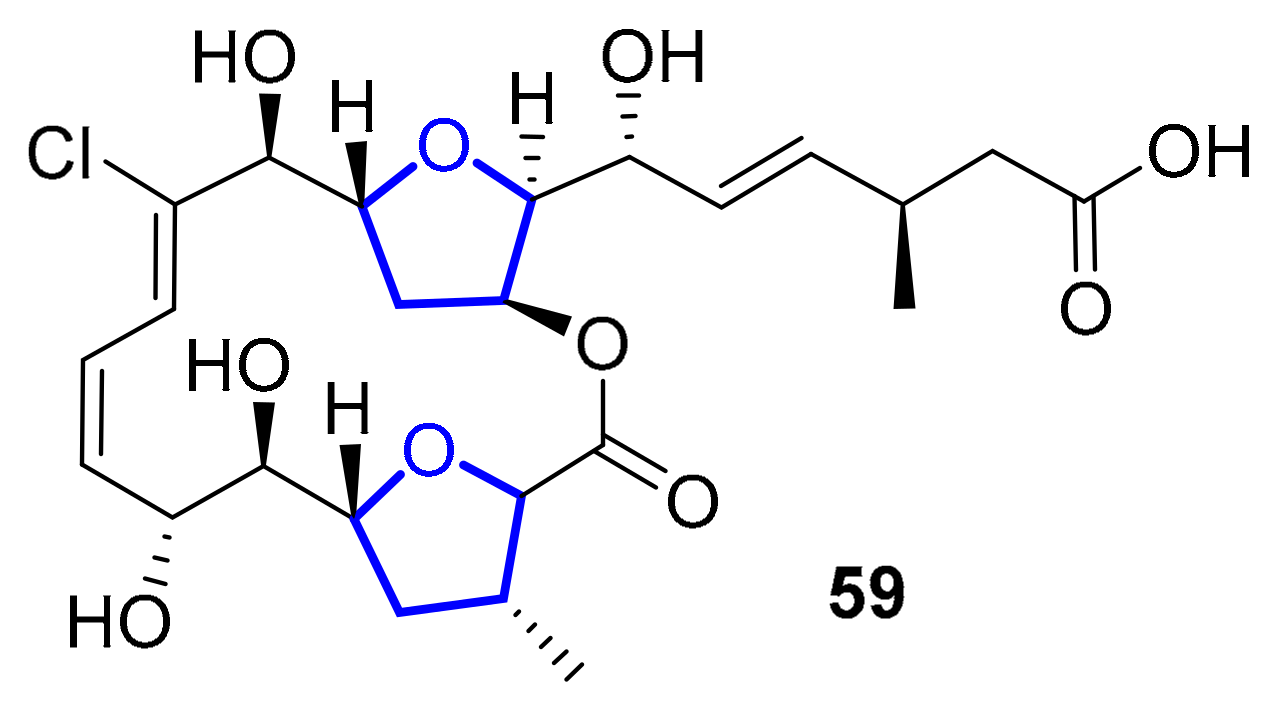
2.1.3. Chagosensine
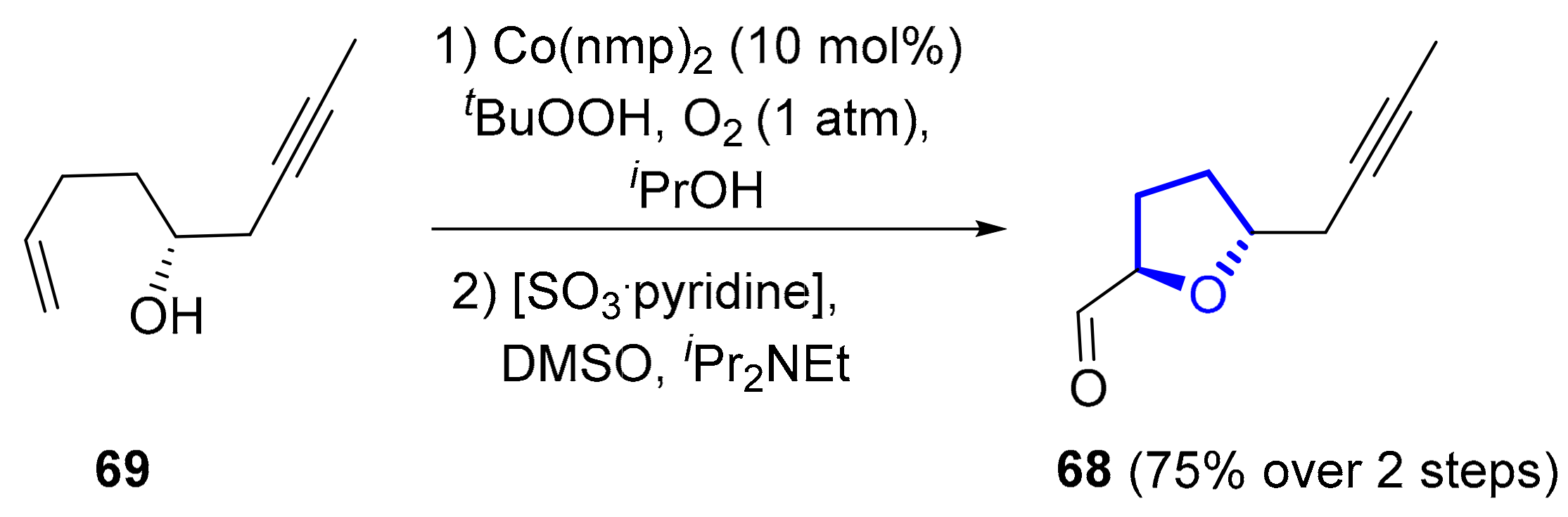
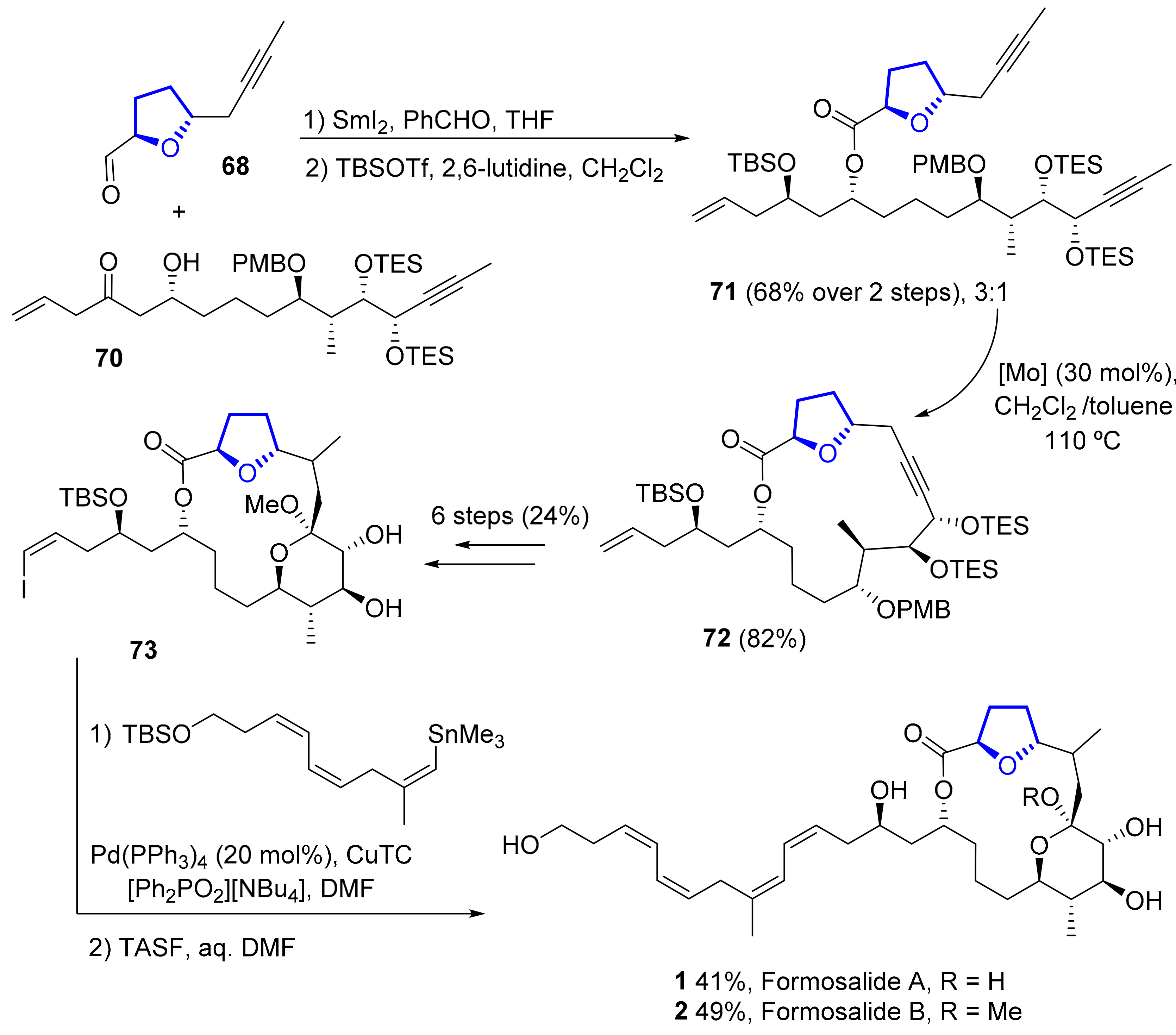
2.1.4. Formosalides
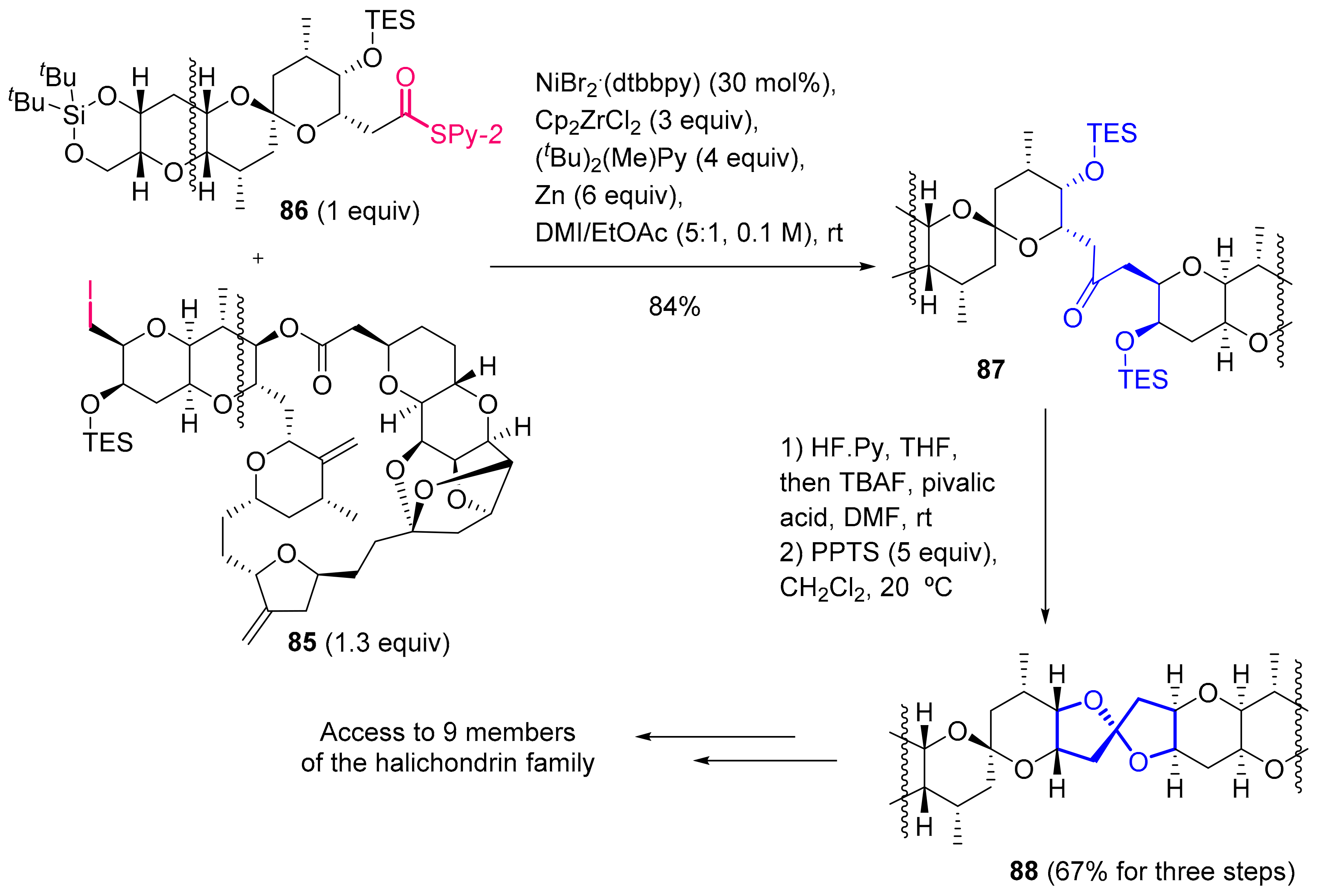
2.1.5. Halichondrins
2.1.6. Iriomoteolides
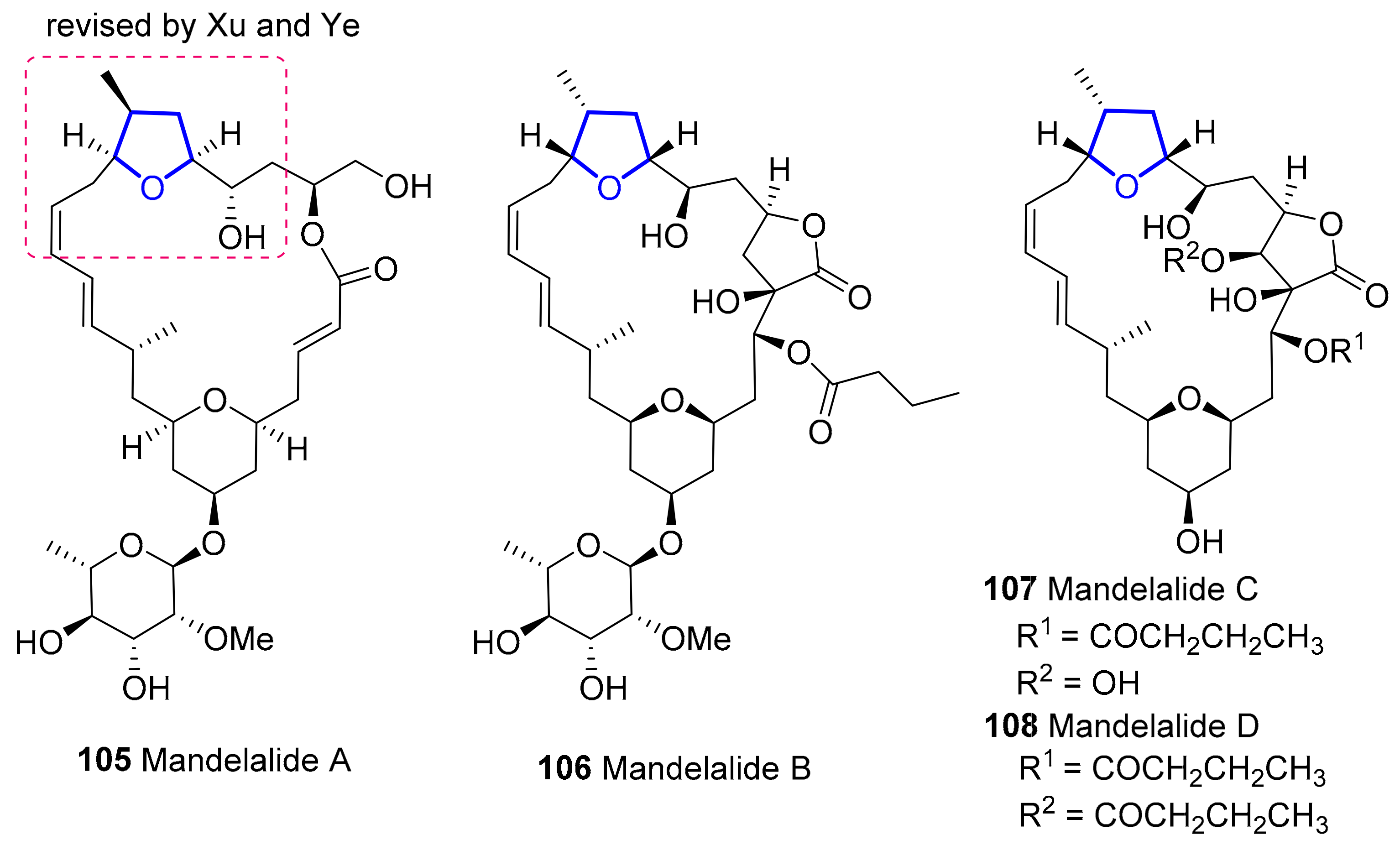
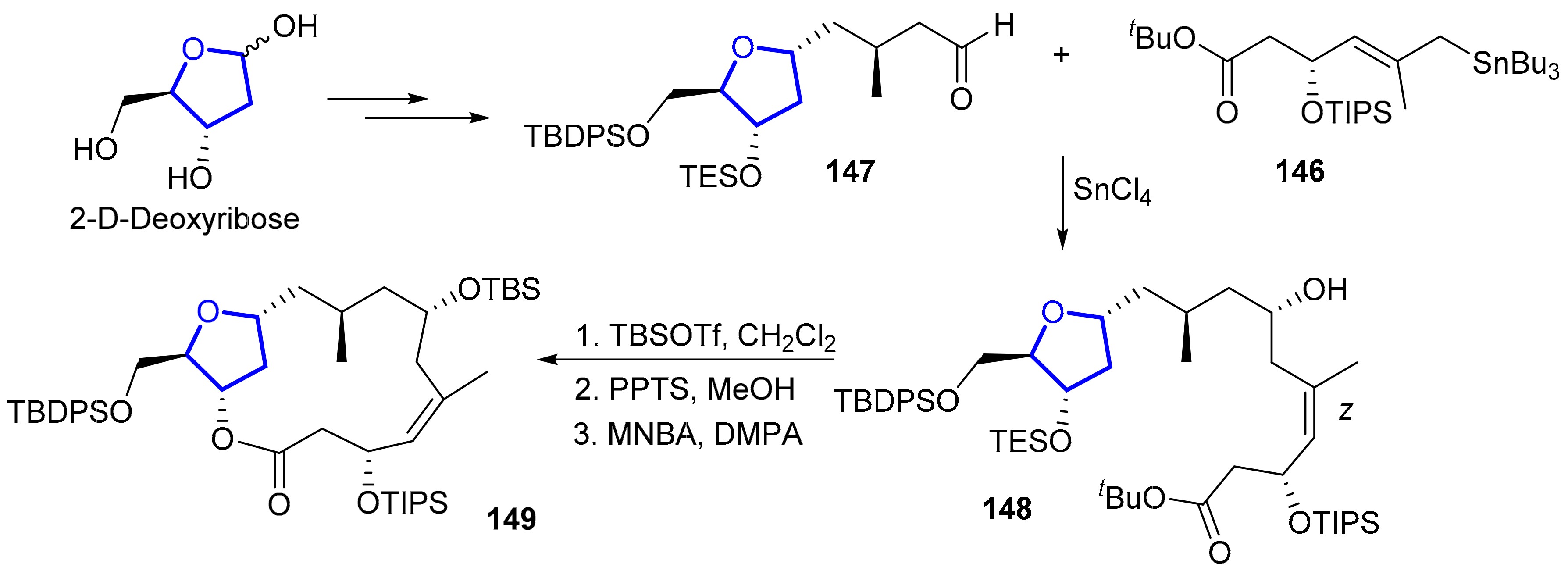
2.1.7. Mandelalides
2.1.8. Mangromicins
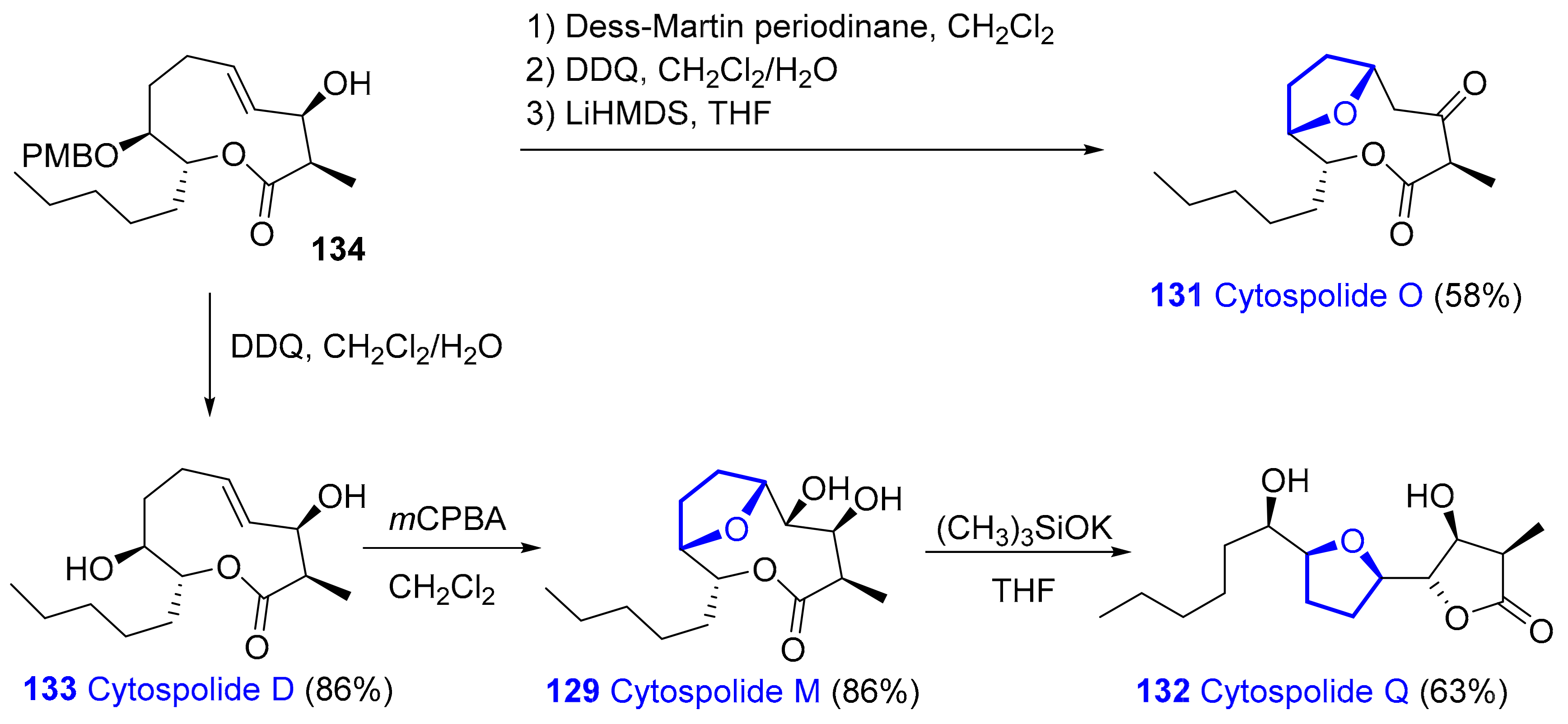
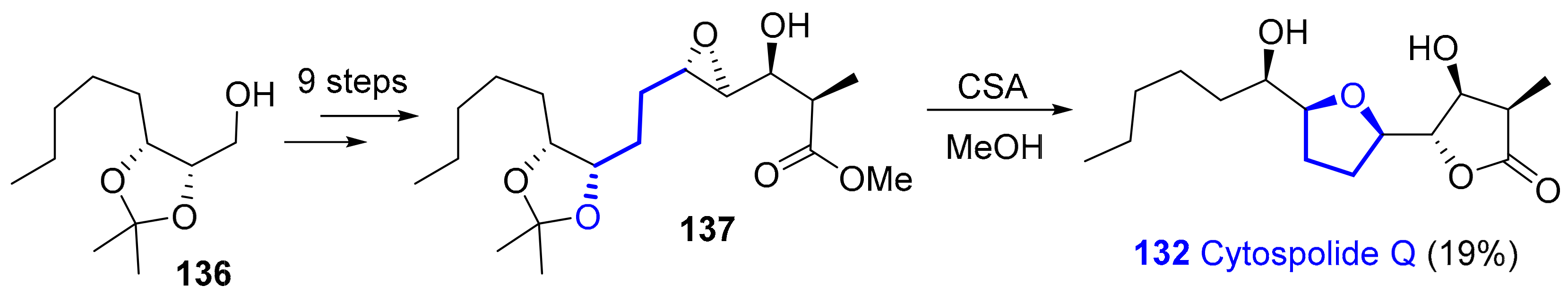
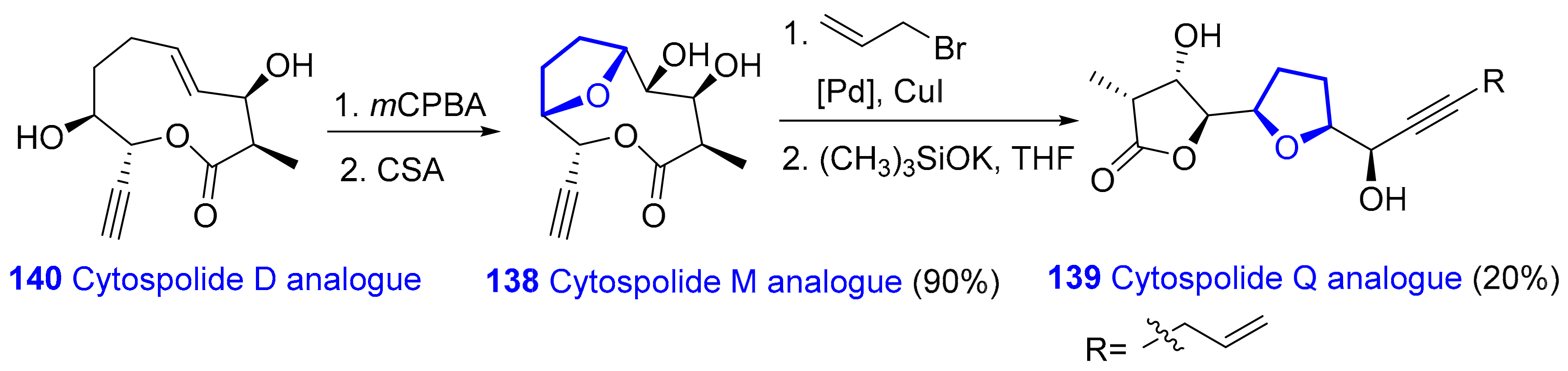
2.1.9. Nonalides: Cytospolides
2.1.10. Oscillariolide
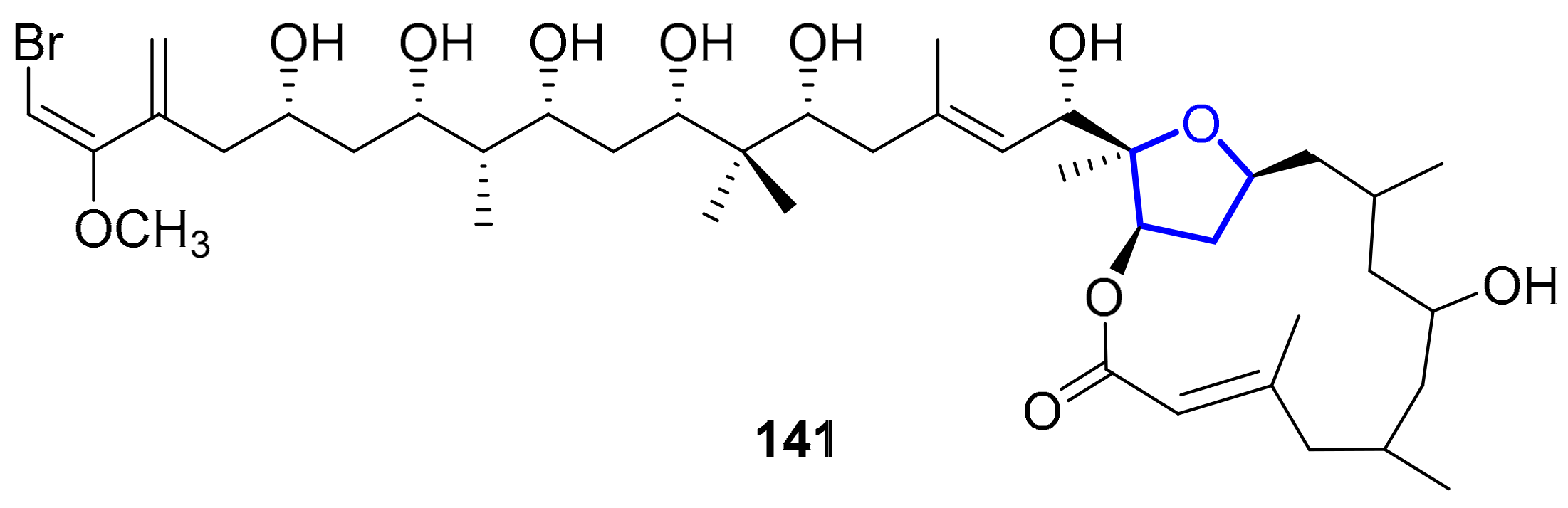
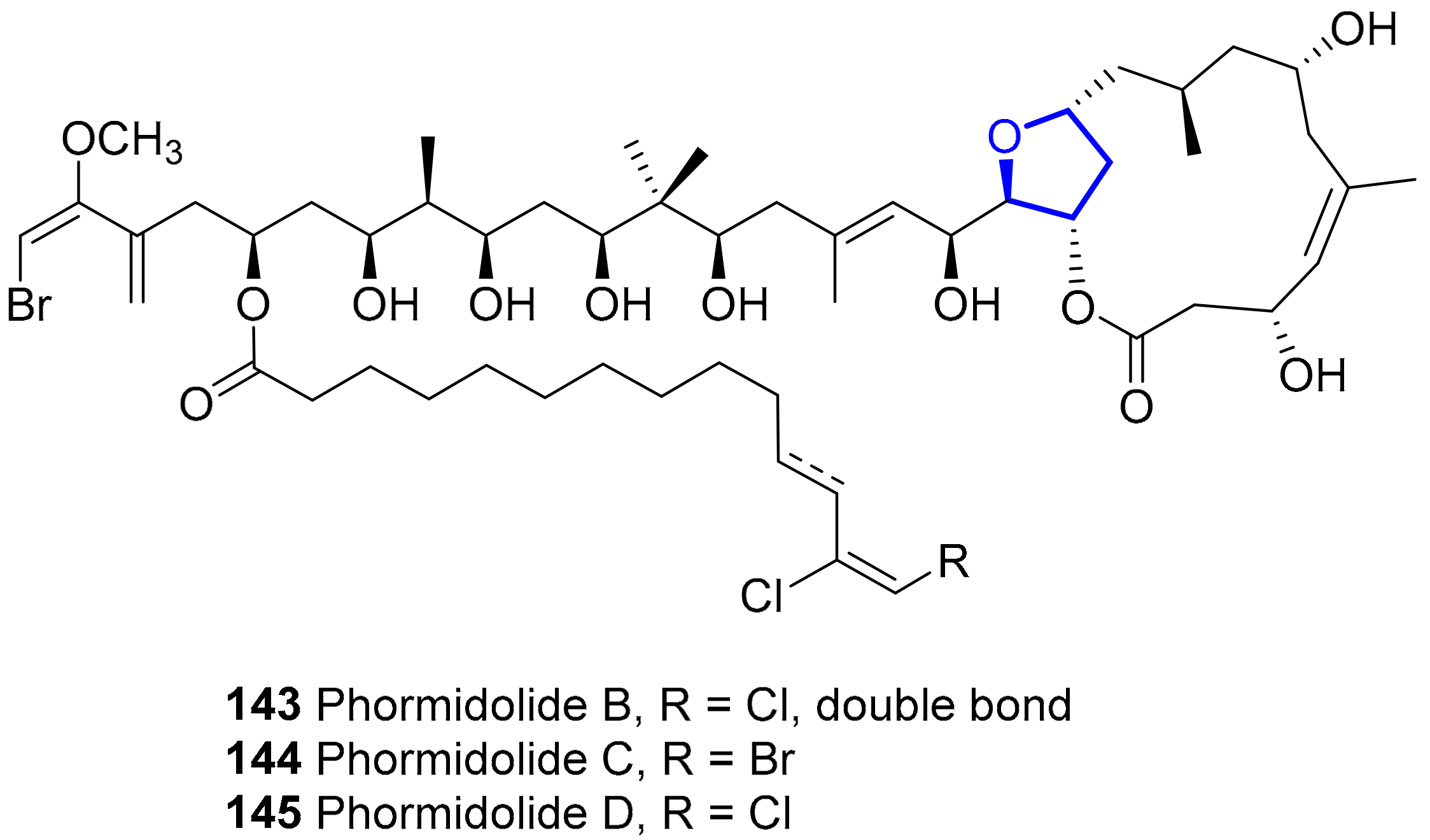
2.1.11. Phormidolides
2.2. Linear Polyketides
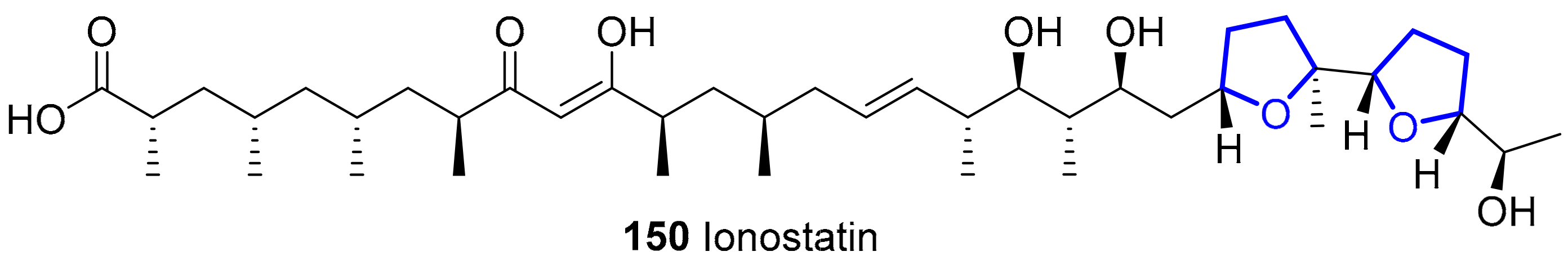
Ionostatin
2.3. Polycyclic Polyketides
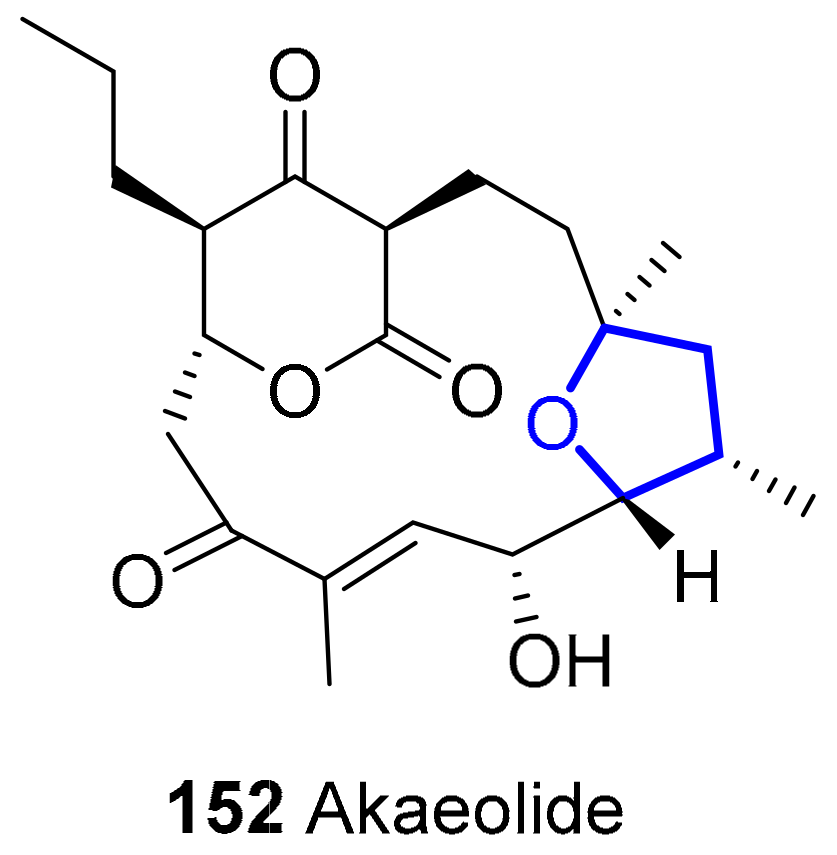
Akaeolide
2.4. Acetogenin Metabolites
Obtusallenes
2.5. Polyhydroxyl
Amphezonol A
2.6. Bycyclic
2.6.1. Asperpentenone
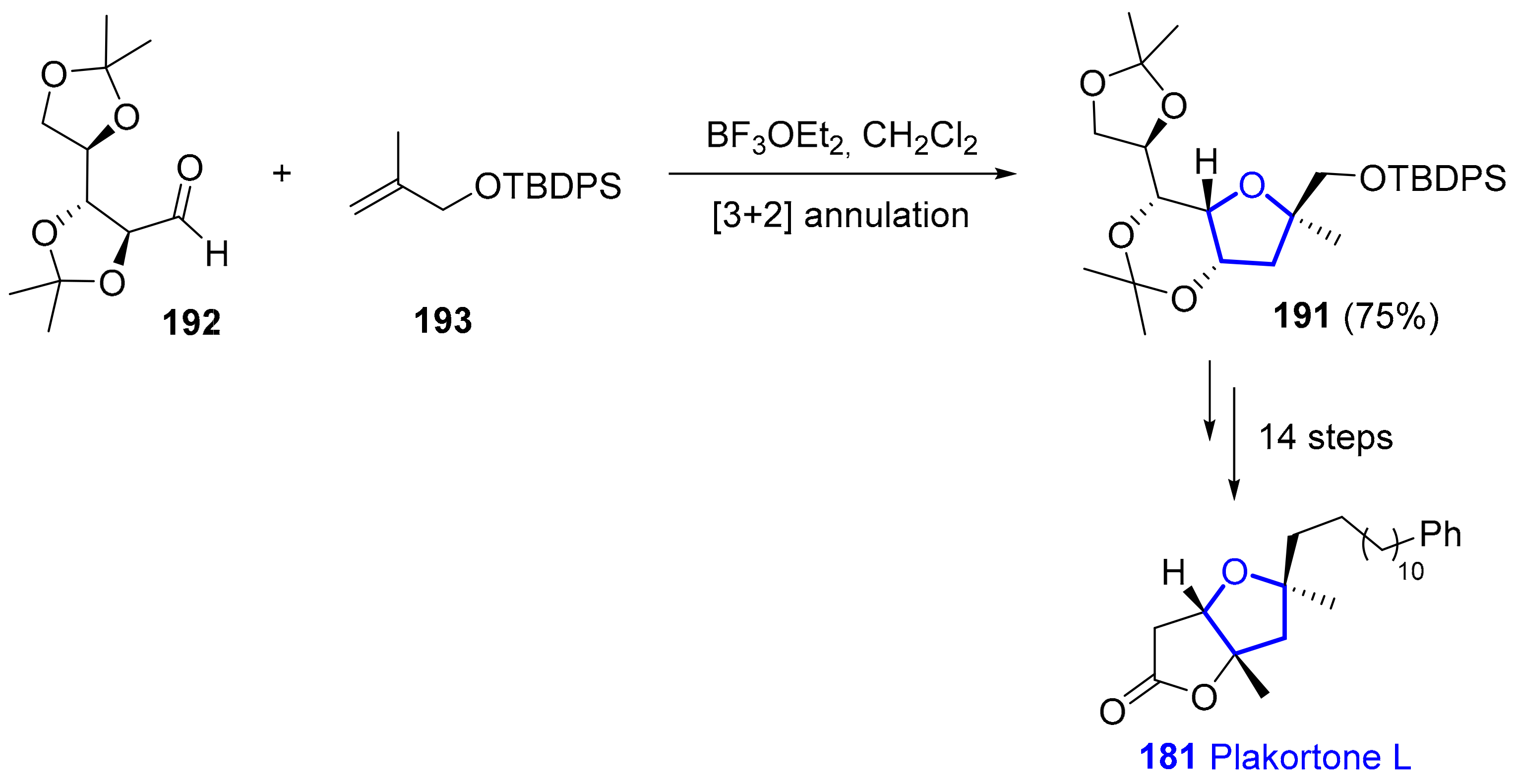
2.6.2. Plakortones
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Fuwa, H. Structure determination, correction, and disproof of marine macrolide natural products by chemical synthesis. Org. Chem. Front. 2021, 8, 3990–4023. [Google Scholar] [CrossRef]
- Lorente, A.; Makowski, K.; Albericio, F.; Álvarez, M. Bioactive Marine Polyketides as Potential and Promising Drugs. Ann. Mar. Biol. Res. 2014, 1, 1003. [Google Scholar]
- Kobayashi, J.; Kubota, T. Bioactive Macrolides and Polyketides from Marine Dinoflagellates of the Genus Amphidinium. J. Nat. Prod. 2007, 70, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Parenty, A.; Moreau, A.X.; Campagne, J.-M. Macrolactonizations in the Total Synthesis of Natural Products. Chem. Rev. 2006, 106, 911–939. [Google Scholar] [CrossRef] [PubMed]
- Nasir, N.M.; Ermanis, K.; Clarke, P.A. Strategies for the construction of tetrahydropyran rings in the synthesis of natural products. Org. Biomol. Chem. 2014, 12, 3323–3335. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H. Contemporary Strategies for the Synthesis of Tetrahydropyran Derivatives: Application to Total Synthesis of Neopeltolide, a Marine Macrolide Natural Product. Mar. Drugs 2016, 14, 65. [Google Scholar] [CrossRef] [PubMed]
- Barbero, H.; Díez-Poza, C.; Barbero, A. The Oxepane Motif in Marine Drugs. Mar. Drugs 2017, 15, 361. [Google Scholar] [CrossRef]
- Fernandes, R.A.; Pathare, R.S.; Gorve, D.A. Advances in Total Synthesis of Some 2,3,5-Trisubstituted Tetrahydrofuran Natural Products. Chem. Asian J. 2020, 15, 2815–2837. [Google Scholar] [CrossRef]
- Fernandes, R.A.; Gorve, D.A.; Pathare, R.S. Emergence of 2,3,5-trisubstituted tetrahydrofuran natural products and their synthesis. Org. Biomol. Chem. 2020, 18, 7002–7025. [Google Scholar] [CrossRef]
- Lorente, A.; Lamariano-Merketegi, J.; Albericio, F.; Álvarez, M. Tetrahydrofuran-Containing Macrolides: A Fascinating Gift from the Deep Sea. Chem. Rev. 2013, 113, 4567–4610. [Google Scholar] [CrossRef]
- Kigoshi, H.; Hayakawa, I. Marine cytotoxic macrolides haterumalides and biselides, and related natural products. Chem. Rec. 2007, 7, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Shabir, G.; Saeed, A. A Comparative Study of Synthetic Approaches Towards Total Synthesis of Mandelalide A, An Anti-Lung Cancer Metabolite from Lissoclinum Ascidian. Curr. Org. Chem. 2018, 22, 101–127. [Google Scholar] [CrossRef]
- Zhang, H.; Zou, J.; Yan, X.; Chen, J.; Cao, X.; Wu, J.; Liu, Y.; Wang, T. Marine-Derived Macrolides 1990–2020: An Overview of Chemical and Biological Diversity. Mar. Drugs 2021, 19, 180. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, G.; Gomes, B.A.; Luongo, E.; Torres, M.C.M.; Santos, E.A.; Cutignano, A.; Pessoa, O.D.L.; Costa-Lotufo, L.V.; Fontana, A. Dinoflagellate-Related Amphidinolides from the Brazilian Octocoral Stragulum bicolor. J. Nat. Prod. 2016, 79, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Valot, G.; Mailhol, D.; Regens, C.S.; O’Malley, D.P.; Godineau, E.; Takikawa, H.; Philipps, P.; Fürstner, A. Concise Total Syntheses of Amphidinolides C and F. Chem. A Eur. J. 2015, 21, 2398–2408. [Google Scholar] [CrossRef]
- Ferrié, L.; Fenneteau, J.; Figadère, B. Total Synthesis of the Marine Macrolide Amphidinolide F. Org. Lett. 2018, 20, 3192–3196. [Google Scholar] [CrossRef] [PubMed]
- Akwaboah, D.C.; Wu, D.; Forsyth, C.J. Stereoselective Synthesis of the C1–C9 and C11–C25 Fragments of Amphidinolides C, C2, C3, and F. Org. Lett. 2017, 19, 1180–1183. [Google Scholar] [CrossRef]
- Su, Y.-X.; Dai, W.-M. Synthesis of the C18–C26 tetrahydrofuran-containing fragment of amphidinolide C congeners via tandem asymmetric dihydroxylation and S N 2 cyclization. Tetrahedron 2018, 74, 1546–1554. [Google Scholar] [CrossRef]
- Namirembe, S.; Yan, L.; Morken, J.P. Studies toward the Synthesis of Amphidinolide C1: Stereoselective Construction of the C(1)–C(15) Segment. Org. Lett. 2020, 22, 9174–9177. [Google Scholar] [CrossRef]
- Kim, C.H.; An, H.J.; Shin, W.K.; Yu, W.; Woo, S.K.; Jung, S.K.; Lee, E. Total Synthesis of (−)-Amphidinolide E. Angew. Chem. Int. Ed. 2006, 45, 8019–8021. [Google Scholar] [CrossRef]
- Kim, C.H.; An, H.J.; Shin, W.K.; Yu, W.; Woo, S.K.; Jung, S.K.; Lee, E. Stereoselective Synthesis of (−)-Amphidinolide E. Chem. Asian J. 2008, 3, 1523–1534. [Google Scholar] [CrossRef]
- Va, P.; Roush, W.R. Total Synthesis of Amphidinolide E. J. Am. Chem. Soc. 2006, 128, 15960–15961. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Va, P.; Roush, W.R. Synthesis of 2-epi-Amphidinolide E: An Unexpected and Highly Selective C(2) Inversion during an Esterification Reaction. Org. Lett. 2007, 9, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Va, P.; Roush, W.R. Total synthesis of amphidinolide E and amphidinolide E stereoisomers. Tetrahedron 2007, 63, 5768–5796. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bosch, L.; Mola, L.; Petit, E.; Saladrigas, M.; Esteban, J.; Costa, A.M.; Vilarrasa, J. Formal Total Synthesis of Amphidinolide E. J. Org. Chem. 2017, 82, 11021–11034. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, D.; Andreou, T.; Costa, A.M.; Meyer, K.G.; Williams, D.R.; Barasoain, I.; Díaz, J.F.; Lucena-Agell, D.; Vilarrasa, J.; Sánchez-Pérez, D. Total Synthesis of Amphidinolide K, a Macrolide That Stabilizes F-Actin. J. Org. Chem. 2015, 80, 8511–8519. [Google Scholar] [CrossRef]
- Sasaki, M.; Kawashima, Y.; Fuwa, H. Studies toward the Total Synthesis of Amphidinolide N: Stereocontrolled Synthesis of the C13–C29 Segment. Heterocycles 2015, 90, 579. [Google Scholar] [CrossRef]
- Kawashima, Y.; Toyoshima, A.; Fuwa, H.; Sasaki, M. Toward the Total Synthesis of Amphidinolide N: Synthesis of the C8–C29 Fragment. Org. Lett. 2016, 18, 2232–2235. [Google Scholar] [CrossRef]
- Fujishima, Y.; Ogura, Y.; Towada, R.; Enomoto, M.; Kuwahara, S. Stereoselective synthesis of the C17–C29 fragment of amphidinolide N. Tetrahedron Lett. 2016, 57, 5240–5242. [Google Scholar] [CrossRef]
- Ohta, M.; Kato, S.; Sugai, T.; Fuwa, H. Cobalt-Catalyzed Hartung–Mukaiyama Cyclization of γ-Hydroxy Olefins: Stereocontrolled Synthesis of the Tetrahydrofuran Moiety of Amphidinolide N. J. Org. Chem. 2021, 86, 5584–5615. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, K.; Kuppusamy, S.; Yasui, Y.; Okano, T.; Matsumoto, Y.; Gupta, N.R.; Takahashi, Y.; Kubota, T.; Kobayashi, J.; Hayashi, Y. Total Synthesis of the 7,10-Epimer of the Proposed Structure of Amphidinolide N, Part I: Synthesis of the C1-C13 Subunit. Chem. A Eur. J. 2016, 22, 3282–3286. [Google Scholar] [CrossRef]
- Ochiai, K.; Kuppusamy, S.; Yasui, Y.; Harada, K.; Gupta, N.R.; Takahashi, Y.; Kubota, T.; Kobayashi, J.; Hayashi, Y. Total Synthesis of the 7,10-Epimer of the Proposed Structure of Amphidinolide N, Part II: Synthesis of C17-C29 Subunit and Completion of the Synthesis. Chem. A Eur. J. 2016, 22, 3287–3291. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Bai, W.-J.; Stivala, C.E.; Hohn, C.; Poock, C.; Heinrich, M.; Xu, S.; Rey, J. Enantioselective Synthesis of des-Epoxy-Amphidinolide N. J. Am. Chem. Soc. 2018, 140, 17316–17326. [Google Scholar] [CrossRef]
- Tsuda, M.; Akakabe, M.; Minamida, M.; Kumagai, K.; Tsuda, M.; Konishi, Y.; Tominaga, A.; Fukushi, E.; Kawabata, J. Structure and Stereochemistry of Amphidinolide N Congeners from Marine Dinoflagellate Amphidinium Species. Chem. Pharm. Bull. 2021, 69, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Endo, T.; Kobayashi, J. Amphidinolide T, Novel 19-Membered Macrolide from Marine Dinoflagellate Amphidinium sp. J. Org. Chem. 2000, 65, 1349–1352. [Google Scholar] [CrossRef]
- Kobayashi, J.; Kubota, T.; Endo, A.T.; Tsuda, M. Amphidinolides T2, T3, and T4, New 19-Membered Macrolides from the Dinoflagellate Amphidinium sp. and the Biosynthesis of Amphidinolide T1. J. Org. Chem. 2001, 66, 134–142. [Google Scholar] [CrossRef]
- Kubota, T.; Endo, T.; Tsuda, M.; Shiro, M.; Kobayashi, J. Amphidinolide T5, a new 19-membered macrolide from a dinoflagellate and X-ray structure of amphidinolide T1. Tetrahedron 2001, 57, 6175–6179. [Google Scholar] [CrossRef]
- Clark, J.S.; Romiti, F. Total Syntheses of Amphidinolides T1, T3, and T4. Angew. Chem. 2013, 125, 10256–10259. [Google Scholar] [CrossRef]
- Ueda, K.; Hu, Y. Haterumalide B: A new cytotoxic macrolide from an Okinawan ascidian Lissoclinum sp. Tetrahedron Lett. 1999, 40, 6305–6308. [Google Scholar] [CrossRef]
- Takada, N.; Sato, H.; Suenaga, K.; Arimoto, H.; Yamada, K.; Ueda, K.; Uemura, D. Isolation and structures of haterumalides NA, NB, NC, ND, and NE, novel macrolides from an Okinawan Sponge Ircinia sp. Tetrahedron Lett. 1999, 40, 6309–6312. [Google Scholar] [CrossRef]
- Teruya, T.; Suenaga, K.; Maruyama, S.; Kurotaki, M.; Kigoshi, H. Biselides A–E: Novel polyketides from the Okinawan ascidian Didemnidae sp. Tetrahedron 2005, 61, 6561–6567. [Google Scholar] [CrossRef]
- Hayakawa, I.; Kigoshi, H.; Okamura, M.; Suzuki, K.; Shimanuki, M.; Kimura, K.; Yamada, T.; Ohyoshi, T. Total Synthesis of Biselide A, A Cytotoxic Macrolide of Marine Origin. Synthesis 2017, 49, 2958–2970. [Google Scholar] [CrossRef]
- Challa, V.R.; Kwon, D.; Taron, M.; Fan, H.; Kang, B.; Wilson, D.; Haeckl, F.P.J.; Keerthisinghe, S.; Linington, R.G.; Britton, R. Total synthesis of biselide A. Chem. Sci. 2021, 12, 5534–5543. [Google Scholar] [CrossRef] [PubMed]
- Satoh, Y.; Kawamura, D.; Yamaura, M.; Ikeda, Y.; Ochiai, Y.; Hayakawa, I.; Kigoshi, H. Synthetic studies toward biselides. Part 1: Synthesis of the core carbon framework of biselides A, B, and E using Stille coupling. Tetrahedron Lett. 2012, 53, 1390–1392. [Google Scholar] [CrossRef]
- Satoh, Y.; Yamada, T.; Onozaki, Y.; Kawamura, D.; Hayakawa, I.; Kigoshi, H. Synthetic studies toward biselides. Part 2: Synthesis of the macrolactone part of biselides A and B using allylic oxidation. Tetrahedron Lett. 2012, 53, 1393–1396. [Google Scholar] [CrossRef]
- Hayakawa, I.; Suzuki, K.; Okamura, M.; Funakubo, S.; Onozaki, Y.; Kawamura, D.; Ohyoshi, T.; Kigoshi, H. Total Synthesis of Biselide E, a Marine Polyketide. Org. Lett. 2017, 19, 5713–5716. [Google Scholar] [CrossRef]
- Řezanka, T.; Hanuš, L.; Dembitsky, V.M. Chagosensine, a New Chlorinated Macrolide from the Red Sea Sponge Leucetta chagosensis. Eur. J. Org. Chem. 2003, 2003, 4073–4079. [Google Scholar] [CrossRef]
- Heinrich, M.; Murphy, J.J.; Ilg, M.K.; Letort, A.; Flasz, J.; Philipps, P.; Fürstner, A. Total Synthesis of Putative Chagosensine. Angew. Chem. Int. Ed. 2018, 57, 13575–13581. [Google Scholar] [CrossRef]
- Heinrich, M.; Murphy, J.J.; Ilg, M.K.; Letort, A.; Flasz, J.T.; Philipps, P.; Fürstner, A. Chagosensine: A Riddle Wrapped in a Mystery Inside an Enigma. J. Am. Chem. Soc. 2020, 142, 6409–6422. [Google Scholar] [CrossRef]
- Lu, C.-K.; Chen, Y.-M.; Wang, S.-H.; Wu, Y.-Y.; Cheng, Y.-M. Formosalides A and B, cytotoxic 17-membered ring macrolides from a marine dinoflagellate Prorocentrum sp. Tetrahedron Lett. 2009, 50, 1825–1827. [Google Scholar] [CrossRef]
- Schulthoff, S.; Hamilton, J.Y.; Heinrich, M.; Kwon, Y.; Wirtz, C.; Fürstner, A. The Formosalides: Structure Determination by Total Synthesis. Angew. Chem. Int. Ed. 2021, 60, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Gajula, S.; Reddy, A.V.V.; Reddy, D.P.; Yadav, J.S.; Mohapatra, D.K. Stereoselective Synthesis of the C1–C16 Fragment of the Purported Structure of Formosalide B. ACS Omega 2020, 5, 10217–10224. [Google Scholar] [CrossRef]
- Hirata, Y.; Uemura, D. Halichondrins-antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef]
- Li, J.; Yan, W.; Kishi, Y. Unified Synthesis of C1–C19 Building Blocks of Halichondrins via Selective Activation/Coupling of Polyhalogenated Nucleophiles in (Ni)/Cr-Mediated Reactions. J. Am. Chem. Soc. 2015, 137, 6226–6231. [Google Scholar] [CrossRef]
- Yan, W.; Li, Z.; Kishi, Y. Selective Activation/Coupling of Polyhalogenated Nucleophiles in Ni/Cr-Mediated Reactions: Synthesis of C1–C19 Building Block of Halichondrin Bs. J. Am. Chem. Soc. 2015, 137, 6219–6225. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-S.; Dong, C.-G.; Kim, J.T.; Guo, H.; Huang, J.; Tiseni, P.S.; Kishi, Y. New Syntheses of E7389 C14−C35 and Halichondrin C14−C38 Building Blocks: Double-Inversion Approach. J. Am. Chem. Soc. 2009, 131, 15636–15641. [Google Scholar] [CrossRef]
- Dong, C.-G.; Henderson, J.A.; Kaburagi, Y.; Sasaki, T.; Kim, D.-S.; Kim, J.T.; Urabe, D.; Guo, H.; Kishi, Y. New Syntheses of E7389 C14−C35 and Halichondrin C14−C38 Building Blocks: Reductive Cyclization and Oxy-Michael Cyclization Approaches. J. Am. Chem. Soc. 2009, 131, 15642–15646. [Google Scholar] [CrossRef]
- Yahata, K.; Ye, N.; Iso, K.; Naini, S.R.; Yamashita, S.; Ai, Y.; Kishi, Y. Unified Synthesis of Right Halves of Halichondrins A–C. J. Org. Chem. 2017, 82, 8792–8807. [Google Scholar] [CrossRef]
- Yahata, K.; Ye, N.; Ai, Y.; Iso, K.; Kishi, Y. Unified, Efficient, and Scalable Synthesis of Halichondrins: Zirconium/Nickel-Mediated One-Pot Ketone Synthesis as the Final Coupling Reaction. Angew. Chem. 2017, 129, 10936–10940. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Pan, S.; Shelke, Y.; Das, D.; Ye, Q.; Lu, Y.; Sau, S.; Bao, R.; Rigol, S. A Reverse Approach to the Total Synthesis of Halichondrin B. J. Am. Chem. Soc. 2021, 143, 9267–9276. [Google Scholar] [CrossRef]
- Towle, M.J.; Salvato, K.A.; Budrow, J.; Wels, B.F.; Kuznetsov, G.; Aalfs, K.K.; Welsh, S.; Zheng, W.; Seletsky, B.M.; Palme, M.H.; et al. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B. Cancer Res. 2001, 61. [Google Scholar]
- Cigler, T.; Vahdat, L.T. Eribulin mesylate for the treatment of breast cancer. Expert Opin. Pharmacother. 2010, 11, 1587–1593. [Google Scholar] [CrossRef] [PubMed]
- Swami, U.; Shah, U.; Goel, S. Eribulin in Cancer Treatment. Mar. Drugs 2015, 13, 5016–5058. [Google Scholar] [CrossRef]
- Tsuda, M.; Oguchi, K.; Iwamoto, R.; Okamoto, Y.; Kobayashi, J.; Fukushi, E.; Kawabata, J.; Ozawa, T.; Masuda, A.; Kitaya, A.Y.; et al. Iriomoteolide-1a, a Potent Cytotoxic 20-Membered Macrolide from a Benthic Dinoflagellate Amphidinium Species. J. Org. Chem. 2007, 72, 4469–4474. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, K.; Tsuda, M.; Masuda, A.; Fukushi, E.; Kawabata, J. Iriomoteolide-2a, a Cytotoxic 23-Membered Macrolide from Marine Benthic Dinoflagellate Amphidinium Species. Heterocycles 2015, 91, 265. [Google Scholar] [CrossRef]
- Akakabe, M.; Kumagai, K.; Tsuda, M.; Konishi, Y.; Tominaga, A.; Kaneno, D.; Fukushi, E.; Kawabata, J.; Masuda, A.; Tsuda, M. Iriomoteolides-10a and 12a, Cytotoxic Macrolides from Marine Dinoflagellate Amphidinium Species. Chem. Pharm. Bull. 2016, 64, 1019–1023. [Google Scholar] [CrossRef]
- Akakabe, M.; Kumagai, K.; Tsuda, M.; Konishi, Y.; Tominaga, A.; Tsuda, M.; Fukushi, E.; Kawabata, J. Iriomoteolide-13a, a cytotoxic 22-membered macrolide from a marine dinoflagellate Amphidinium species. Tetrahedron 2014, 70, 2962–2965. [Google Scholar] [CrossRef]
- Sakamoto, K.; Hakamata, A.; Tsuda, M.; Fuwa, H. Total Synthesis and Stereochemical Revision of Iriomoteolide-2a. Angew. Chem. Int. Ed. 2018, 57, 3801–3805. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Hakamata, A.; Iwasaki, A.; Suenaga, K.; Tsuda, M.; Fuwa, H. Total Synthesis, Stereochemical Revision, and Biological Assessment of Iriomoteolide-2a. Chem. A Eur. J. 2019, 25, 8528–8542. [Google Scholar] [CrossRef]
- Sikorska, J.; Hau, A.M.; Anklin, C.; Parker-Nance, S.; Davies-Coleman, M.; Ishmael, J.E.; McPhail, K.L. Mandelalides A–D, Cytotoxic Macrolides from a New Lissoclinum Species of South African Tunicate. J. Org. Chem. 2012, 77, 6066–6075. [Google Scholar] [CrossRef]
- Lei, H.; Yan, J.; Yu, J.; Liu, Y.; Wang, Z.; Xu, Z.; Ye, T. Total Synthesis and Stereochemical Reassignment of Mandelalide A. Angew. Chem. 2014, 126, 6651–6655. [Google Scholar] [CrossRef]
- Nazari, M.; Serrill, J.D.; Sikorska, J.; Ye, T.; Ishmael, J.E.; McPhail, K.L. Discovery of Mandelalide E and Determinants of Cytotoxicity for the Mandelalide Series. Org. Lett. 2016, 18, 1374–1377. [Google Scholar] [CrossRef]
- Nazari, M.; Serrill, J.D.; Wan, X.; Nguyen, M.H.; Anklin, C.; Gallegos, D.A.; Smith, A.B.; Ishmael, J.E.; McPhail, K.L. New Mandelalides Expand a Macrolide Series of Mitochondrial Inhibitors. J. Med. Chem. 2017, 60, 7850–7862. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.H.; Imanishi, M.; Kurogi, T.; Smith, A.B., III. Total Synthesis of (−)-Mandelalide A Exploiting Anion Relay Chemistry (ARC): Identification of a Type II ARC/CuCN Cross-Coupling Protocol. J. Am. Chem. Soc. 2016, 138, 3675–3678. [Google Scholar] [CrossRef]
- Nguyen, M.H.; Imanishi, M.; Kurogi, T.; Wan, X.; Ishmael, J.E.; McPhail, K.L.; Smith, A.B., III. Synthetic Access to the Mandelalide Family of Macrolides: Development of an Anion Relay Chemistry Strategy. J. Org. Chem. 2018, 83, 4287–4306. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.M.; Yamini, V.; Singarapu, K.K.; Ghosh, S. Synthesis of Proposed Aglycone of Mandelalide A. Org. Lett. 2014, 16, 2658–2660. [Google Scholar] [CrossRef] [PubMed]
- Yamini, V.; Reddy, K.M.; Krishna, A.S.; Lakshmi, J.K.; Ghosh, S. Formal total synthesis of mandelalide A. J. Chem. Sci. 2019, 131, 25. [Google Scholar] [CrossRef]
- AnkiReddy, P.; AnkiReddy, S.; Sabitha, G. Synthetic Studies toward the Revised Aglycone of Mandelalide A. ChemistrySelect 2017, 2, 1032–1036. [Google Scholar] [CrossRef]
- Brütsch, T.M.; Bucher, P.; Altmann, K.-H. Total Synthesis and Biological Assessment of Mandelalide A. Chem. A Eur. J. 2016, 22, 1292–1300. [Google Scholar] [CrossRef]
- Nakashima, T.; Iwatsuki, M.; Ochiai, J.; Kamiya, Y.; Nagai, K.; Matsumoto, A.; Ishiyama, A.; Otoguro, K.; Shiomi, K.; Takahashi, Y.; et al. Mangromicins A and B: Structure and antitrypanosomal activity of two new cyclopentadecane compounds from Lechevalieria aerocolonigenes K10-0216. J. Antibiot. 2013, 67, 253–260. [Google Scholar] [CrossRef]
- Nakashima, T.; Kamiya, Y.; Iwatsuki, M.; Takahashi, Y.; Omura, S. Mangromicins, six new anti-oxidative agents isolated from a culture broth of the actinomycete, Lechevalieria aerocolonigenes K10-0216. J. Antibiot. 2014, 67, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Takada, H.; Yamada, T.; Hirose, T.; Ishihara, T.; Nakashima, T.; Takahashi, Y.K.; Omura, S.; Sunazuka, T. Total Synthesis and Determination of the Absolute Configuration of Naturally Occurring Mangromicin A, with Potent Antitrypanosomal Activity. Org. Lett. 2017, 19, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Sun, P.; Li, T.; Kurtán, T.; Mándi, A.; Antus, S.; Krohn, K.; Draeger, S.; Schulz, B.; Yi, Y.; et al. Bioactive Nonanolide Derivatives Isolated from the Endophytic Fungus Cytospora sp. J. Org. Chem. 2011, 76, 9699–9710. [Google Scholar] [CrossRef]
- Ehrlich, G.; Stark, C.B.W. Total Synthesis of Cytospolide D and Its Biomimetic Conversion to Cytospolides M, O, and Q. Org. Lett. 2016, 18, 4802–4805. [Google Scholar] [CrossRef]
- Chatterjee, S.; Mandal, G.H.; Goswami, R.K. Total Synthesis of Cytospolide Q. ACS Omega 2018, 3, 7350–7357. [Google Scholar] [CrossRef]
- Ehrlich, G.; Stark, C.B.W. Synthesis of Cytospolide Analogues and Late-State Diversification Thereof. J. Org. Chem. 2019, 84, 3132–3147. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Matsuda, H.; Makabe, K.; Yamaguchi, K. Oscillariolide, a novel macrolide from a blue-green alga Oscillatoria sp. Tetrahedron Lett. 1991, 32, 2391–2394. [Google Scholar] [CrossRef]
- Williamson, R.T.; Boulanger, A.; Vulpanovici, A.; Roberts, M.A.; Gerwick, W.H. Structure and Absolute Stereochemistry of Phormidolide, a New Toxic Metabolite from the Marine Cyanobacterium Phormidium sp. J. Org. Chem. 2002, 67, 7927–7936. [Google Scholar] [CrossRef]
- Lam, N.Y.S.; Muir, G.; Challa, V.R.; Britton, R.; Paterson, I. A counterintuitive stereochemical outcome from a chelation-controlled vinylmetal aldehyde addition leads to the configurational reassignment of phormidolide A. Chem. Commun. 2019, 55, 9717–9720. [Google Scholar] [CrossRef]
- Ndukwe, I.E.; Wang, X.; Lam, N.Y.S.; Ermanis, K.; Alexander, K.L.; Bertin, M.J.; Martin, G.E.; Muir, G.; Paterson, I.; Britton, R.; et al. Synergism of anisotropic and computational NMR methods reveals the likely configuration of phormidolide A. Chem. Commun. 2020, 56, 7565–7568. [Google Scholar] [CrossRef]
- Lorente, A.; Gil, A.; Fernández, R.; Cuevas, C.; Albericio, F.; Álvarez, M. Phormidolides B and C, Cytotoxic Agents from the Sea: Enantioselective Synthesis of the Macrocyclic Core. Chem. Eur. J. 2015, 21, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Lorente, A.; Albericio, F.; Alvarez, M. Stereoselective Allylstannane Addition for a Convergent Synthesis of a Complex Molecule. Org. Lett. 2015, 17, 6246–6249. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Giarrusso, M.; Lamariano-Merketegi, J.; Lorente, A.; Albericio, F.; Álvarez, M. Toward the Synthesis of Phormidolides. ACS Omega 2018, 3, 2351–2362. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Lamariano-Merketegi, J.; Lorente, A.; Albericio, F.; Álvarez, M. Enantioselective Synthesis of the Polyhydroxylated Chain of Oscillariolide and Phormidolides A–C. Org. Lett. 2016, 18, 4485–4487. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Lamariano-Merketegi, J.; Lorente, A.; Albericio, F.; Álvarez, M. Synthesis of (E)-4-Bromo-3-methoxybut-3-en-2-one, the Key Fragment in the Polyhydroxylated Chain Common to Oscillariolide and Phormidolides A-C. Chem. A Eur. J. 2016, 22, 7033–7035. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.C.; Winter, J.M.; Cullum, R.; Li, Z.; Fenical, W. Complementary Genomic, Bioinformatics, and Chemical Approaches Facilitate the Absolute Structure Assignment of Ionostatin, a Linear Polyketide from a Rare Marine-Derived Actinomycete. ACS Chem. Biol. 2020, 15, 2507–2515. [Google Scholar] [CrossRef]
- Liu, H.; Lin, S.; Jacobsen, K.M.; Poulsen, T.B. Chemical Syntheses and Chemical Biology of Carboxyl Polyether Ionophores: Recent Highlights. Angew. Chem. Int. Ed. 2019, 58, 13630–13642. [Google Scholar] [CrossRef]
- Kevin, D.A., II; Meujo, D.A.F.; Hamann, M.T. Polyether ionophores: Broad-spectrum and promising biologically active molecules for the control of drug-resistant bacteria and parasites. Expert Opin. Drug Discov. 2009, 4, 109–146. [Google Scholar] [CrossRef]
- Rutkowski, J.; Brzezinski, B. Structures and Properties of Naturally Occurring Polyether Antibiotics. BioMed Res. Int. 2013, 2013, 162513. [Google Scholar] [CrossRef]
- Toeplitz, B.K.; Cohen, A.I.; Funke, P.T.; Parker, W.L.; Gougoutas, J.Z. Structure of ionomycin-A novel diacidic polyether antibiotic having high affinity for calcium ions. J. Am. Chem. Soc. 1979, 101, 3344–3353. [Google Scholar] [CrossRef]
- Huczyński, A. Polyether ionophores—Promising bioactive molecules for cancer therapy. Bioorganic Med. Chem. Lett. 2012, 22, 7002–7010. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y.; Zhou, T.; Sato, S.; Matsumoto, T.; Yu, L.; Oku, N. Akaeolide, a Carbocyclic Polyketide from Marine-Derived Streptomyces. Org. Lett. 2013, 15, 5678–5681. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Komaki, H.; Ichikawa, N.; Hosoyama, A.; Sato, S.; Igarashi, Y. Biosynthesis of Akaeolide and Lorneic Acids and Annotation of Type I Polyketide Synthase Gene Clusters in the Genome of Streptomyces sp. NPS554. Mar. Drugs 2015, 13, 581–596. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Hartshorn, M.P.; McLennan, T.J.; Munro, M.H.G.; Robinson, W.T.; Yorke, S.C. Thyrsiferol: A squalene-derived metabolite of Laurencia thyrsifera. Tetrahedron Lett. 1978, 19, 69–72. [Google Scholar] [CrossRef]
- Juang, S.-H.; Chiang, C.-Y.; Liang, F.-P.; Chan, H.-H.; Yang, J.-S.; Wang, S.-H.; Lin, Y.-C.; Kuo, P.-C.; Shen, M.-R.; Thang, T.D.; et al. Mechanistic Study of Tetrahydrofuran- acetogenins In Triggering Endoplasmic Reticulum Stress Response-apotoposis in Human Nasopharyngeal Carcinoma. Sci. Rep. 2016, 6, 39251. [Google Scholar] [CrossRef]
- van Lint, M.J.; Hall, M.; Faber, K.; van Spanning, R.J.M.; Ruijter, E.; Orru, R.V.A. Stereoselective Chemoenzymatic Cascade Synthesis of the bis-THF Core of Acetogenins. Eur. J. Org. Chem. 2019, 2019, 1092–1101. [Google Scholar] [CrossRef]
- Öztunç, A.; Imre, S.; Lotter, H.; Wagner, H. Two C15 bromoallenes from the red alga Laurencia obtusa. Phytochemistry 1991, 30, 255–257. [Google Scholar] [CrossRef]
- Guella, G.; Chiasera, G.; Mancini, I.; Öztunç, A.; Pietra, F. Twelve-Membered O-Bridged Cyclic Ethers of Red Seaweeds in the Genus Laurencia Exist in Solution as Slowly Interconverting Conformers. Chem. A Eur. J. 1997, 3, 1223–1231. [Google Scholar] [CrossRef]
- Guella, G.; Mancini, I.; Öztunç, A.; Pietra, F. Conformational Bias in Macrocyclic Ethers and Observation of High Solvolytic Reactivity at a Masked Furfuryl (=2-Furylmethyl) C-Atom. Helv. Chim. Acta 2000, 83, 336–348. [Google Scholar] [CrossRef]
- Braddock, D.C.; Rzepa, H.S. Structural Reassignment of Obtusallenes V, VI, and VII by GIAO-Based Density Functional Prediction. J. Nat. Prod. 2008, 71, 728–730. [Google Scholar] [CrossRef]
- Braddock, D.C.; Millan, D.S.; Pérez-Fuertes, Y.; Pouwer, R.H.; Sheppard, R.N.; Solanki, S.; White, A.J.P. Bromonium Ion Induced Transannular Oxonium Ion Formation−Fragmentation in Model Obtusallene Systems and Structural Reassignment of Obtusallenes V−VII. J. Org. Chem. 2009, 74, 1835–1841. [Google Scholar] [CrossRef] [PubMed]
- Braddock, D.C.; Bhuva, R.; Millan, D.S.; Pérez-Fuertes, Y.; Roberts, C.A.; Sheppard, R.N.; Solanki, S.; Stokes, E.S.E.; White, A.J.P. A Biosynthetically-Inspired Synthesis of the Tetrahydrofuran Core of Obtusallenes II and IV. Org. Lett. 2007, 9, 445–448. [Google Scholar] [CrossRef]
- Arthuis, M.; Beaud, R.; Gandon, V.; Roulland, E. Counteranion-Directed Catalysis in the Tsuji-Trost Reaction: Stereocontrolled Access to 2,5-Disubstituted 3-Hydroxy-Tetrahydrofurans. Angew. Chem. Int. Ed. 2012, 51, 10510–10514. [Google Scholar] [CrossRef]
- Clarke, J.; Bonney, K.J.; Yaqoob, M.; Solanki, S.; Rzepa, H.S.; White, A.J.P.; Millan, D.S.; Braddock, D.C. Epimeric Face-Selective Oxidations and Diastereodivergent Transannular Oxonium Ion Formation Fragmentations: Computational Modeling and Total Syntheses of 12-Epoxyobtusallene IV, 12-Epoxyobtusallene II, Obtusallene X, Marilzabicycloallene C, and Marilzabicycloallene D. J. Org. Chem. 2016, 81, 9539–9552. [Google Scholar] [CrossRef]
- Kubota, T.; Sakuma, Y.; Shimbo, K.; Tsuda, M.; Nakano, M.; Uozumi, Y.; Kobayashi, J. Amphezonol A, a novel polyhydroxyl metabolite from marine dinoflagellate Amphidinium sp. Tetrahedron Lett. 2006, 47, 4369–4371. [Google Scholar] [CrossRef]
- Chen, W.; Liu, H.; Long, J.; Tao, H.; Lin, X.; Liao, S.; Yang, B.; Zhou, X.; Liu, Y.; Wang, J. Asperpentenone A, A novel polyketide isolated from the deep-sea derived fungus Aspergillus sp. SCSIO 41024. Phytochem. Lett. 2020, 35, 99–102. [Google Scholar] [CrossRef]
- Patil, A.D.; Freyer, A.J.; Bean, M.F.; Carte, B.K.; Westley, J.W.; Johnson, R.K.; Lahouratate, P. The plakortones, novel bicyclic lactones from the sponge Plakortis halichondrioides: Activators of cardiac SR-Ca2+-pumping ATPase. Tetrahedron 1996, 52, 377–394. [Google Scholar] [CrossRef]
- Cafieri, F.; Fattorusso, E.; Taglialatela-Scafati, O.; di Rosa, M.; Ianaro, A. Metabolites from the sponge plakortis simplex. II.: Isolation of four bioactive lactone compounds and of a novel related amino acid. Tetrahedron 1999, 55, 13831–13840. [Google Scholar] [CrossRef]
- Chianese, G.; Yu, H.-B.; Yang, F.; Sirignano, C.; Luciano, P.; Han, B.-N.; Khan, S.; Lin, H.-W.; Taglialatela-Scafati, O. PPAR Modulating Polyketides from a Chinese Plakortis simplex and Clues on the Origin of Their Chemodiversity. J. Org. Chem. 2016, 81, 5135–5143. [Google Scholar] [CrossRef]
- Yong, K.W.L.; de Voss, J.J.; Hooper, J.N.A.; Garson, M.J. Configurational Assignment of Cyclic Peroxy Metabolites Provides an Insight into Their Biosynthesis: Isolation of Plakortolides, seco-Plakortolides, and Plakortones from the Australian Marine Sponge Plakinastrella clathrata. J. Nat. Prod. 2011, 74, 194–207. [Google Scholar] [CrossRef]
- Akiyama, M.; Isoda, Y.; Nishimoto, M.; Narazaki, M.; Oka, H.; Kuboki, A.; Ohira, S. Total synthesis and absolute stereochemistry of plakortone E. Tetrahedron Lett. 2006, 47, 2287–2290. [Google Scholar] [CrossRef]
- Semmelhack, M.F.; Hooley, R.J.; Kraml, C.M. Synthesis of Plakortone B and Analogs. Org. Lett. 2006, 8, 5203–5206. [Google Scholar] [CrossRef] [PubMed]
- Paddon-Jones, G.C.; Hungerford, N.L.; Hayes, P.; Kitching, W. Efficient Palladium(II)-Mediated Construction of Functionalized Plakortone Cores. Org. Lett. 1999, 1, 1905–1907. [Google Scholar] [CrossRef]
- Hayes, P.Y.; Kitching, W. Total Synthesis and Absolute Stereochemistry of Plakortone D. J. Am. Chem. Soc. 2002, 124, 9718–9719. [Google Scholar] [CrossRef]
- Hayes, P.Y.; Kitching, W. Synthesis in the Plakortone Series: Plakortone E. Heterocycles 2004, 35, 173–177. [Google Scholar] [CrossRef]
- Xie, X.-G.; Wu, X.-W.; Lee, H.-K.; Peng, X.-S.; Wong, H.N.C. Total Synthesis of Plakortone B. Chem. A Eur. J. 2010, 16, 6933–6941. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-Y.; Tian, X.-Y.; Li, Z.-W.; Peng, X.-S.; Wong, H.N.C. Total Synthesis of Plakortide E and Biomimetic Synthesis of Plakortone B. Chem. A Eur. J. 2011, 17, 5874–5880. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, H.; Sato, S.; Tokudome, K.; Yamada, T. Stereoselective Formation of Tetrahydrofuran Rings via [3 + 2] Annulation: Total Synthesis of Plakortone L. Org. Lett. 2014, 16, 3384–3387. [Google Scholar] [CrossRef]
- Li, J.; Li, C.; Riccio, R.; Lauro, G.; Bifulco, G.; Li, T.-J.; Tang, H.; Zhuang, C.-L.; Ma, H.; Sun, P.; et al. Chemistry and Selective Tumor Cell Growth Inhibitory Activity of Polyketides from the South China Sea Sponge Plakortis sp. Mar. Drugs 2017, 15, 129. [Google Scholar] [CrossRef]































































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Peña, L.; Díez-Poza, C.; González-Andrés, P.; Barbero, A. The Tetrahydrofuran Motif in Polyketide Marine Drugs. Mar. Drugs 2022, 20, 120. https://doi.org/10.3390/md20020120
Fernández-Peña L, Díez-Poza C, González-Andrés P, Barbero A. The Tetrahydrofuran Motif in Polyketide Marine Drugs. Marine Drugs. 2022; 20(2):120. https://doi.org/10.3390/md20020120
Chicago/Turabian StyleFernández-Peña, Laura, Carlos Díez-Poza, Paula González-Andrés, and Asunción Barbero. 2022. "The Tetrahydrofuran Motif in Polyketide Marine Drugs" Marine Drugs 20, no. 2: 120. https://doi.org/10.3390/md20020120
APA StyleFernández-Peña, L., Díez-Poza, C., González-Andrés, P., & Barbero, A. (2022). The Tetrahydrofuran Motif in Polyketide Marine Drugs. Marine Drugs, 20(2), 120. https://doi.org/10.3390/md20020120