2. Results and Discussion
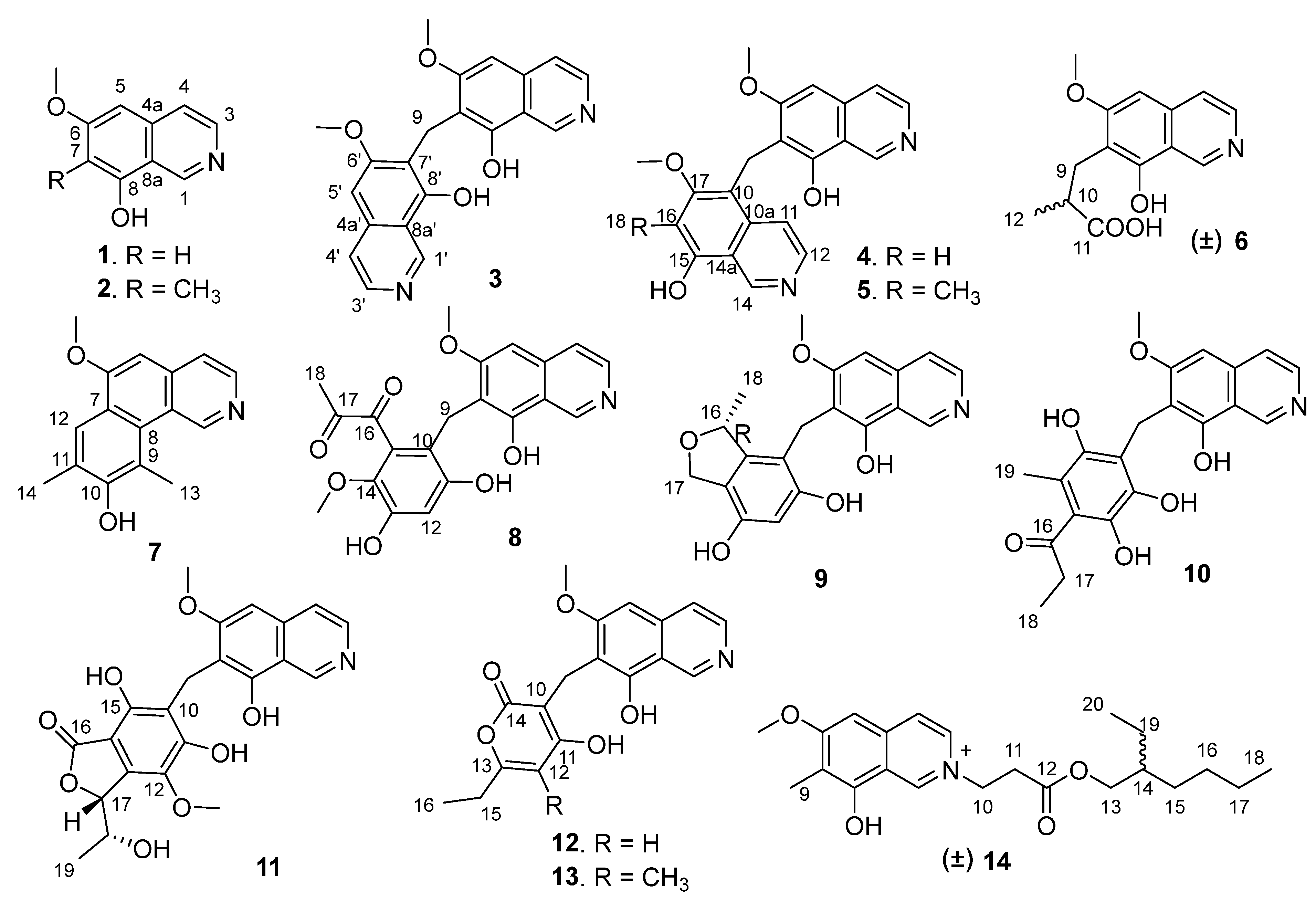
Puniceusine A (
1) has the molecular formula C
10H
9NO
2, as determined by HRESIMS. The
1H NMR spectrum (
Table 1) showed the presence of one methoxy group at
δH 3.96 (3H, s) and five aromatic hydrogens at
δH 9.45 (1H, s), 8.40 (1H, d,
J = 5.7 Hz), 8.06 (1H, d,
J = 6.4 Hz), 7.13 (1H, s), 6.81 (1H, s). The
13C NMR spectrum (
Table 2) showed 10 carbon signals including one methoxy, five aromatic methines, and four aromatic non-protonated carbons. These NMR data were similar to those of 6,8-dimethoxyisoquinolin [
15] and papraline [
16], and the only clear difference between
1 and 6,8-dimethoxyisoquinolin was the disappearance of one oxygenated methyl, which indicated that
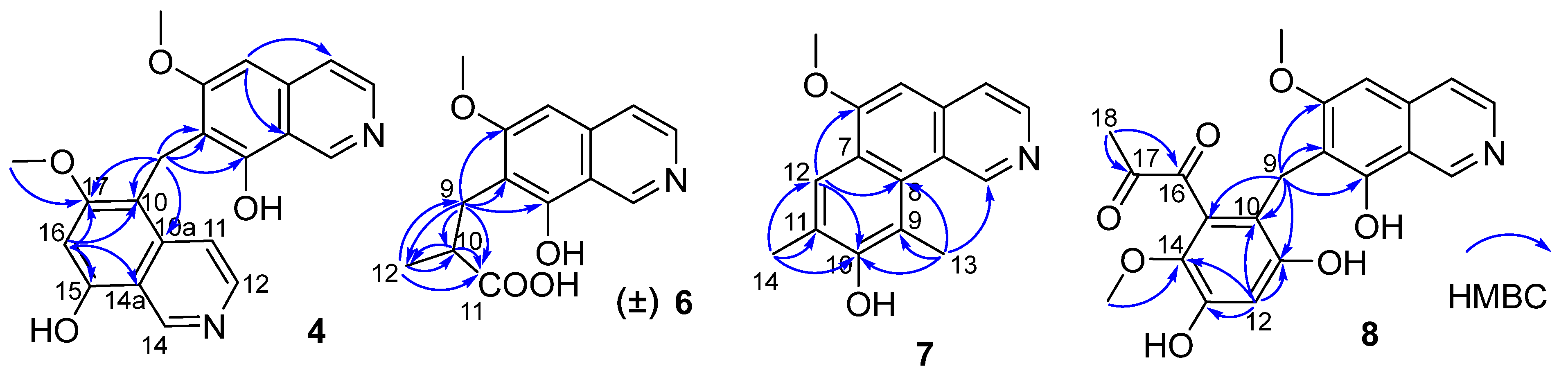
1 was also an isoquinoline alkaloid. This was supported by the HMBC spectrum showing correlations from H-1 to C-3/C-4a/C-8a, H-3 to C-4/C-4a, H-4 to C-3/C-5/C-8a, H-5 to C-4/C-6/C-7/C-8a, and H-7 to C-5/C-6/C-8/C-8a. In addition, the HMBC correlation from δ
H 3.96 (3H, s) to C-6 suggested that a methoxy group was attached at C-6. Thus, the structure of
1 was determined to be 6-methoxy-8-hydroxy-isoquinolin.
Puniceusine B (
2) was assigned the molecular formula C
11H
11NO
2 by HRESIMS. The
1H NMR and
13C NMR data (
Table 1 and
Table 2) showed great similarity to those of
1, and the main difference between them was the additional presence of one methyl (δ
H 2.25, 3H, s; δ
C 9.2) and the disappearance of one aromatic hydrogen in
2. The HMBC correlations from δ
H 2.25 to C-6/C-7/C-8 suggested the additional methyl attached at C-7. Hence, the structure of
2 was determined to be 6-methoxy-7-methyl-8-hydroxy-isoquinolin.
Puniceusine C (
3) was found to have the molecular formula C
21H
18N
2O
4 by HRESIMS that was nearly twice that of
2. The
1H and
13C NMR data (
Table 1 and
Table 2) showed great similarity to those of
2, and the clearest difference between them was the disappearance of a methyl signal and the additional presence of a methylene signal (
δH 4.32, 2H, s; δ
C 19.4) in
3. The
1H NMR spectrum of
3 showed a double increase in the integral areas for H-1, H-3, H-4, H-5, and a methoxy group. The HMBC correlations (
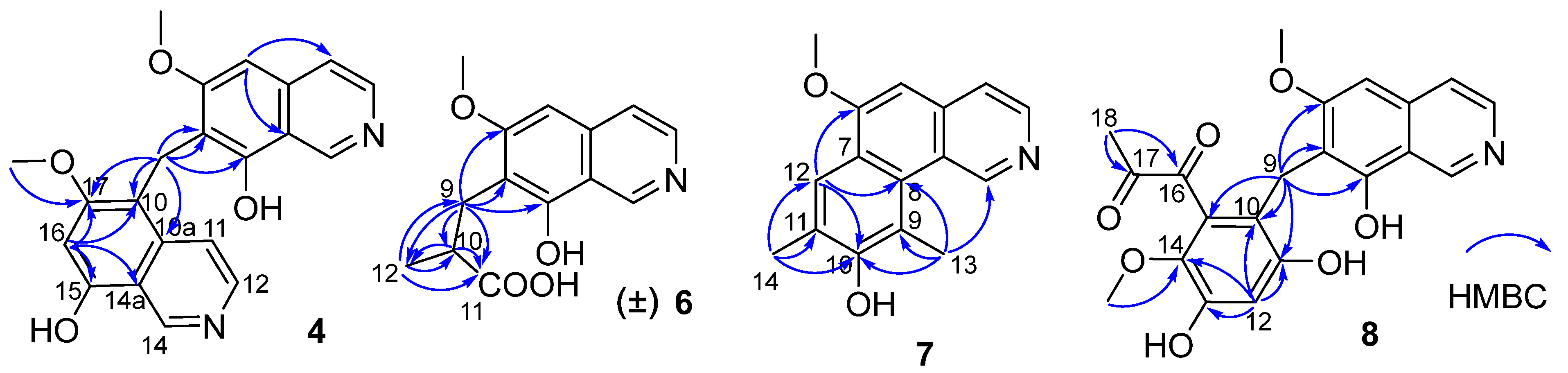
Figure 2) from
δH 4.32 (H
2-9) to C-6/C-7/C-8 suggested a methylene instead of a methyl attached at C-7. These data indicated that
3 was a symmetrical dimer of
1, connected at positions C-7′ and C-7 by a methylene C-9. Thus, the structure of
3 was determined as shown.
Puniceusine D (
4) showed the same molecular formula of C
21H
18N
2O
4 as that of
3 by analysis of its HRESIMS and NMR data (
Table 1 and
Table 2). The
1H NMR spectrum showed the presence of two downfield hydrogens at δ
H 9.59 (1H, s) and 9.52 (1H, s); six aromatic hydrogens at δ
H 8.58 (1H, d,
J = 7.0 Hz), 8.31 (1H, d,
J = 7.8 Hz), 8.29 (1H, d,
J = 7.0 Hz), 8.05 (1H, d,
J = 6.5 Hz), 7.11 (1H, s), and 7.00 (1H, s); two methoxys groups at δ
H 3.79 (3H, s) and 3.98 (3H, s); and one methylene at δ
H 4.47 (2H, s). The
13C NMR spectrum showed 21 carbon signals including one methylene, two methoxyls, eight aromatic methines, and ten aromatic non-protonated carbons. These data showed similarity to those of
1–
3, which indicated that
4 was also a dimer of
1. The HMBC correlations from H-5 to C-4/C-8a, from H-16 to C-10/C-14a/C-15/C-17, and from H
2-9 to C-6/C-7/C-8/C-10/C-10a/C-17 (
Figure 2) suggested that
4 was an asymmetric dimer of
1 connected at positions C-7 and C-10 by a methylene C-9. The two methoxy groups were attached at C-6 and C-17 based on the HMBC correlations of δ
H 3.98 (3H, s) with C-17 and δ
H 3.79 (3H, s) with C-6, respectively. Therefore, the structure of
4 was established as shown.
The molecular formula of puniceusine E (
5) was determined as C
22H
21N
2O
4 by HRESIMS. The
1H and
13C NMR data (
Table 1 and
Table 2) of
5 were greatly similar to those of
4, and the only obvious difference between them was the absence of one aromatic hydrogen and the additional presence of one methyl signal (δ
H 2.38, 3H, s; δ
C 10.8) in
5. The HMBC correlations from H
3-18 (δ
H 2.38) to C-15/C-16/C-17 suggested a methyl located at C-16 instead of a hydrogen. Thus, the structure of
5 was established as shown.
Puniceusine F (
6) had the molecular formula C
14H
15NO
4, as determined by HRESIMS. The
1H and
13C NMR data (
Table 1 and
Table 2) of
6 were similar to those of
1, and the clearest difference between them was the disappearance of one aromatic hydrogen and the additional presence of one methyl (
δH 1.27, d,
J = 7.5 Hz, 3H), one methylene, one methine, and one carboxyl group in
6. The HMBC correlations (
Figure 2) from H
2-9 (
δH 2.97 (dd,
J = 14.0, 5.5 Hz, 1H), 3.17 (dd,
J = 14.0, 8.5 Hz, 1H)) to C-6/C-7/C-8/C-10/C-11/C-12, from H-10 (
δH 2.85, m, 1H) to C-11/C-12, and from H
3-12 (
δH 1.27, d,
J =7.5 Hz, 3H) to C-9/C-10/C-11 suggested an isobutyric acid group attached at C-7 of the isoquinoline nucleus. The optical rotation and measured CD data of
6 were zero, which indicated that
6 was a racemic mixture. This was supported by the HPLC analysis of
6 with a chiral column (CHIRALPAK IA 4.6 mm × 250 mm column) eluting with n-hexane/ethanol/TFA (68:32:0.2,
v/
v) (
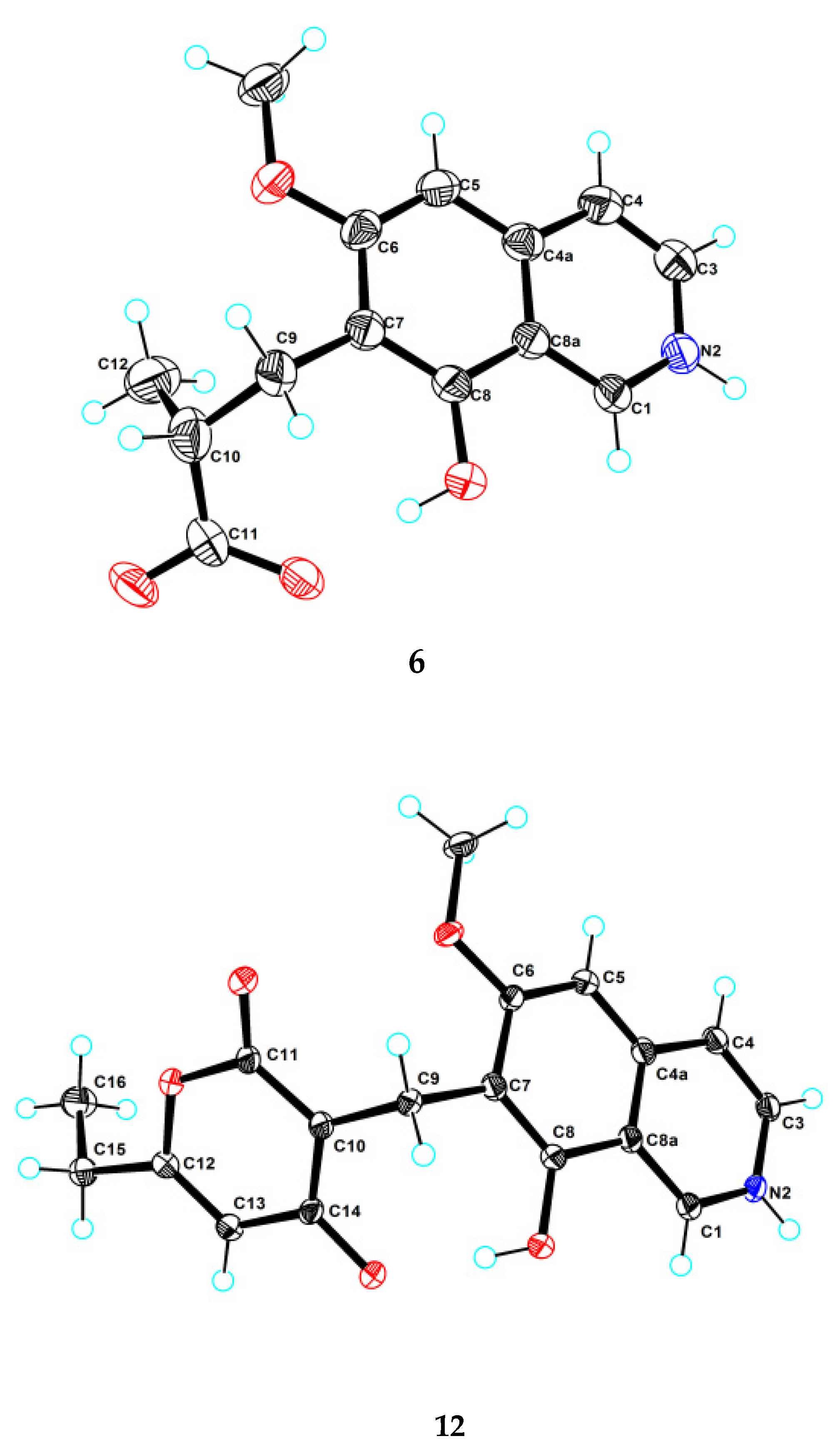
Figure S39). This structure was further confirmed by a single-crystal X-ray diffraction analysis (
Figure 3).
The molecular formula of puniceusine G (
7) was determined to be C
16H
15NO
2, according to its HRESIMS. The
1H and
13C NMR data (
Table 1 and
Table 2) of
7 were similar to those of
1, and the clearest difference between them was the additional presence of two double bonds (
δC 112.0 (C, C-9), 115.0 (CH, C-12), 120.7 (C, C-11), 148.7 (C, C-10)) and two methyls (
δH 2.51 (3H, s, H-14), 2.93 (3H, s, H-13);
δC 8.0, 8.4) in
7. The HMBC correlations (
Figure 2) from H-12 to C-6/C-8/C-10, from H
3-13 to C-1/C-8/C-9/C-10, and from H
3-14 to C-10/C-11/C-12, suggested a 3,5-dimethyl-4-hydroxyphenyl group attached at the isoquinoline nucleus by sharing C-7 and C-8 to form a benzo[
h]isoquinoline unit. Therefore, the structure of
7 was established as shown.
Puniceusine H (
8) had the molecular formula of C
21H
19NO
7 as determined by its HRESIMS. The
1H and
13C NMR data (
Table 2 and
Table 3) of
8 showed similarity to those of
4–
6 with the presence of characteristic chemical shifts for a methylene C-9 (δ
H 4.01 (2H, s), δ
C 19.3 (CH
2)). The HMBC correlations (
Figure 2) from H
2-9 to C-6/C-7/C-8/C-10/C-11/C-15, from H-12 (δ
H 6.58, 1H, s) to C-10/C-11/C-13/C-14, and from H
3-19 (δ
H 3.52, 3H, s) to C-14 suggested a 2,4-dihydroxy-5-methoxyphenyl fragment connected with isoquinoline unit by a methylene at position C-7. In addition, the HMBC correlations from H
3-18 (δ
H 2.30, 3H, s) to C-16 (δ
C 195.1, C)/C-17 (δ
C 196.9, C) suggested the presence of a 1,2-propanedione group that was exclusively assigned to attach at C-15 of the benzene ring based on the above data. Thus, the structure of
8 was established as shown.
Puniceusine I (
9) had the molecular formula of C
20H
19NO
5 according to its HRESIMS with 12 degrees of unsaturation. The
1H and
13C NMR data (
Table 3 and
Table 4) of
9 were similar to those of
8, and the clearest difference between them was the disappearance of two keto carbons and one methoxy and the additional presence of one oxygenated methylene (δ
H 4.84 (1H, dd,
J = 11.5, 2.5 Hz), 4.69 (1H, dd,
J = 11.5 Hz), δ
C 68.9, CH
2) and one oxygenated methine (δ
H 5.22 (1H, m), δ
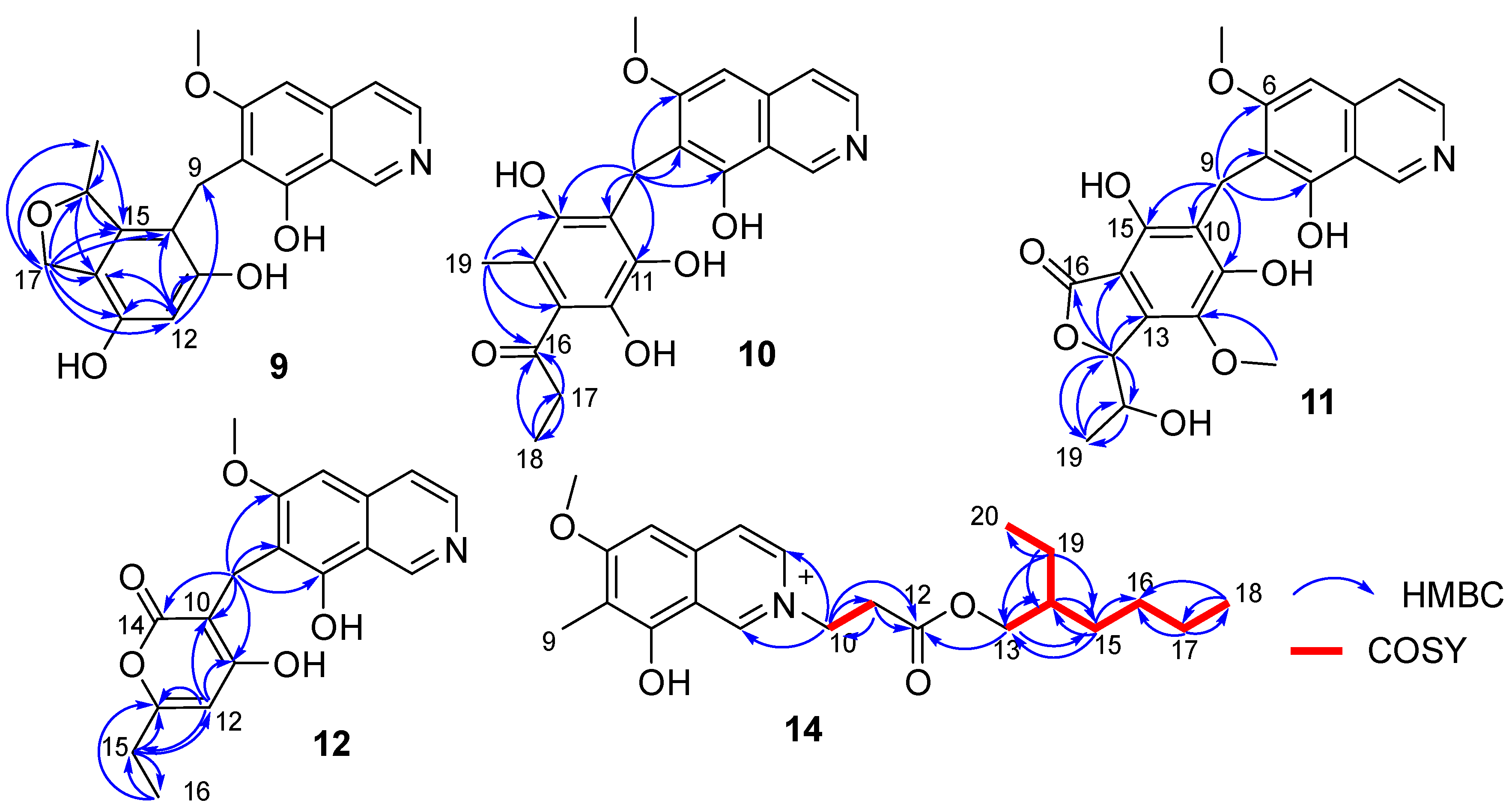
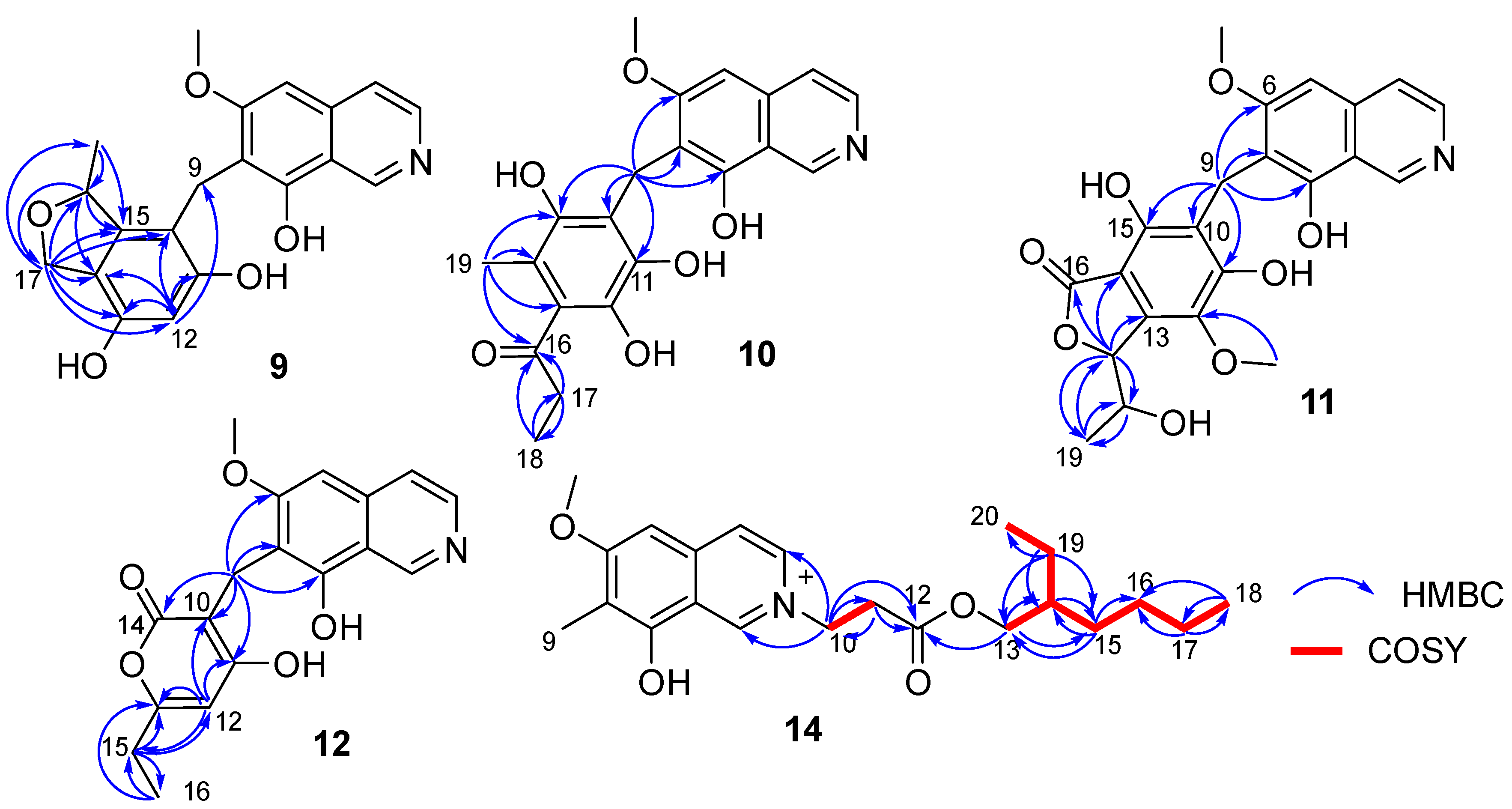
C 79.3, CH). A detailed analysis of HSQC and HMBC spectra proved that
9 also contained a 1,2,4,5,6-pentasubstituted-benzyl attached at C-7 of isoquinoline nucleus. In addition, the HMBC correlations from H-16 to C-14/C-15/C-17, from H
2-17 to C-10/C-11/C-12/C-13/C-14/C-15/C-16/C-18, from H
3-18 (δ
H 1.35, d,
J = 6.2 Hz, 3H) to C-15/C-16 (
Figure 4), suggested a 2-methyl-2,5-dihydrofuran ring connected with the benzene ring via C-14 and C-15. Therefore, the 2D structure of
9 was established as shown. The absolute configuration of
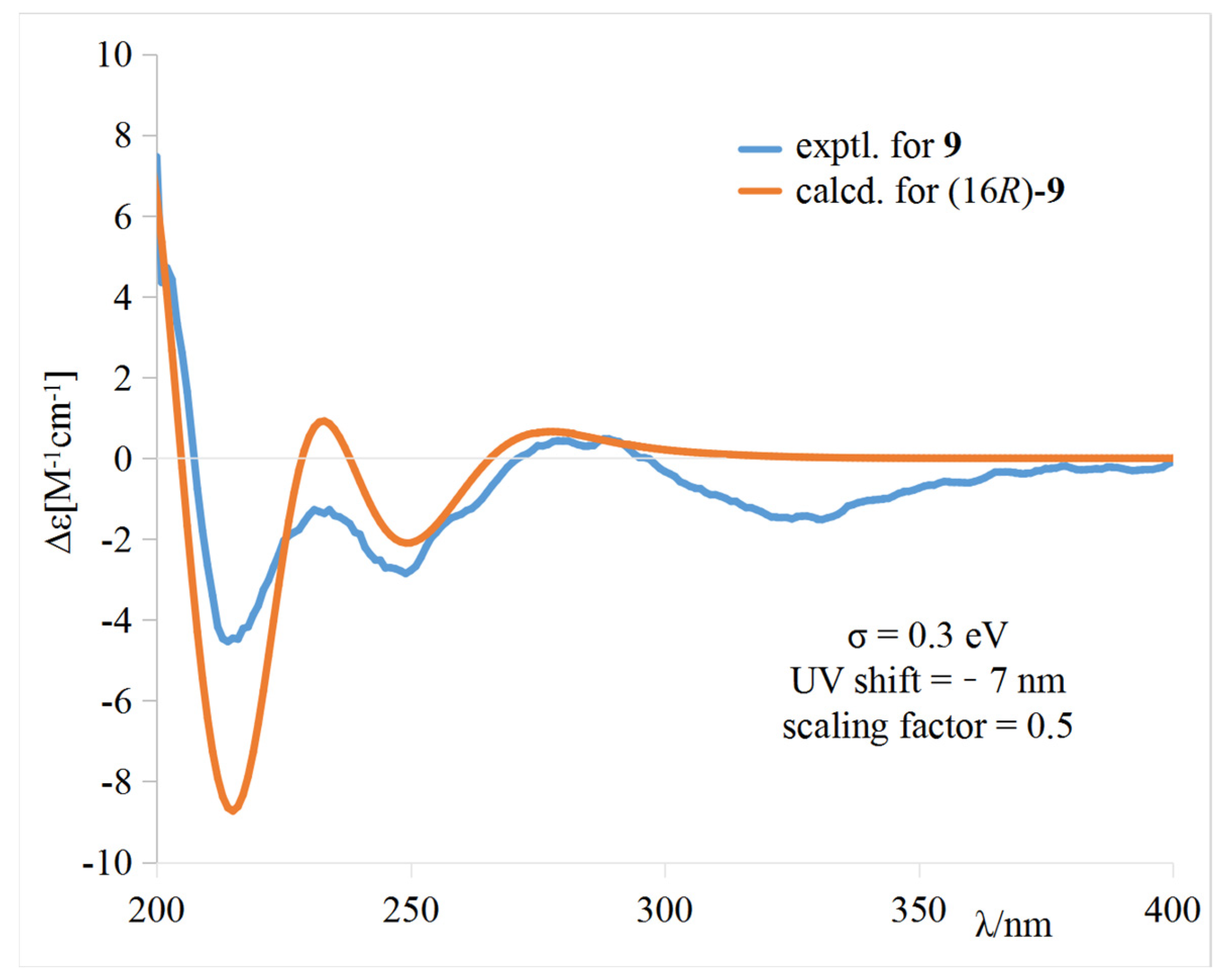
9 was further determined by electronic circular dichroism (ECD) calculations (
Tables S1–S3).
The calculated weighted ECD spectrum of (16
R)-
9 agreed well with the experimental ECD spectrum of
9 (
Figure 5), leading to the assignment of the absolute configuration at C-16.
Puniceusine J (
10) had the molecular formula of C
21H
21NO
6, as determined by its HRESIMS. The
1H and
13C NMR data of
10 (
Table 3 and
Table 4) were very similar to those of
8 and
9. A detailed analysis of HSQC and HMBC spectra proved that
10 also contained a benzyl attached at position C-7 of isoquinoline unit. The HMBC correlations of H
2-9 with C-6/C-7/C-8/C-10/C-11 (δ
C 170.0, C)/C-15 (δ
C 171.6, C) (
Figure 4) suggested two hydroxyl groups attached at C-11 and C-15, respectively. In addition, the HMBC correlations from H
3-19 (δ
H 1.92, 3H, s) to C-13/C-14/C-15 suggested a methyl attached at C-14. Furthermore, the HMBC correlations (
Figure 4) from H
2-17 (δ
H 2.58, 2H, q,
J = 7.3 Hz) to C-16/C-18, from H
3-18 (δ
H 1.02, 3H, t,
J = 7.3 Hz) to C-16/C-17, and from H
3-19 to C-16 suggested an 1-acetonyl group attached at C-13. Lastly, the chemical shift of C-12 (δ
C 163.1, C) indicated a hydroxy group attached at C-12. Therefore, the structure of
10 was established as shown.
Puniceusine K (
11) was found to have the molecular formula C
22H
21NO
8 by HRESIMS. The
1H and
13C NMR data of
11 (
Table 3 and
Table 4) were very similar to those of
10. A detailed analysis of HSQC and HMBC spectra proved that
11 also contained a hexasubstituted benzyl attached at position C-7 of isoquinoline unit. The HMBC correlations of H
2-9 with C-6/C-7/C-8/C-10/C-11 (δ
C 157.1, C)/C-15 (δ
C 153.0, C) suggested two hydroxyl groups attached at C-11 and C-15, respectively. Additionally, the HMBC correlations (
Figure 4) from H-17 (δ
H 5.56, d,
J = 2.5 Hz) to C-13/C-14/C-16/C-18/C-19, from H-18 (δ
H 4.36, qd,
J = 6.5, 2.5 Hz) to C-19, and from H
3-19 (δ
H 0.94, d,
J = 6.5 Hz, 3H) to C-17/C-18, suggested a 5-hydroxy-2-hexene-4-lactone group attached on the benzene ring via C-13 and C-14. In addition, the HMBC correlation of δ
H 3.76 (s, 3H) with C-12 (δ
C 136.8, C) suggested a methoxy group attached at C-12 of the benzene ring. The assignment of the substituent groups at positions C-12, C-13 and C-14 of the benzene ring was further supported by comparison of the
1H and
13C NMR data of the same isobenzofuran moiety in
11, embeurekol C [
17] and acetophthalidin [
18]. In addition, the small
3JHH value (2.5 Hz) between H-17 and H-18 in
11 was closely similar to that of embeurekol C [
17]. Furthermore, the specific rotation value of
11 ([α]
−9.1 (
c 0.1, CH
3OH)) was close to that of embeurekol C ([α]
−17 (
c 0.05, CH
3OH)) [
17], and the experimental ECD spectrum of
11 (
Figure S76) was greatly similar to that of embeurekol C [
17]. These data suggested that the absolute configuration of
11 was also 17
R, 18
S for that of embeurekol C.
Puniceusine L (
12) had a molecular formula of C
18H
17NO
5 on the basis of its HRESIMS and NMR data. Its
1H and
13C NMR data (
Table 3 and
Table 4) showed a similarity to those of
8–
11. A detailed analysis of HSQC and HMBC spectra suggested that
12 contained the same isoquinoline unit as
8–
11. In addition, considering the molecular formula and unsaturation degrees of
12, the HMBC correlations from H
2-9 to C-6/C-7/C-8/C-10/C-11/C-14, from H-12 to C-10/C-11/C-13/C-15, from H
2-15 to C-12/C-13/C-16, and from H
3-16 to C-13/C-15 (
Figure 4), suggested a 6-ethyl-4-hydroxy-2
H-pyran-2-one unit attached at the methylene C-9 of isoquinoline unit. The above assignment was further confirmed by a single crystal X-ray diffraction analysis (
Figure 3).
Puniceusine M (
13) had the molecular formula of C
19H
19NO
5 on the basis of its HRESIMS. The
1H and
13C NMR data (
Table 3 and
Table 4) were very similar to those of
12. The only difference between them was the disappearance of one aromatic hydrogen and the additional presence of a methyl (δ
H 2.01 (3H, s), δ
C 9.9). The HMBC correlations from H
3-17 (δ
H 2.01) to C-11/C-12/C-13 suggested the additional methyl attached at C-12. Therefore, the structure of
13 was established as shown.
Puniceusine N (
14) had the molecular formula C
22H
32NO
4+, as determined by HRESIMS. The
1H and
13C NMR (
Table 3 and
Table 4) data of
14 showed a similarity to those of
2, and the clearest difference between them was the additional presence of two methyls, seven methylenes (one oxygenated), one methine, and one carboxyl in
14. Detailed analysis of the HMBC and COSY spectra proved that
14 contained the same isoquinoline unit as that of
2. In addition, combining with the COSY correlation of H
2-10 (
δH 4.88, t,
J = 6.3 Hz) with H
2-11 (
δH 3.18, t,
J = 6.3 Hz) (
Figure 4), the HMBC correlations from H
2-10 to C-1/C-3/C-11/C-12 (
δC 171.8, C), and from H
2-11 to C-9/C-12 (
Figure 4), suggested a -CH
2-CH
2-COO- group attached at the nitrogen atom of isoquinoline unit. Furthermore, the sequential COSY correlations of H
2-13/H-14/H
2-15/H
2-16/H
2-17/H
2-18, and H-14/H
2-19/H
3-20 (
Figure 4), together with the HMBC correlations from H
2-13 to C-12/C-14/C-15, from H
2-15 to C-13/C-14/C-16, from H
2-17 to C-16/C-18, from H
3-18 to C-16/C-17, from H
2-19 to C-13/C-14/C-15/C-20, and from H
3-20 to C-14/C-19 (
Figure 4), suggested that the 2-ethylhexanol group connected with the carboxyl of the -CH
2-CH
2-COO- group to form an ester. The optical rotation and measured CD data of
14 were zero, which indicated
14 was a racemic mixture. However, an HPLC analysis of
14 with a chiral column (CHIRALPAK IA and IB, respectively, 4.6 mm × 250 mm column), eluting with n-hexane/ethanol/TFA (
Figures S99 and S100), showed a big trailing peak. The reason for this could be that the two kinds of chiral columns were not suitable for the chiral separation of
14. Thus, the structure of
14 was determined as shown.
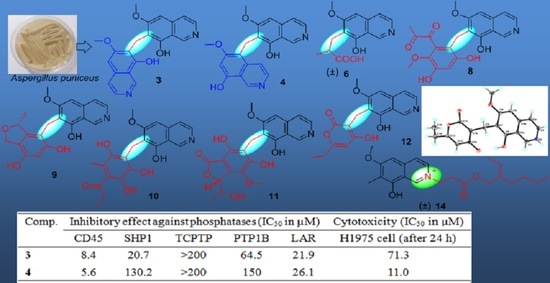
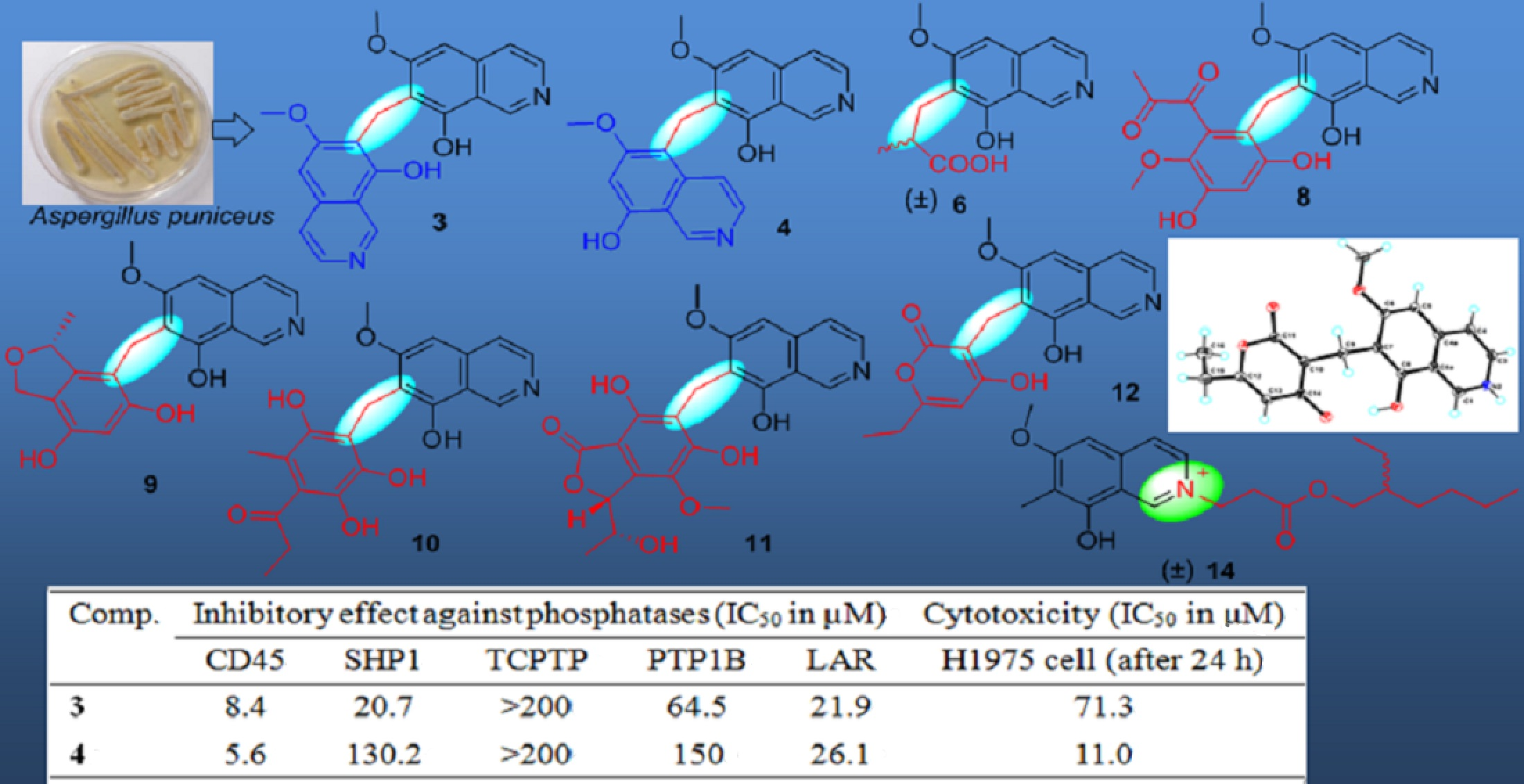
All of the 14 compounds were evaluated for their enzyme inhibitory activity against five PTPs including CD45, SHP1, TCPTP, PTP1B and LAR, cytotoxicity towards human lung adenocarcinoma cell line H1975, and antibacterial activity. The results of protein phosphatase inhibition assays (
Table 5) showed that only
3 and
4 selectively exhibited significant inhibitory activity against CD45 with IC
50 values of 8.4 and 5.6 µM, respectively, and
1,
8,
9,
10,
12 and
13 showed a mild inhibitory activity against several PTPs. A cytotoxicity assay (
Table 5) showed that only
4 had a moderate cytotoxicity towards H1975 cell lines with an IC
50 value of 11.0 µM. The analysis of the relationship of their structures, enzyme inhibitory activity and cytotoxicity displayed that the substituents at C-7 of the isoquinoline nucleus could greatly affect their bioactivity. In addition, antibacterial assays exhibited that
14 had medium antibacterial activity towards
Staphylococcus aureus, methicillin-resistant
S. aureus (MRSA), and
Escherichia coli, with MIC values of 100 µg/mL, and
4 could inhibit the growth of
E. coli with a MIC value of 100 µg/mL, while other compounds did not show clear antibacterial activity towards the three indicators. The results indicated that -C=N
+ unit was an active center for the antibacterial activity of
14.
3. Experimental Section
3.1. General Experimental Procedure
The procedures were the same as previously reported [
13,
14].
3.2. Fungal Material
The strain Aspergillus puniceus SCSIO z021 was isolated from a deep-sea sediment of Okinawa Trough (27°34.01′ N and 126°55.59′ E, ~1589 depth), which was located approximately 4.7 km from an active hydrothermal vent. The strain (GenBank accession number KX258801) was identified as Aspergillus puniceus through DNA extraction, ITS sequence amplification and sequence alignment, which has a 99% similarity to A. puniceus (GenBank accession number GU456970). The strain A. puniceus SCSIO z021 was deposited in the RNAM Center, South China Sea Institute of Oceanology, Chinese Academy of Science.
3.3. Fermentation and Extraction
The fungus strain was cultivated on potato glucose agar (PDA) plate containing 3% sea salt at 28 °C for 7 days. The spores were selected and transferred to a complex culture medium (glucose 1%, D-mannitol 2%, maltose 2%, corn meal 0.05%, monosodium glutamate 1%, KH2PO4 0.05%, MgSO4·7H2O 0.03%, yeast extract 0.3%, sea salt 3%) to obtain a spore suspension that was cultured in a shaker at 28 °C for 3 days at a rotating speed of 180 rmp. The fungus was cultured in 1 L Erlenmeyer flasks each containing 300 mL of 3# medium (glucose 1%, D-mannitol 2%, maltose 2%, corn meal 0.05%, monosodium glutamate 1%, KH2PO4 0.05%, MgSO4·7H2O 0.03%, yeast extract 0.3%, sea salt 3%) at 28 °C for 33 days under static condition. After fermentation, the broth and mycelia were separated with gauze. The broth was extracted with XAD-16 resin and sequentially eluted with H2O and EtOH to obtain crude extract (61.7 g). The mycelia was extracted three times with acetone, and further extracted three times with EtOAc to yield a crude extract (48.2 g).
3.4. Isolation and Purification
The combined extracts (109.9 g) were subjected to a normal-phase silica gel column eluting with a gradient of dichloromethane (DCM)/MeOH (100:0, 98:2, 95:5, 95:5, 90:10, 80:20, 70:30, 50:50, 0:100) to give nine subfractions (Fr.1–Fr.9) based on TLC analysis. Fr.4 (6.6 g) was separated by ODS column using MeOH-H2O-TFA (5:95:0.02 to 100:0:0.02) as eluent to afford 10 subfractions (Fr.4.1–Fr.4.10). Fr.4.1 was separated by Sephadex LH-20 eluting with MeOH followed by semipreparative HPLC (MeOH/H2O/TFA, 28:72:0.03, 3 mL/min) to yield 1 (11.6 mg, tR = 15.8 min). Fr.4.3 was subjected to Sephadex LH-20 using MeOH as mobile phase, which was further purified by semipreparative HPLC (CH3CN/H2O/TFA, 17:83:0.03, 3 mL/min) to yield 2 (30.7 mg, tR = 16.3 min). Fr.4.5 was isolated by Sephadex LH-20 eluting with MeOH, then purified by semipreparative HPLC (CH3CN/H2O/TFA, 25:75:0.03, 3 mL/min) to yield 7 (2.9 mg, tR = 25.9 min). Fr.4.6 was separated by Sephadex LH-20 with a mobile phase of MeOH, and then further purified by semipreparative HPLC (MeOH/H2O/TFA, 45:55:0.03, 3 mL/min) to yield 10 (2.8 mg, tR = 16.1 min) and 12 (15.2 mg, tR = 14.5 min). Fr.4.8 was isolated by silica gel column eluting with a gradient of CH2Cl2/MeOH (100:0, 80:1, 60:1, 40:1, 10:1, 5:1, 1:1, 0:100) to obtain three fractions, then Fr.4.8.1 was further purified by semipreparative HPLC (CH3CN/H2O/TFA, 49:51:0.03, 3 mL/min) to yield 14 (29.4 mg, tR = 20.2 min). Fr.6 (11.0 g) was separated by ODS column using MeOH-H2O-TFA (5:95:0.02 to 100:0:0.02) as mobile phase to yield eight subfractions (Fr.6.1–Fr.6.8). Fr.6.3 was purified by semipreparative HPLC (CH3CN/H2O/TFA, 21:79:0.03, 3 mL/min) to obtain 6 (4.0 mg, tR = 19.5 min), 3 (17.6 mg, tR = 18.4 min) and 4 (12.2 mg, tR = 19.7 min). Fr.6.6 was isolated by semipreparative HPLC (CH3CN/H2O/TFA, 24:76:0.03, 3 mL/min) to yield 11 (7.8 mg, tR = 16.0 min) and 5 (4.3 mg, tR = 14.4 min). Fr.6.7 was isolated by Sephadex LH-20 with MeOH as mobile phase, then further purified by semipreparative HPLC (CH3CN/H2O/TFA, 28:72:0.03, 3 mL/min) to yield 8 (4.1 mg, tR = 39.2 min) and 9 (2.8 mg, tR = 18.4 min). Fr.6.8 was separated by Sephadex LH-20, eluting with MeOH and further purified by semipreparative HPLC (CH3CN/H2O/TFA, 35:65:0.03, 3 mL/min) to get 13 (10.3 mg, tR = 18.0 min).
Puniceusine A (
1): white acicular crystal; UV (CH
3OH)
λmax (log
ε) 204 (1.42), 208 (1.42), 241 (1.56), 258 (1.58), 357 (0.74) nm;
1H and
13C NMR,
Table 1 and
Table 2; HRESIMS
m/
z 176.0709 [M + H]
+ (calcd for C
10H
10NO
2, 176.0706).
Puniceusine B (
2): pale yellow powder; UV (CH
3OH)
λmax (log
ε) 206 (1.61), 245 (1.75), 260 (1.83), 305 (0.73), 360 (0.84) nm;
1H and
13C NMR,
Table 1 and
Table 2; HRESIMS
m/
z 190.0864 [M + H]
+ (calcd for C
11H
12NO
2, 190.0863).
Puniceusine C (
3): pale yellow powder; UV (CH
3OH)
λmax (log
ε) 205 (1.87), 244 (1.98), 262 (2.04), 332 (1.07) nm; IR (film)
νmax 3415, 1678, 1436, 1382, 1321, 1203, 1184, 1130, 1022, 954, 900, 840, 800, 723 cm
−1;
1H and
13C NMR,
Table 1 and
Table 2; HRESIMS
m/
z 363.1339 [M + H]
+ (calcd for C
21H
19N
2O
4, 363.1349).
Puniceusine D (
4): pale yellow powder; UV (CH
3OH)
λmax (log
ε) 207 (1.86), 243 (1.88), 260 (1.88) nm; IR (film)
νmax 3402, 1678, 1643, 1566, 1384, 1342, 1197, 1132, 1045, 989, 954, 842, 800, 723 cm
−1;
1H and
13C NMR,
Table 1 and
Table 2; HRESIMS
m/
z 363.1339 [M + H]
+ (calcd for C
21H
19N
2O
4, 363.1338).
Puniceusine E (
5): pale yellow powder; UV (CH
3OH)
λmax (log
ε) 206 (2.07), 248 (1.83), 265 (2.00), 337 (1.06) nm; IR (film)
νmax 1703, 1678, 1365, 1178, 0012, 835, 800, 721 cm
−1;
1H and
13C NMR,
Table 1 and
Table 2; HRESIMS
m/
z 377.1496 [M + H]
+ (calcd for C
22H
21N
2O
4, 377.1486).
Puniceusine F (
6): colorless crystals; [α]
0 (
c 0.10, CH
3OH); UV (CH
3OH)
λmax (log
ε) 206 (1.63), 244 (1.76), 261 (1.71) nm; IR (film)
νmax 3419, 1703, 1681, 1363, 1201, 1180, 1134, 837, 800, 721 cm
−1;
1H and
13C NMR,
Table 1 and
Table 2; HRESIMS
m/
z 262.1076 [M + H]
+ (calcd for C
14H
16NO
4, 262.1074).
Puniceusine G (
7): yellow powder; UV (CH
3OH)
λmax (log
ε) 200 (1.26), 246 (1.46) nm; IR (film)
νmax 3412, 1680, 1440, 1195, 1136, 1028, 844, 800, 725 cm
−1;
1H and
13C NMR,
Table 1 and
Table 2; HRESIMS
m/
z 254.1182 [M + H]
+ (calcd for C
16H
16NO
2, 254.1176).
Puniceusine H (
8): pale yellow powder; UV (CH
3OH)
λmax (log
ε) 191 (1.61), 204 (2.08), 242 (1.06), 272 (1.12) nm; IR (film)
νmax 3367, 1680, 1456, 1417, 1394, 1201, 1139, 1020, 839, 802, 721 cm
−1;
1H and
13C NMR,
Table 2 and
Table 3; HRESIMS
m/
z 398.1244 [M + H]
+ (calcd for C
21H
20NO
7, 398.1234).
Puniceusine I (
9): yellow powder; [α]
+ 60.7 (
c 0.10, CH
3OH); UV (CH
3OH)
λmax (log
ε) 205 (1.30), 237 (0.90), 244 (0.92), 276 (0.63) nm; ECD (CH
3OH)
λmax (Δ
ε) 201 (+4.35), 202 (+4.71), 214 (−4.54), 232 (−1.32), 246 (−2.71), 279 (+0.44), 322 (−1.45) nm; IR (film)
νmax 3402, 1699, 1681, 1361, 1201, 1136, 837, 800, 721 cm
−1;
1H and
13C NMR,
Table 3 and
Table 4; HRESIMS
m/
z 354.1331 [M + H]
+ (calcd for C
20H
20NO
5, 354.1336).
Puniceusine J (
10): pale yellow powder; UV (CH
3OH)
λmax (log
ε) 195 (2.08), 208 (1.06), 243 (1.12) nm; IR (film)
νmax 3406, 1678, 1392, 1321, 1203, 1132, 840, 800, 723 cm
−1;
1H and
13C NMR,
Table 3 and
Table 4; HRESIMS
m/
z 384.1455 [M + H]
+ (calcd for C
21H
22NO
6, 384.1442).
Puniceusine K (
11): pale yellow powder; [α]
− 155.6 (
c 0.10, CH
3OH); UV (CH
3OH)
λmax (log
ε) 205 (1.30), 237 (0.90), 244 (0.92), 276 (0.63) nm; ECD (CH
3OH)
λmax (Δ
ε) 215 (−2.39), 229 (+0.45), 238 (+0.29), 255 (+0.67), 304 (−0.18) nm; IR (film)
νmax 3390, 1674, 1435, 1371, 1319, 1199, 1134, 840, 800, 723 cm
−1;
1H and
13C NMR,
Table 3 and
Table 4; HRESIMS
m/
z 428.1348 [M + H]
+ (calcd for C
22H
22NO
8, 428.1340).
Puniceusine L (
12): colorless crystals; UV (CH
3OH)
λmax (log
ε) 206 (2.14), 264 (2.04), 366 (1.12) nm; IR (film)
νmax 3080, 1678, 1643, 1396, 1319, 1197, 1130, 839, 798, 721 cm
−1;
1H and
13C NMR,
Table 3 and
Table 4; HRESIMS
m/
z 328.1195 [M + H]
+ (calcd for C
18H
18NO
5, 328.1179).
Puniceusine M (
13): pale yellow powder; UV (CH
3OH)
λmax (log
ε) 207 (2.08), 244 (1.74), 267 (1.84) nm; IR (film)
νmax 3404, 1678, 1394, 1319, 1201, 1176, 1138, 1026, 837, 800, 721 cm
−1;
1H and
13C NMR,
Table 3 and
Table 4; HRESIMS
m/
z 342.1332 [M + H]
+ (calcd for C
19H
20NO
5, 342.1336).
Puniceusine N (
14): pale yellow powder; [α]
0 (
c 0.10, CH
3OH); UV (CH
3OH)
λmax (log
ε) 206 (1.89), 232 (1.61), 266 (2.08), 309 (1.06), 364 (1.12) nm; IR (film)
νmax 3423, 1680, 1363, 1195, 1182, 1128, 839, 800, 719 cm
−1;
1H and
13C NMR,
Table 3 and
Table 4; HRESIMS
m/
z 374.2329 [M]
+ (calcd for C
22H
32NO
4, 374.2326).
3.5. X-ray Crystallographic Analysis of 6 and 12
The crystal data were obtained on a Rigaku MicroMax 007 diffractometer (Rigaku Corporation, Tokyo, Japan)with Cu Kα radiation and a graphite monochromator. The crystal structures of 6 and 12 were solved by direct methods with the SHELXTL and refined by full-matrix, least-squares techniques. Crystallographic data for 6 and 12 were deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers, CCDC 2112471 and 2112479, respectively.
Crystal data for 6: C14H14.0375NO4, FW = 260.30; colorless crystal from MeOH; crystal size = 0.15 × 0.12 × 0.1 mm3; T = 100.00 (10) K; monoclinic, space group P21/c (no. 14); unit cell parameters: a = 4.82780 (10) Å, b = 13.4963 (3) Å, c = 22.6665 (7) Å, α = 90°, β = 96.025 (3)°, γ = 90°, V = 1468.73 (6) Å3, Z = 4, Dcalc = 1.177 g/cm3, F (000) = 548.0, μ (CuKα) = 0.724 mm−1; 7185 reflections measured (7.634° ≤ 2θ ≤ 147.79°), 2852 unique (Rint = 0.0197, Rsigma = 0.0255), which were used in all calculations. The final R1 was 0.0487 (I > 2σ(I)) and wR2 was 0.1406 (all data).
Crystal data for 12: C18H17NO5, FW = 327.32; colorless crystal from MeOH; crystal size = 0.13 × 0.12 × 0.1 mm3; T = 100.00 (10) K; triclinic, space group P-1 (no. 2); unit cell parameters: a = 7.6037 (4) Å, b = 9.9253 (5) Å, c = 10.5261 (6) Å, α = 70.543 (5)°, β = 85.628 (4)°, γ = 81.605 (4)°, V = 740.68(7) Å3, Z = 2, Dcalc = 1.468 g/cm3, F (000) = 344.0, μ (CuKα) = 0.897 mm−1; 7019 reflections measured (8.914° ≤ 2θ ≤ 148.202°), 2880 unique (Rint = 0.0259, Rsigma = 0.0317) which were used in all calculations. The final R1 was 0.0419 (I > 2σ(I)) and wR2 was 0.1150 (all data).
3.6. ECD Calculations
The ECD calculation for
9 was performed by Gaussian 16 program package. The procedures were the same as described in our previous study [
13,
14]. Briefly, the conformational search was performed by a MMFF model, then the conformers with lower relative energies (<10 kcal/mol) were subjected to geometry optimization with the DFT method at the B3LYP/6-311G(d) level. Vibrational frequency calculations were carried out at the same level to evaluate their relative thermal (ΔE) and free energies (ΔG) at 298.15 K. The geometry-optimized conformers were further calculated at the M06-2X/def2-TZVP level and the solvent (methanol) effects were taken into consideration by using SMD. The optimized conformers with a Boltzmann distribution of more than 1% population were subjected to ECD calculation, which were performed by TDDFT methodology at the PBE1PBE/TZVP level. The ECD spectrum was generated by the software SpecDis using a Gaussian band shape with 0.3 eV exponential half-width from dipole-length dipolar and ratational strengths. The calculated spectrum of
9 was generated from the low-energy conformers according to the Boltzmann distribution of each conformer in MeOH solution. Details regarding optimized conformation geometries, thermodynamic parameters, and Boltzmann distributions (
Tables S1–S3) of all conformations are provided in the
Supporting Information.
3.7. Protein Phosphatase Inhibition Assays
The same methods as described in our previous study [
13,
14] were applied to test the inhibition activity of compounds
1–
14 against five human protein tyrosine phosphatases (CD45, SHP1, TCPTP, PTP1B and LAR).
3.8. Cytotoxicity
Cytotoxic activity was evaluated using human lung adenocarcinoma cell line H1975 by CCK-8 method. Briefly, each of the test compounds was dissolved in DMSO and further diluted to give final concentrations of 80, 40, 20, 10, 5, 2.5, and 1.25 µg/mL, respectively. H1975 cells (5 × 103 cells/plate) were seeded in 96-well plates and treated with compounds at the indicated concentration for 24 h, and then 10 μL CCK-8 reagent was added to each well, and the plates were incubated at 37 °C for another 4 h. Next, the optical density was measured at a wavelength of 450 nm with the Bio-Rad (Hercules, CA, USA) microplate reader. Dose–response curves were generated, and the IC50 values were calculated from the linear portion of log dose–response curves.
3.9. Antibacterial Assays
Antibacterial activities of 1–14 against E. coli, S. aureus and MRSA were evaluated using the 2-fold dilution assay in 96-microwell plates. Briefly, all the indicator bacteria were cultured on Luria−Bertani (LB) agar plates at 37 °C for 12 h, and then a single colony was picked into LB liquid medium and cultivated on a rotary shaker at 37 °C for 12 h. Then, the bacterial suspensions with LB medium were diluted until the difference of the OD600 values between the bacterial suspensions and the medium was 0.01~0.02. Each of the tested compounds was dissolved in DMSO to give an initial concentration of 5 mg/mL, and further diluted with the bacterial suspensions by twofold serial dilution to give a final concentration of 100, 50, 25, 12.5, 6.25, and 3.125 μg/mL, respectively. The 96-well plates were incubated at 37 °C for 12 h. MIC value was determined as the lowest concentration with no visible bacterial growth. Ampicillin was used as the positive control and DMSO as the negative control. All experiments were performed three times.



 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}