
Thioester-Containing Benzoate Derivatives with α-Glucosidase Inhibitory Activity from the Deep-Sea-Derived Fungus Talaromyces indigoticus FS688


Abstract
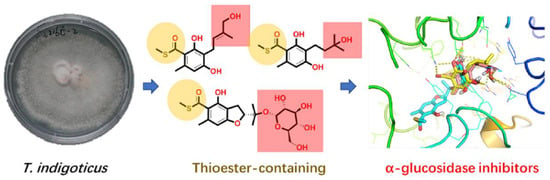
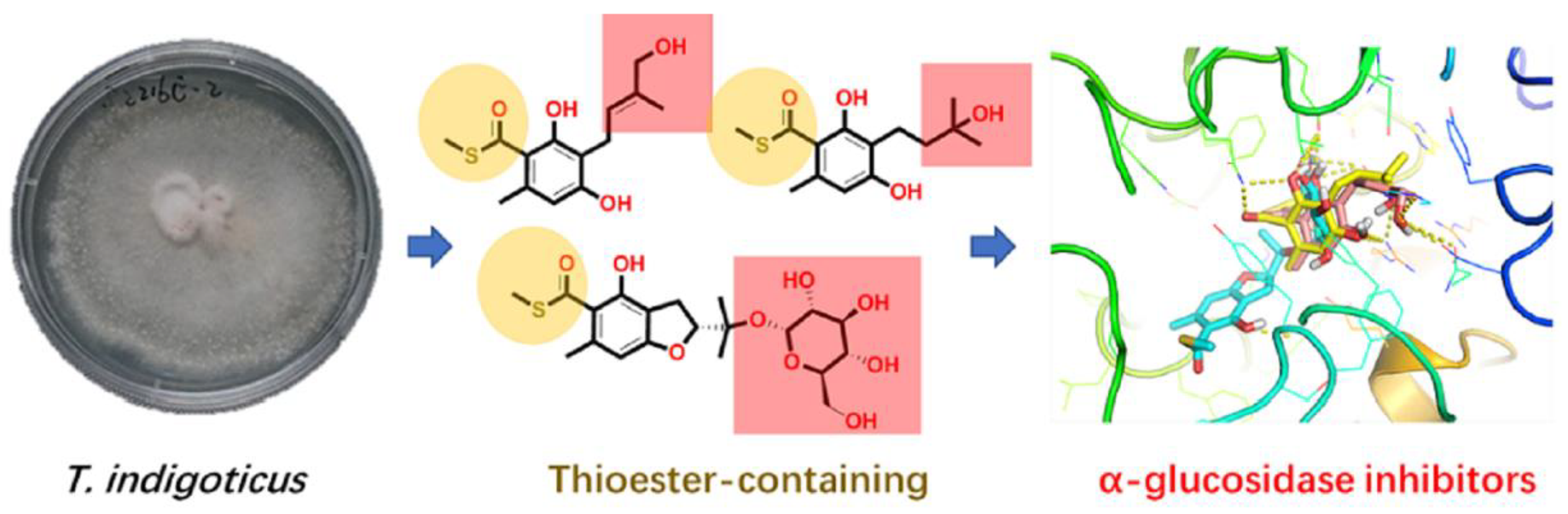
:
1. Introduction
2. Results
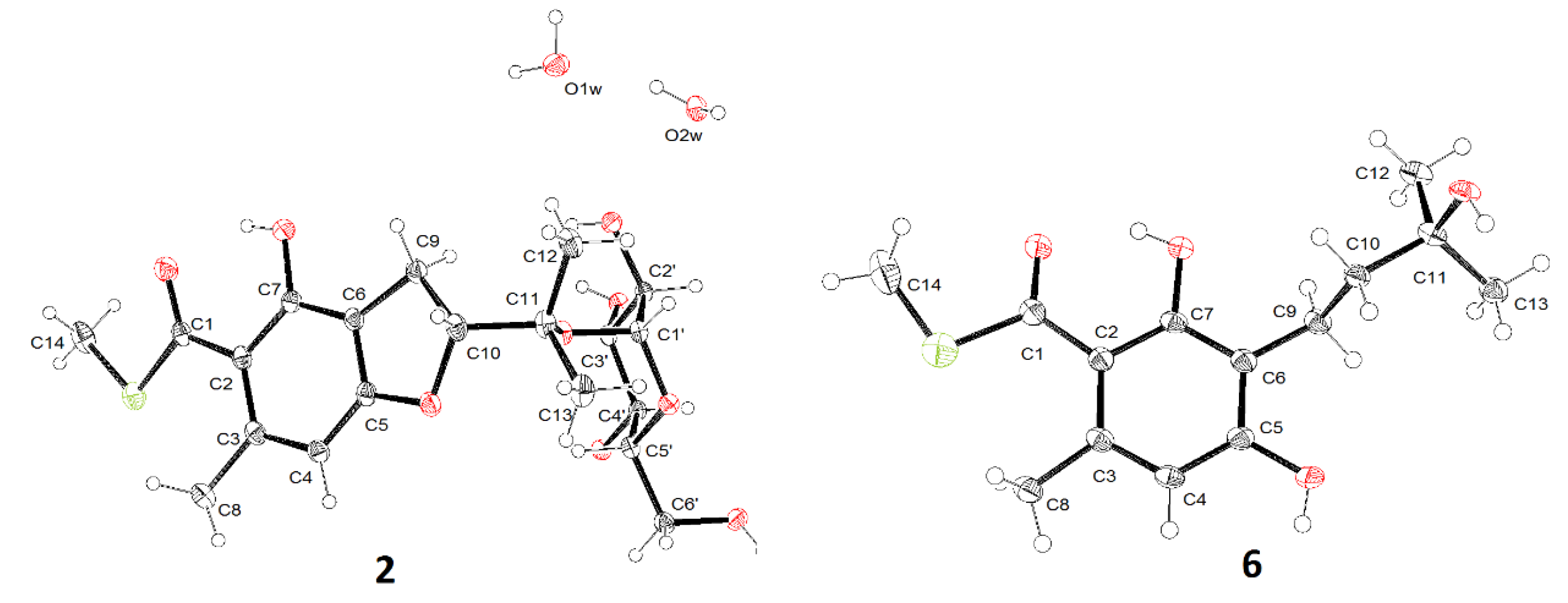
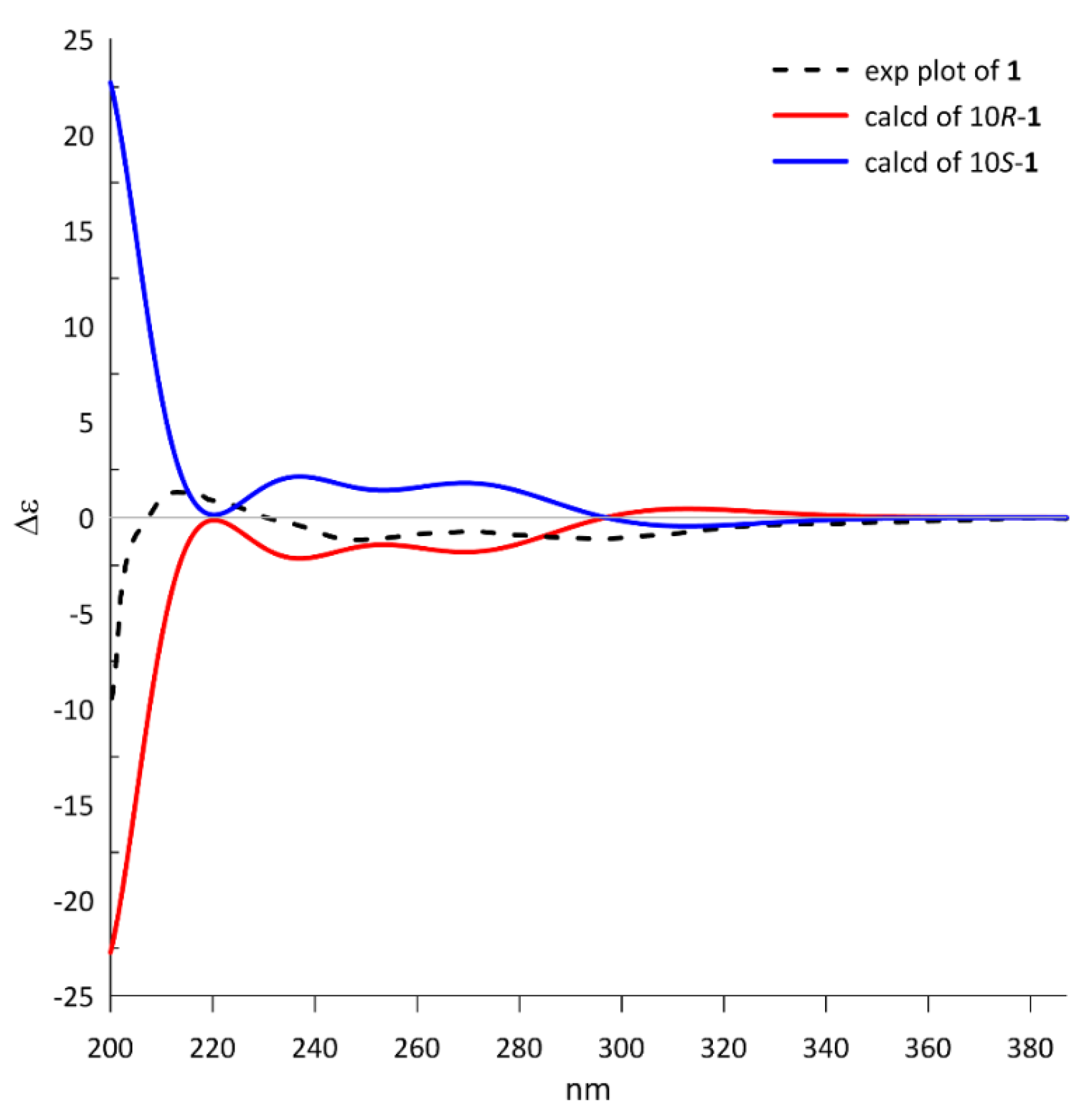
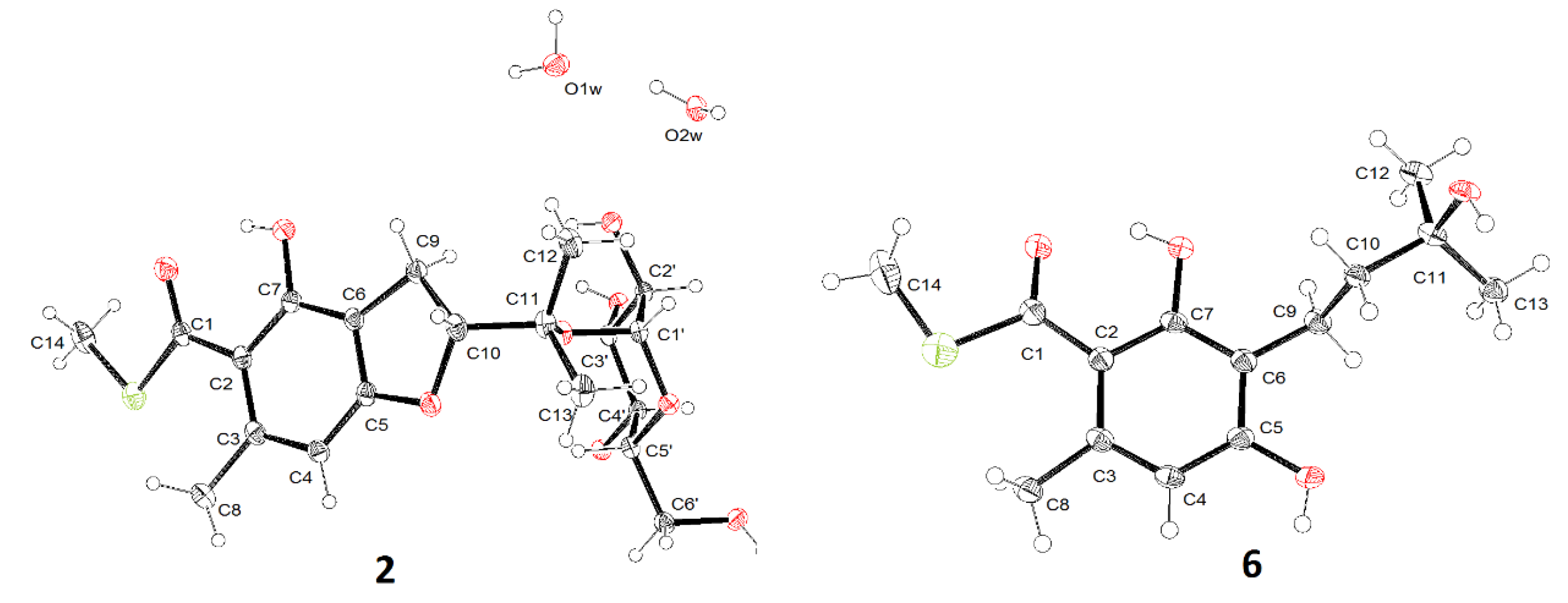
2.1. Structure Identification
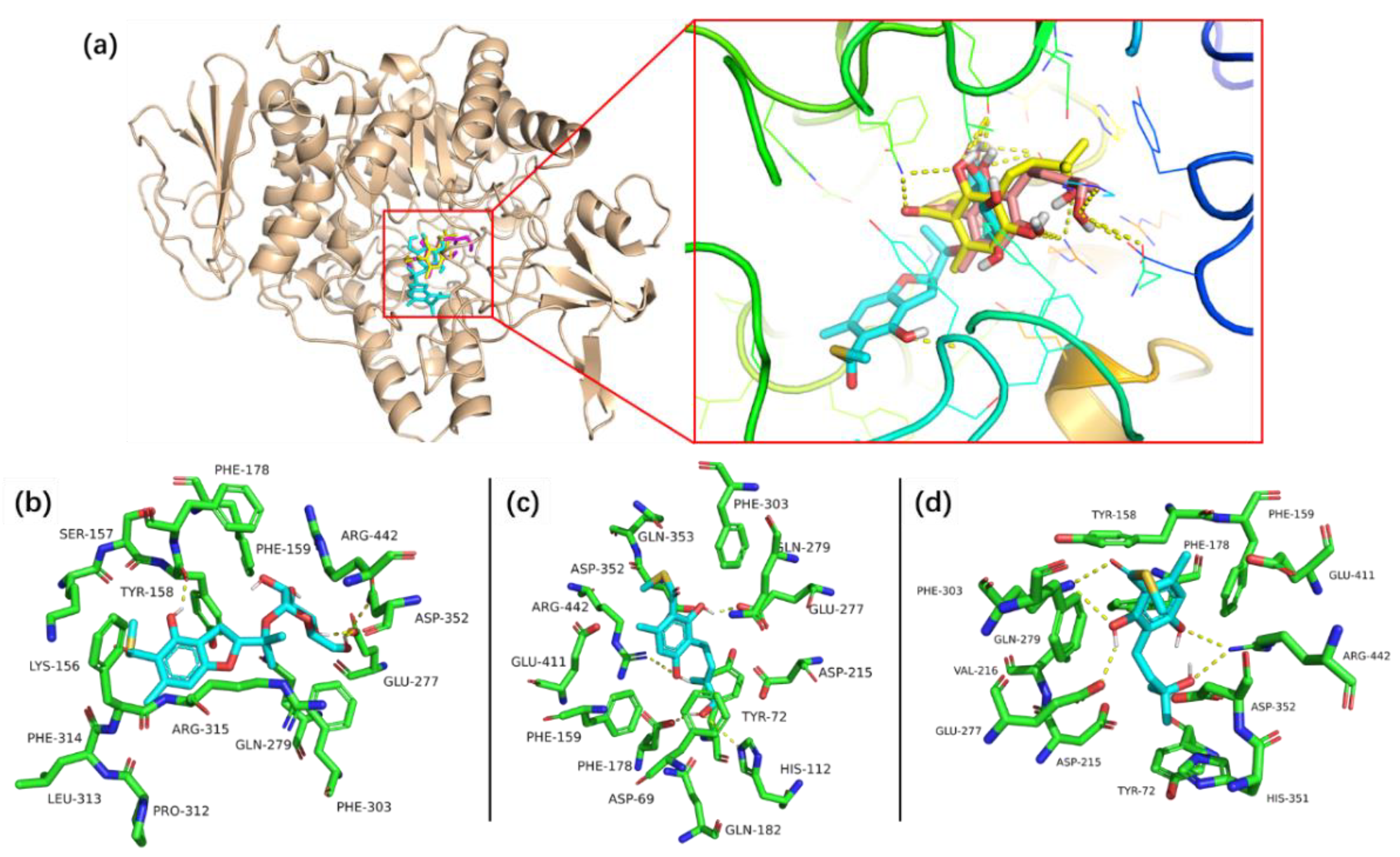
2.2. Bioassays and Molecular Docking
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation, Extraction, and Isolation
3.4. Details of Quantum Chemical Calculations
3.5. α-Glucosidase Inhibition Assay
3.6. Docking Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ilardi:, E.A.; Vitaku, E.; Njardarson, J.T. Data-mining for sulfur and fluorine: An evaluation of pharmaceuticals to reveal opportunities for drug design and discovery. J. Med. Chem. 2014, 57, 2832–2842. [Google Scholar] [CrossRef]
- Feng, M.; Tang, B.; Liang, S.H.; Jiang, X. Sulfur containing scaffolds in drugs: Synthesis and application in medicinal chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216. [Google Scholar] [CrossRef]
- Scott, K.A.; Njardarson, J.T. Analysis of US FDA-approved drugs containing sulfur atoms. Top. Curr. Chem. 2018, 376, 5. [Google Scholar] [CrossRef] [PubMed]
- Milito, A.; Brancaccio, M.; D’Argenio, G.; Castellano, I. Natural sulfur-containing compounds: An alternative therapeutic strategy against liver fibrosis. Cells 2019, 8, 1356. [Google Scholar] [CrossRef] [Green Version]
- Brosnan, J.T.; Brosnan, M.E. The sulfur-containing amino acid: An overview. J. Nutri. 2006, 136, 1636–1640. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Cao, L.; Zhang, W.; Lu, R.; Bian, J.-S.; Nie, X. Therapeutic potential of sulfur-containing natural products in inflammatory diseases. Pharmacol. Therapeut. 2020, 216, 107678. [Google Scholar] [CrossRef]
- Pan, C.; Kuranaga, T.; Liu, C.; Lu, S.; Shinzato, N.; Kakeya, H. Thioamycolamides A–E, sulfur-containing cycliclipopeptides produced by the rare Actinomycete Amycolatopsis sp. Org. Lett. 2020, 8, 3014–3017. [Google Scholar] [CrossRef]
- Abdalla, M.A.; Mühling, K.H. Plant-derived sulfur containing natural products produced as a response to biotic and abiotic stresses: A review of their structural diversity and medicinal importance. J. App. Bot. Food Qual. 2019, 92, 204–215. [Google Scholar]
- Jimenez, C. Marine sulfur-containing natural products. Stud. Nat. Prod. Chem. 2001, 25, 811–917. [Google Scholar]
- Shin, J.; Fenical, W. Isolation of gliovictin from the marine deuteromycete Asteromyces cruciatus. Phytochemistry 1987, 26, 3347. [Google Scholar] [CrossRef]
- Hai, Y.; Wei, M.-Y.; Wang, C.-Y.; Gu, Y.-C.; Shao, C.-L. The intriguing chemistry and biology of sulfur-containing natural products from marine microorganisms (1987–2020). Mar. Life Sci. Technol. 2021, 3, 488–518. [Google Scholar] [CrossRef]
- Wang, Y.-T.; Xue, Y.-R.; Liu, C.-H. A brief review of bioactive metabolites derived from deep-sea fungi. Mar. Drugs 2015, 13, 4594–4616. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Xia, G.; Chen, S.; Liu, Y.; Li, H.; She, Z. Eurothiocin A and B, sulfur-containing benzofurans from a soft coral-derived fungus Eurotium rubrum SH-823. Mar. Drugs 2014, 12, 3669–3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, B.; Ahn, S.; Noh, M.; Shin, J.; Oh, D.C. Suncheonosides A–D, benzothioate glycosides from a marine-derived Streptomycessp. J. Nat. Prod. 2015, 78, 1390–1396. [Google Scholar] [CrossRef]
- Yang, A.G.; Si, L.L.; Shi, Z.P.; Tian, L.; Liu, D.; Zhou, D.M.; Proksch, P.; Lin, W.H. Nitrosporeusines A and B, unprecedented thioester–bearing alkaloids from the Arctic Streptomyces nitrosporeus. Org. Lett. 2013, 15, 5366–5369. [Google Scholar] [CrossRef]
- Perez Baz, J.; Cañedo, L.M.; Fernández Puentes, J.L.; Silva Elipe, M.V. Thiocoraline, a novel depsipeptide with antitumor activity produced by a marine Micromonospora. II. Physico-chemical properties and structure determination. J. Antibiot. 1997, 50, 738–741. [Google Scholar]
- Taori, K.; Paul, V.J.; Luesch, H. Structure and activity of largazole, a potent antiproliferative agent from the foridian marine cyanobacterium Symploca sp. J. Am. Chem. Soc. 2008, 130, 1806–1807. [Google Scholar] [CrossRef]
- Pereira, A.R.; Etzbach, L.; Engene, N.; Muller, R.; Gerwick, W.H. Molluscicidal metabolites from an assemblage of palmyra atoll cyanobacteria. J. Nat. Prod. 2011, 74, 1175–1181. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Li, P.; Luo, X.; Qian, D.; Tang, X.; Li, G. Two new compounds from the marine sponge derived fungus Penicillium chrysogenum. Nat. Prod. Res. 2019, 34, 2926–2930. [Google Scholar] [CrossRef]
- Yamamoto, K.; Nakayama, A.; Yamamoto, Y.; Tabata, S. Val216 decides the substrate specificity of α-glucosidase in Saccharomyces cerevisiae. Eur. J. Biochem. 2004, 271, 3414–3420. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef]
- Fan, J.; Kuang, Y.; Dong, Z.; Yi, Y.; Zhou, Y.; Li, B.; Qiao, X.; Ye, M. Prenylated Phenolic Compounds from the Aerial Parts of Glycyrrhiza uralensisas PTP1B and α-Glucosidase Inhibitors. J. Nat. Prod. 2020, 83, 814–824. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 3 a | 4 b | |||
|---|---|---|---|---|---|---|
| δC, Type | δH | δC, Type | δH | δC, Type | δH | |
| 1 | 197.8, C | 198.2, C | 197.6, C | |||
| 2 | 116.3, C | 115.4, C | 115.6, C | |||
| 3 | 132.2, C | 138.7, C | 137.1, C | |||
| 4 | 135.8, C | 111.7, CH | 6.22, s | 110.3, CH | 6.23, s | |
| 5 | 156.9, C | 158.9, C | 158.9, C | |||
| 6 | 112.8, C | 112.0, C | 112.7, C | |||
| 7 | 153.2, C | 160.5, C | 159.0, C | |||
| 8 | 15.3, CH3 | 2.59, s | 25.0, CH3 | 2.63, s | 22.4, CH3 | 2.51, s |
| 9 | 28.0, CH2 | 3.17, dd (15.6, 9.7) | 22.1, CH2 | 3.40, d (7.1) | 21.0, CH2 | 3.34, d (7.1) |
| 3.09, dd (15.6, 8.3) | ||||||
| 10 | 91.9, CH | 4.78, dd (9.7, 8.3) | 121.5, CH | 5.25, tq (7.1, 1.4) | 123.8, CH | 5.48, brt (7.1) |
| 11 | 72.0, C | 135.0, C | 134.0, C | |||
| 12 | 24.0, CH3 | 1.24, s | 25.8, CH3 | 1.74,d (1.4) | 67.7, CH2 | 3.89,d (1.4) |
| 13 | 25.7, CH3 | 1.35, s | 17.9, CH3 | 1.81, s | 12.4, CH3 | 1.80, s |
| 14 | 13.1, CH3 | 2.46, s | 13.1, CH3 | 2.45, s | 11.5, CH3 | 2.44, s |
| 4-OMe | 60.4, CH3 | 3.78, s | ||||
| 7-OH | 11.1, s | 12.16, s | ||||
| Position | 2 a | ||||
|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | ||
| 1 | 197.2, C | 11 | 77.9, C | ||
| 2 | 119.8, C | 12 | 20.5, CH3 | 1.30, s | |
| 3 | 137.8, C | 13 | 21.3, CH3 | 1.33, s | |
| 4 | 103.3, CH | 6.19, s | 14 | 11.3, CH3 | 2.43, s |
| 5 | 162.9, C | 1′ | 93.2, CH | 5.20, d (3.8) | |
| 6 | 110.7, C | 2′ | 72.1, CH | 3.35, dd (9.8, 3.8) | |
| 7 | 152.6, C | 3′ | 73.6, CH | 3.54, t (9.8) | |
| 8 | 20.0, CH3 | 2.32, s | 4′ | 70.4, CH | 3.30, m |
| 9 | 27.7, CH3 | 3.11, brd (3.8) | 5′ | 72.1, CH | 3.73, m |
| 3.13, brd (2.4) | 6′ | 61.2, CH2 | 3.69, m | ||
| 10 | 89.2, CH | 4.80, t (8.8) | |||
| Position | 5 a | 6 b | |||
|---|---|---|---|---|---|
| δC, Multiplicities | δH (J in Hz) | δC, Multiplicities | δH (J in Hz) | ||
| 1 | 197.7, C | 1 | 195.2, C | - | |
| 2 | 116.0, C | 2 | 122.7, C | ||
| 3 | 136.5, C | 3 | 137.8, C | ||
| 4 | 110.2, CH | 6.23, s | 4 | 109.1, CH | 6.24, d (2.1) |
| 5 | 156.9, C | 5 | 158.3, C | ||
| 6 | 114.1, C | 6 | 96.8, CH | 6.27, d (2.1) | |
| 7 | 153.2, C | 7 | 157.5, C | ||
| 8 | 22.0, CH3 | 2.48, s | 8 | 19.1, CH3 | 2.24, s |
| 9 | 17.5, CH2 | 2.65, m | 9 | 12.6, CH3 | 2.46, s |
| 10 | 55.9, CH3 | 3.78, s | |||
| 10 | 41.7, CH | 1.63, m | |||
| 11 | 70.4, C | ||||
| 12 | 27.6, CH3 | 1.24, s | |||
| 13 | 27.6, CH3 | 1.24, s | |||
| 14 | 11.5, CH3 | 2.44, s | |||
| Compounds | IC50 (μM) |
|---|---|
| 1 | >100 |
| 2 | 5.4 |
| 3 | >100 |
| 4 | 33.6 |
| 5 | 72.1 |
| 6 | >100 |
| Acarbose | 317.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Li, S.; Hu, J.; Gao, X.; Wang, Y.; Liu, Z.; Zhang, W. Thioester-Containing Benzoate Derivatives with α-Glucosidase Inhibitory Activity from the Deep-Sea-Derived Fungus Talaromyces indigoticus FS688. Mar. Drugs 2022, 20, 33. https://doi.org/10.3390/md20010033
Li M, Li S, Hu J, Gao X, Wang Y, Liu Z, Zhang W. Thioester-Containing Benzoate Derivatives with α-Glucosidase Inhibitory Activity from the Deep-Sea-Derived Fungus Talaromyces indigoticus FS688. Marine Drugs. 2022; 20(1):33. https://doi.org/10.3390/md20010033
Chicago/Turabian StyleLi, Mingqiong, Saini Li, Jinhua Hu, Xiaoxia Gao, Yanlin Wang, Zhaoming Liu, and Weimin Zhang. 2022. "Thioester-Containing Benzoate Derivatives with α-Glucosidase Inhibitory Activity from the Deep-Sea-Derived Fungus Talaromyces indigoticus FS688" Marine Drugs 20, no. 1: 33. https://doi.org/10.3390/md20010033
APA StyleLi, M., Li, S., Hu, J., Gao, X., Wang, Y., Liu, Z., & Zhang, W. (2022). Thioester-Containing Benzoate Derivatives with α-Glucosidase Inhibitory Activity from the Deep-Sea-Derived Fungus Talaromyces indigoticus FS688. Marine Drugs, 20(1), 33. https://doi.org/10.3390/md20010033