
Blue Biotechnology: Computational Screening of Sarcophyton Cembranoid Diterpenes for SARS-CoV-2 Main Protease Inhibition

 ,
,  ,
,  ,
,  , ,
, ,  , , ,
, , ,  ,
, 

 ,
,  and
and 
Abstract
:1. Introduction
2. Results
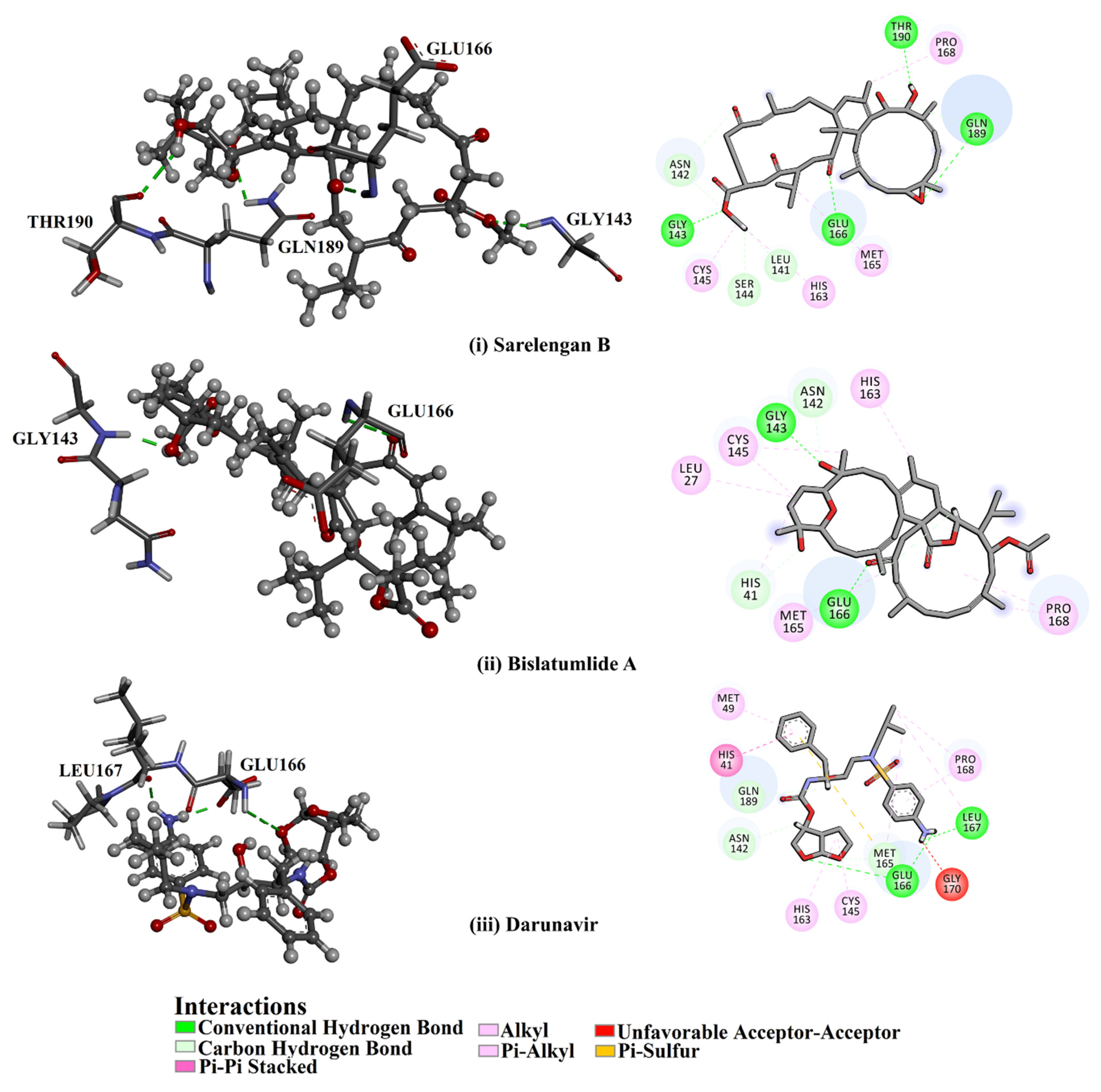
2.1. Molecular Docking
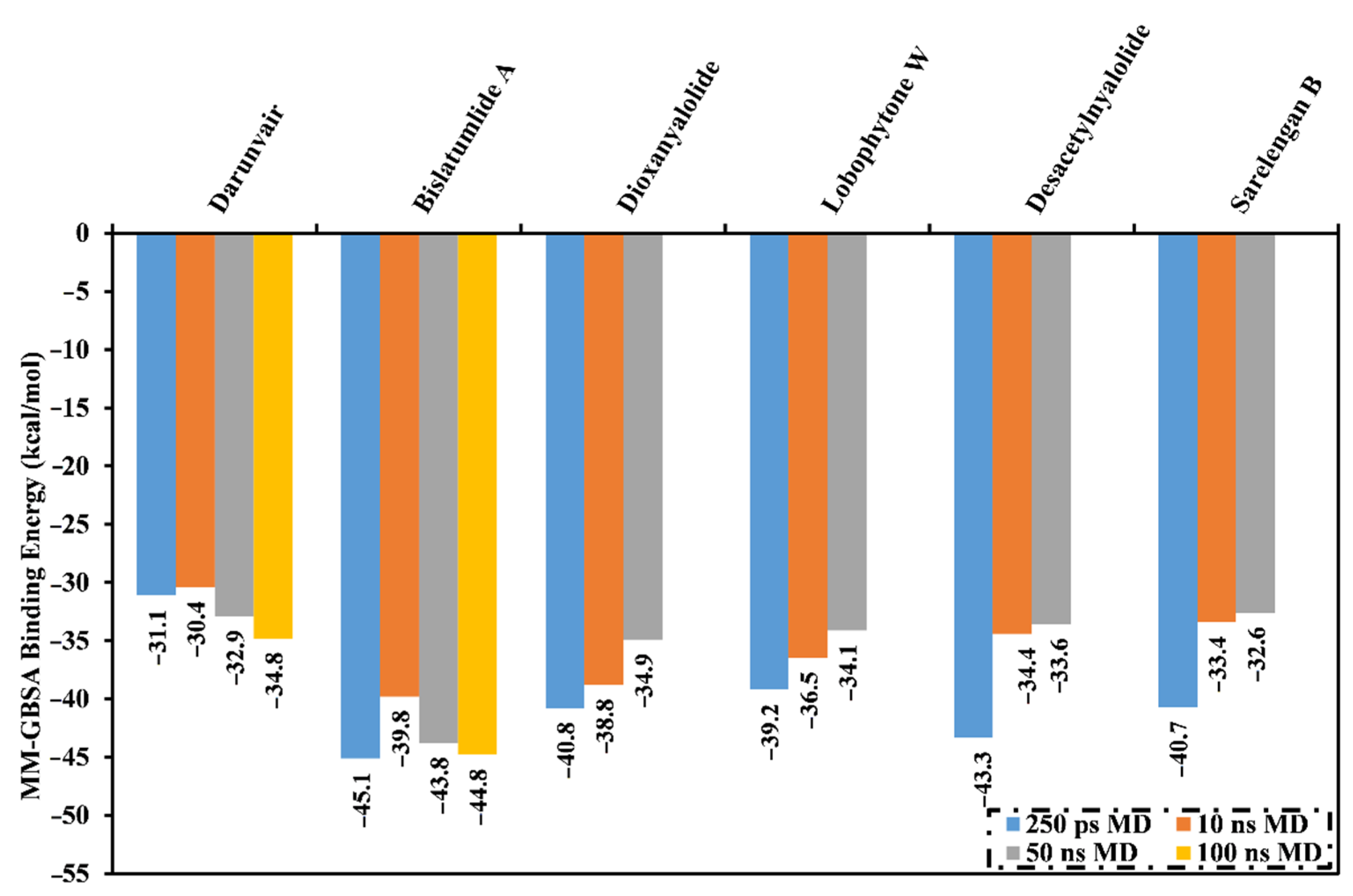
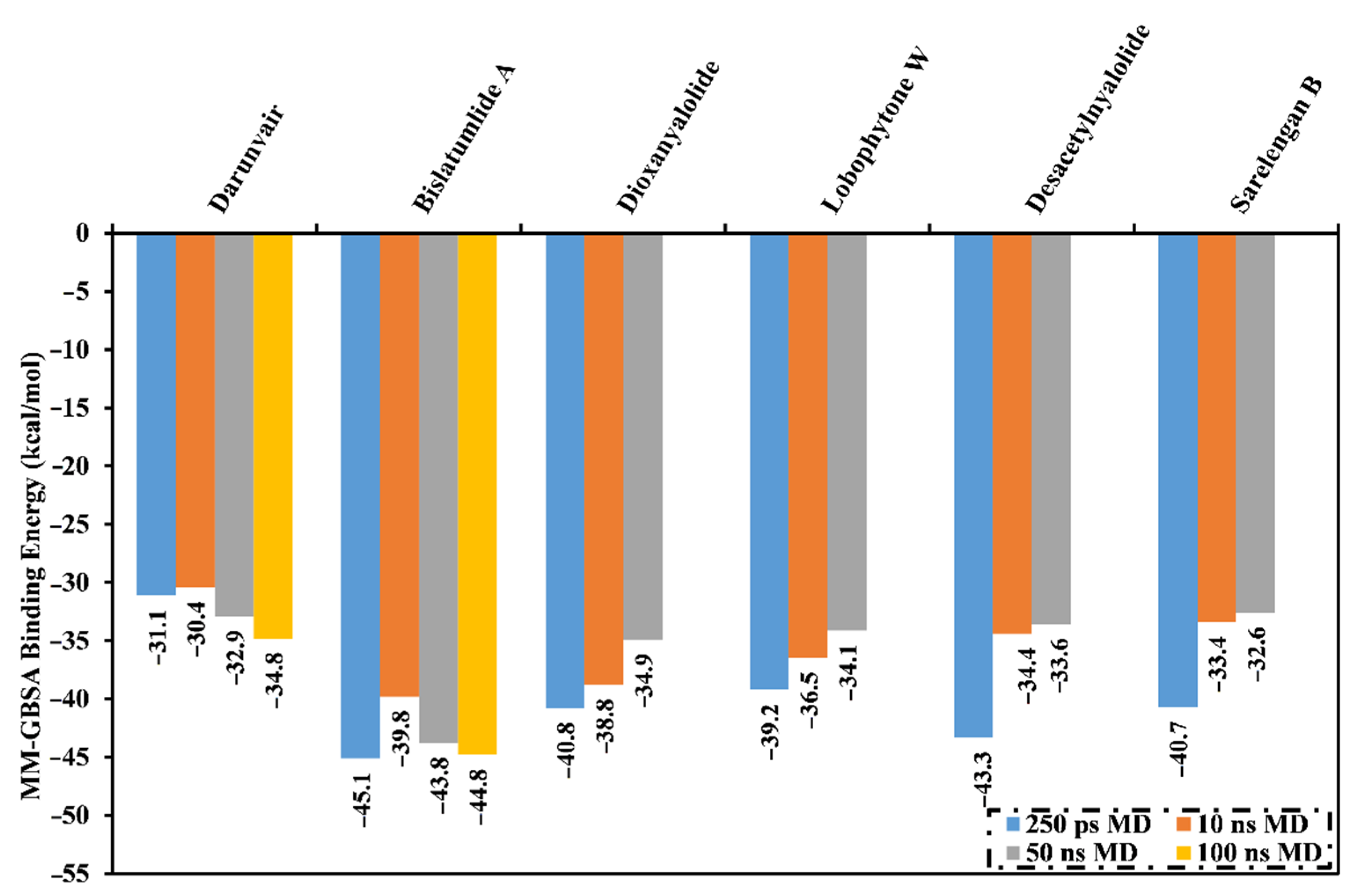
2.2. MD Simulations and Binding Energy Calculations
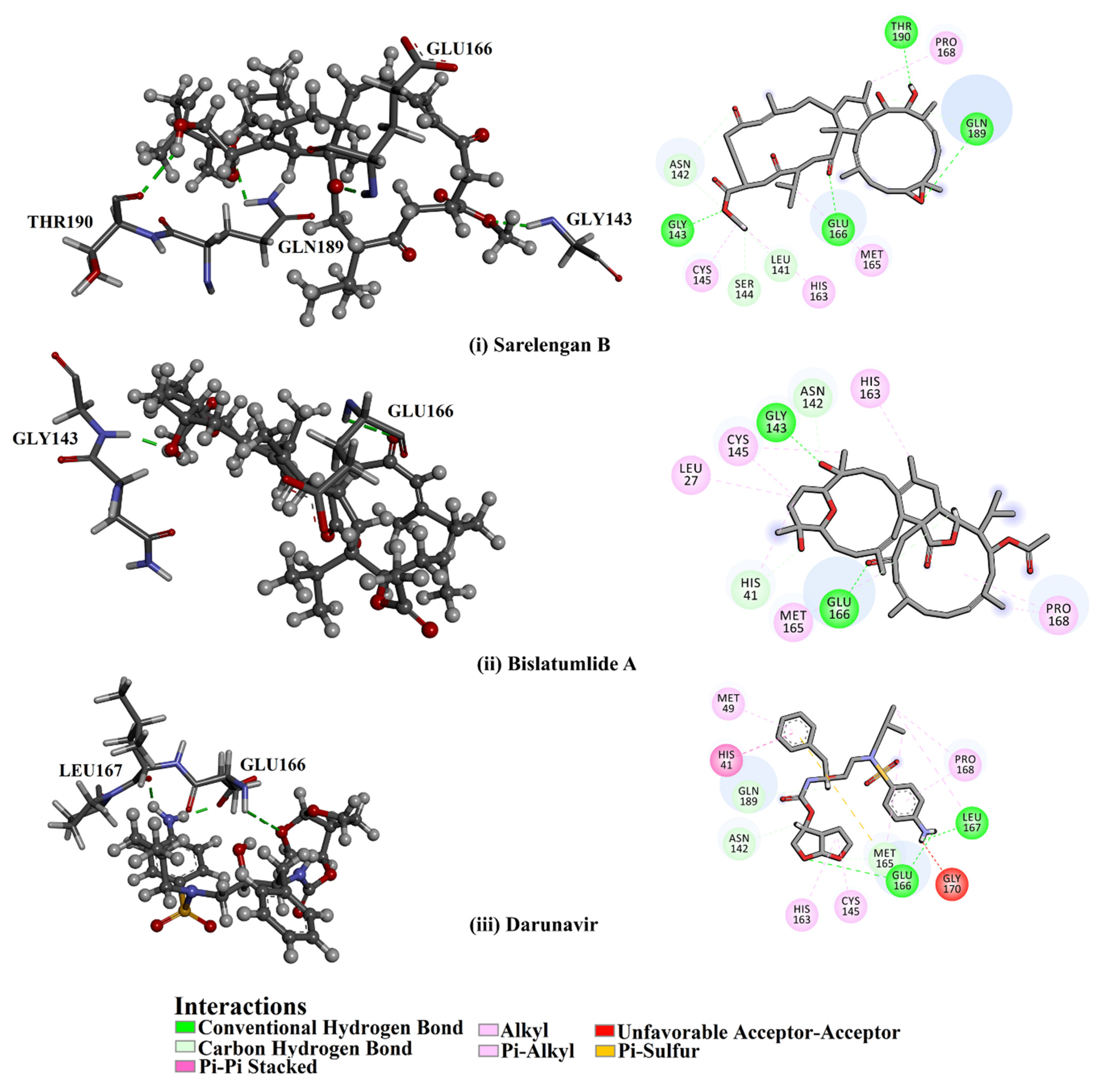
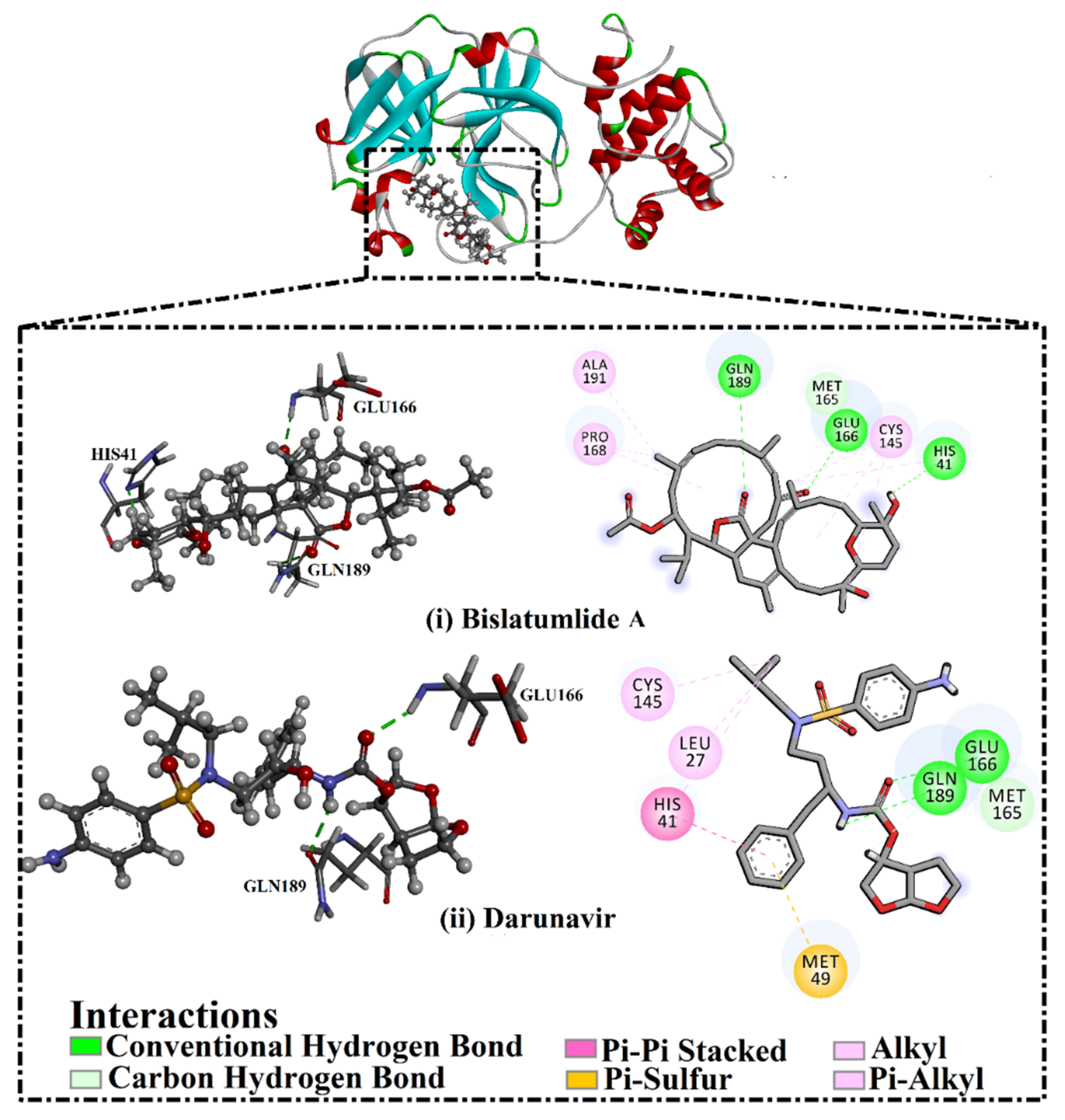
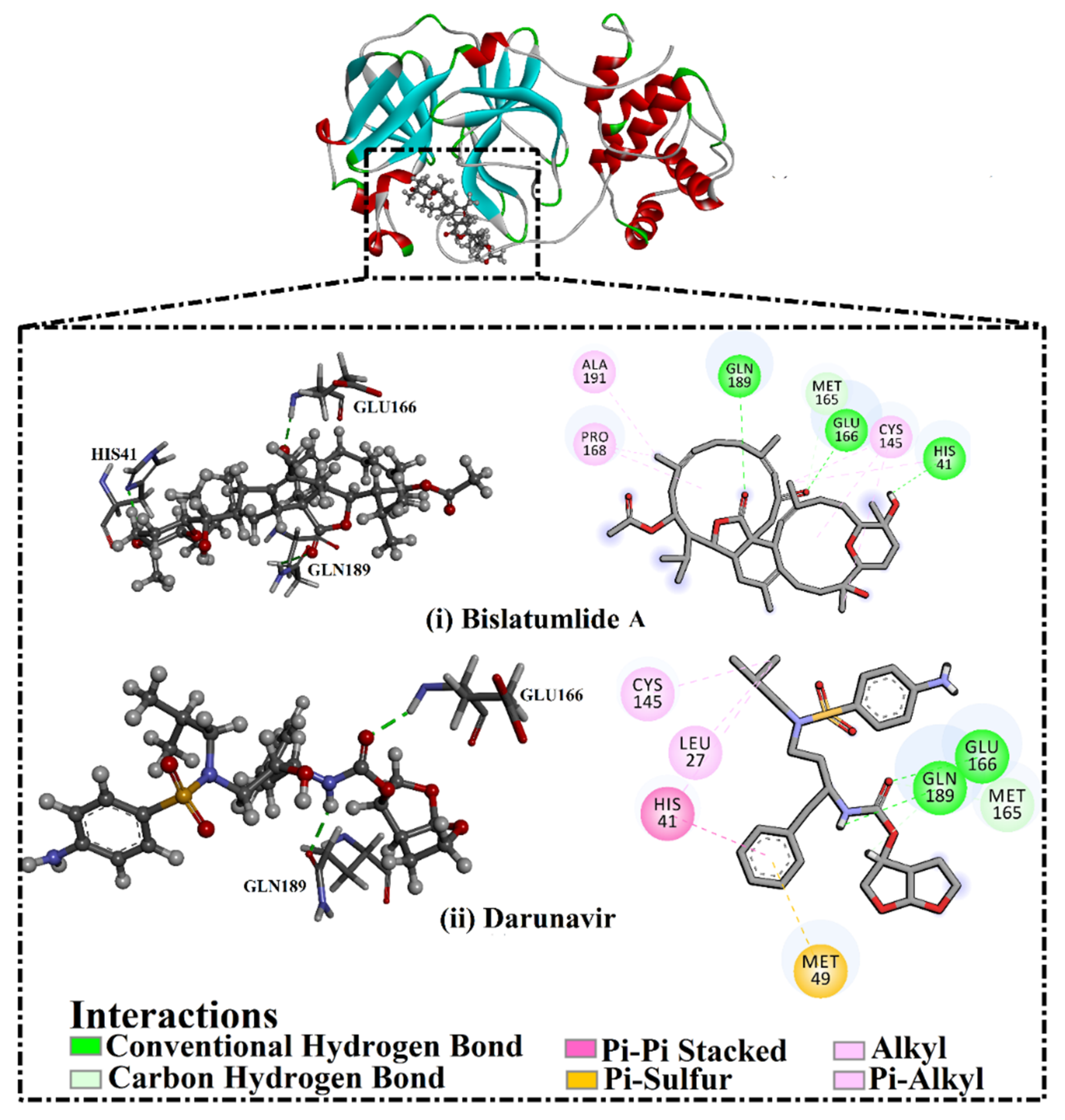
2.3. Post-MD Analyses
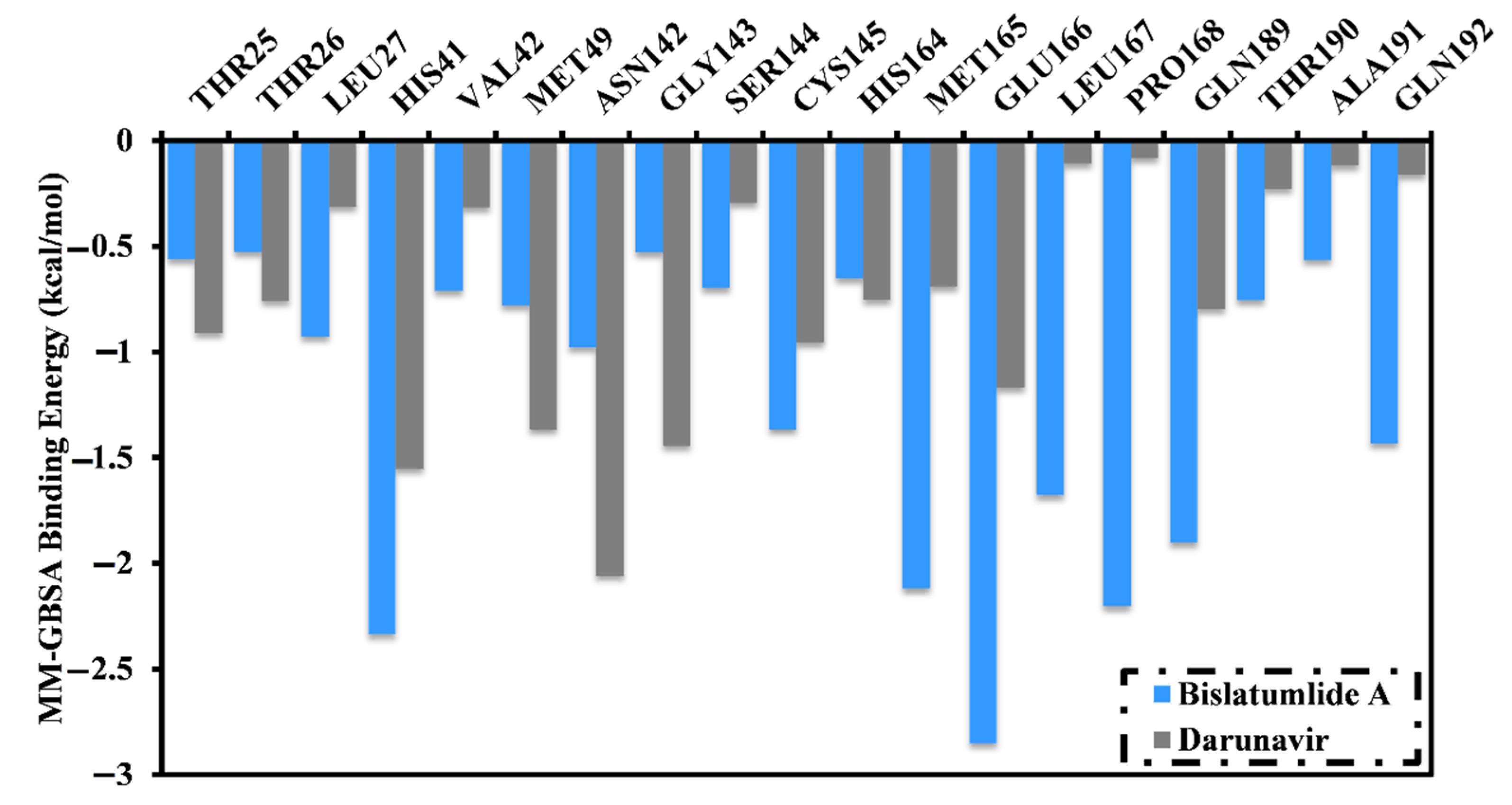
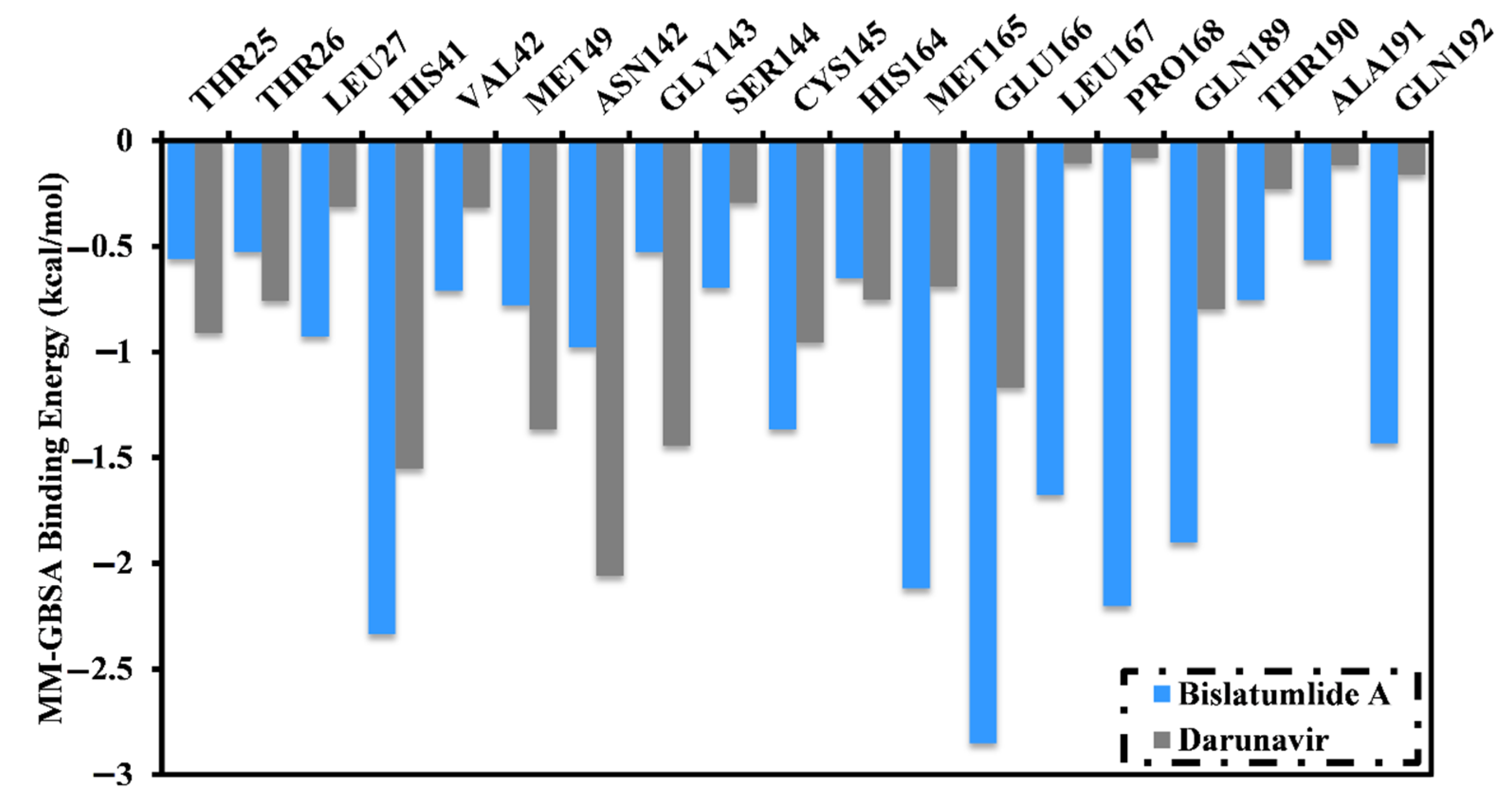
2.3.1. Binding Energy per Frame
2.3.2. Hydrogen Bond Length
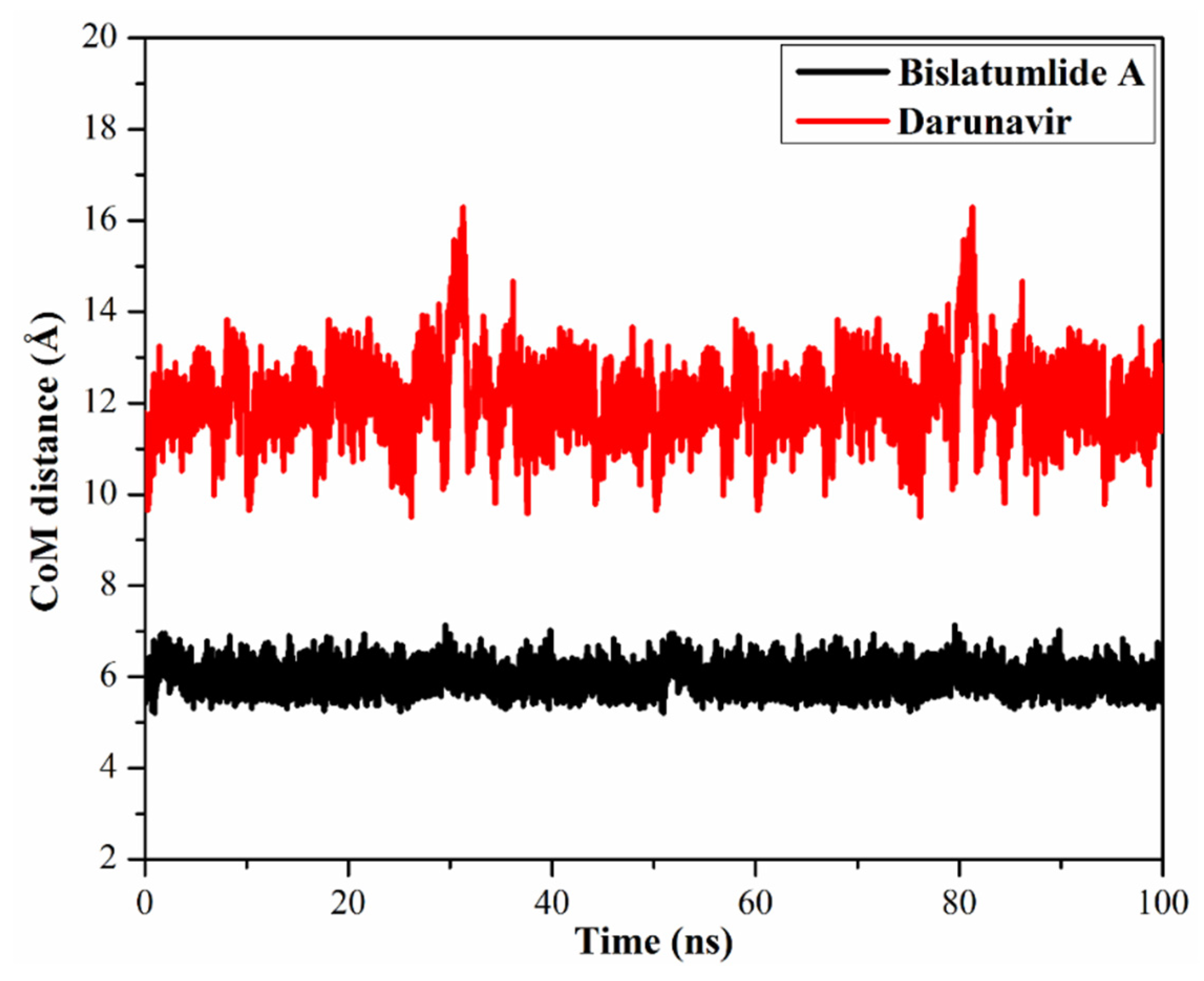
2.3.3. Center-of-Mass Distance
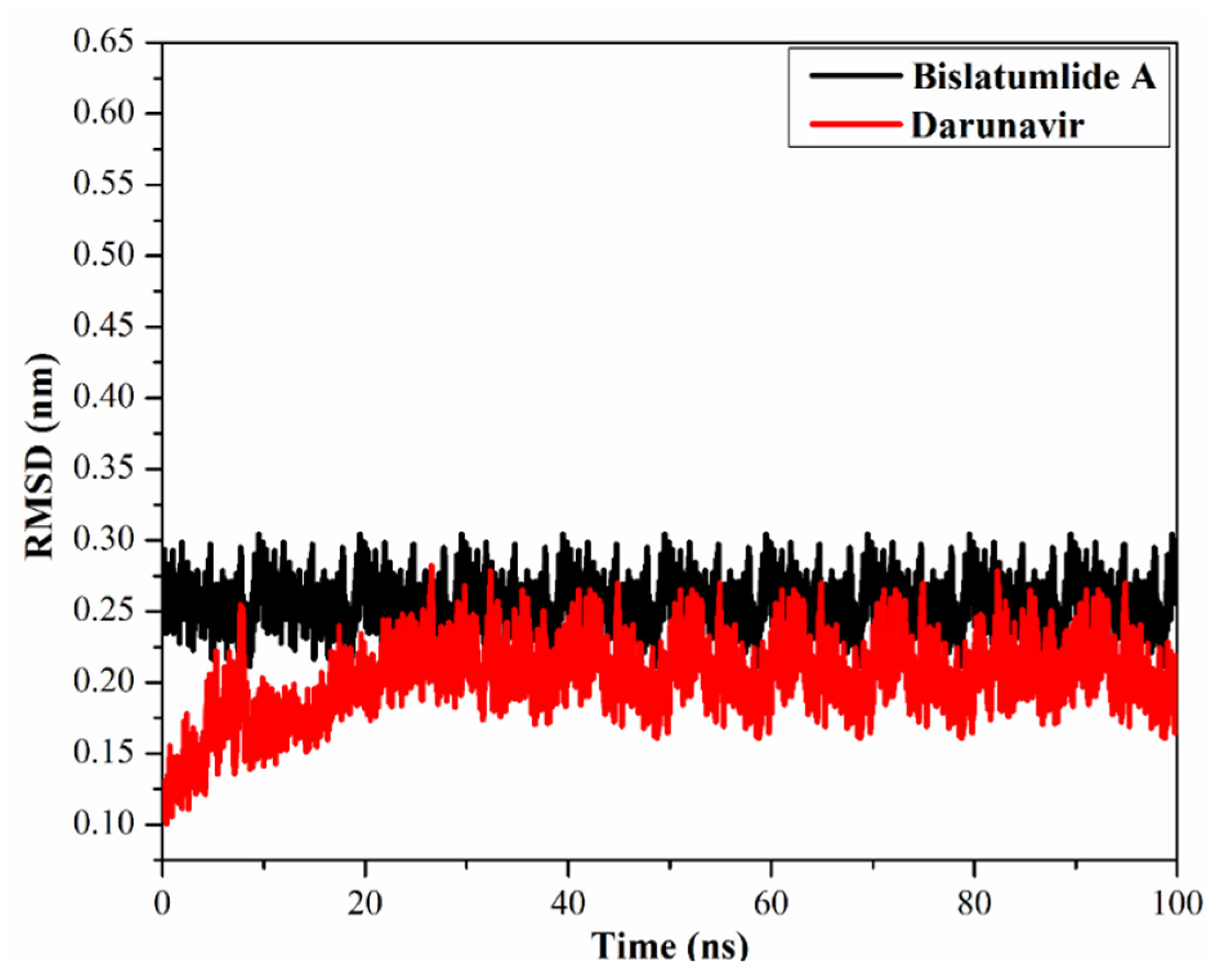
2.3.4. Root-Mean-Square Deviation
2.4. In Silico Drug-Likeness
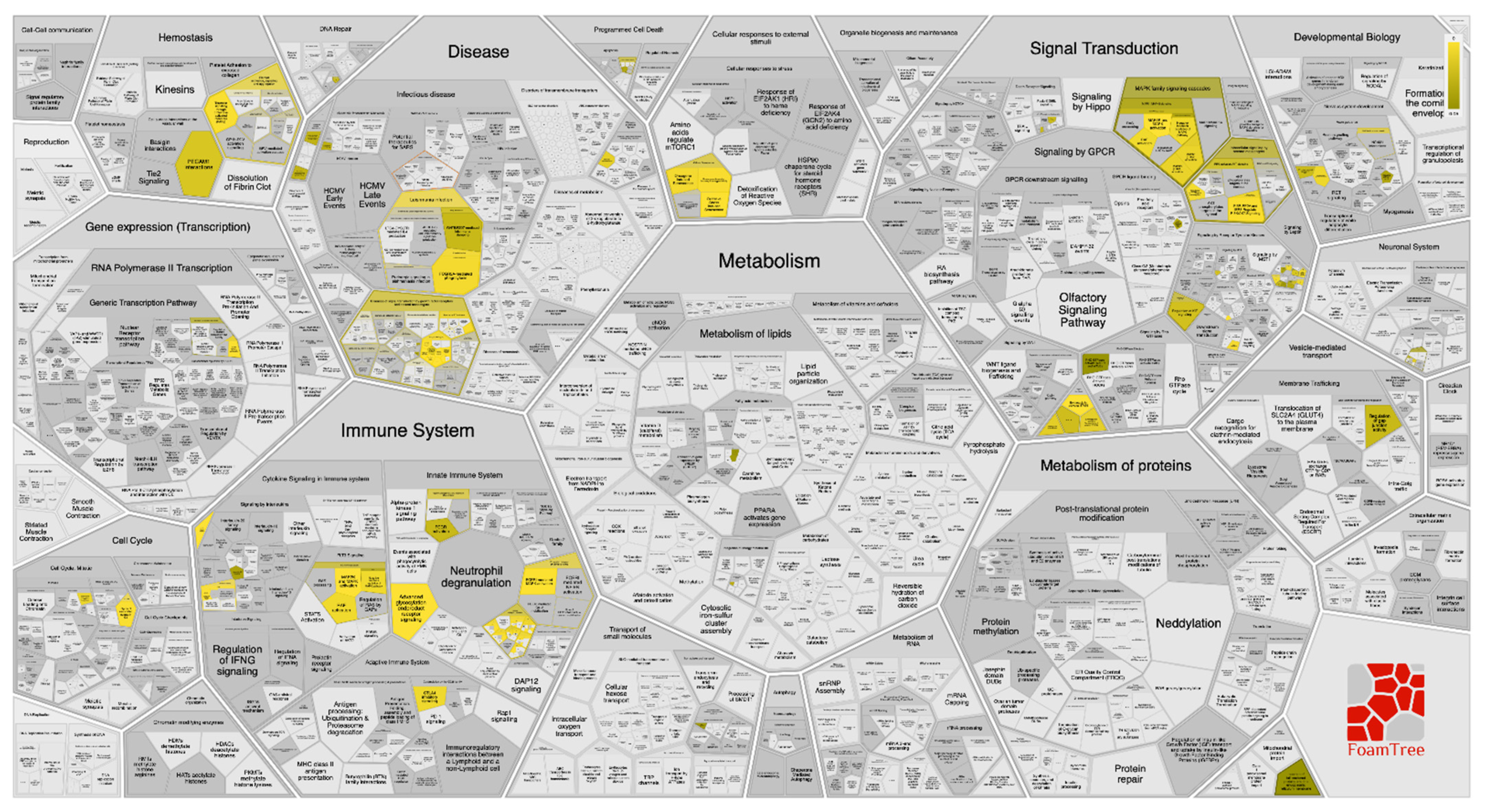
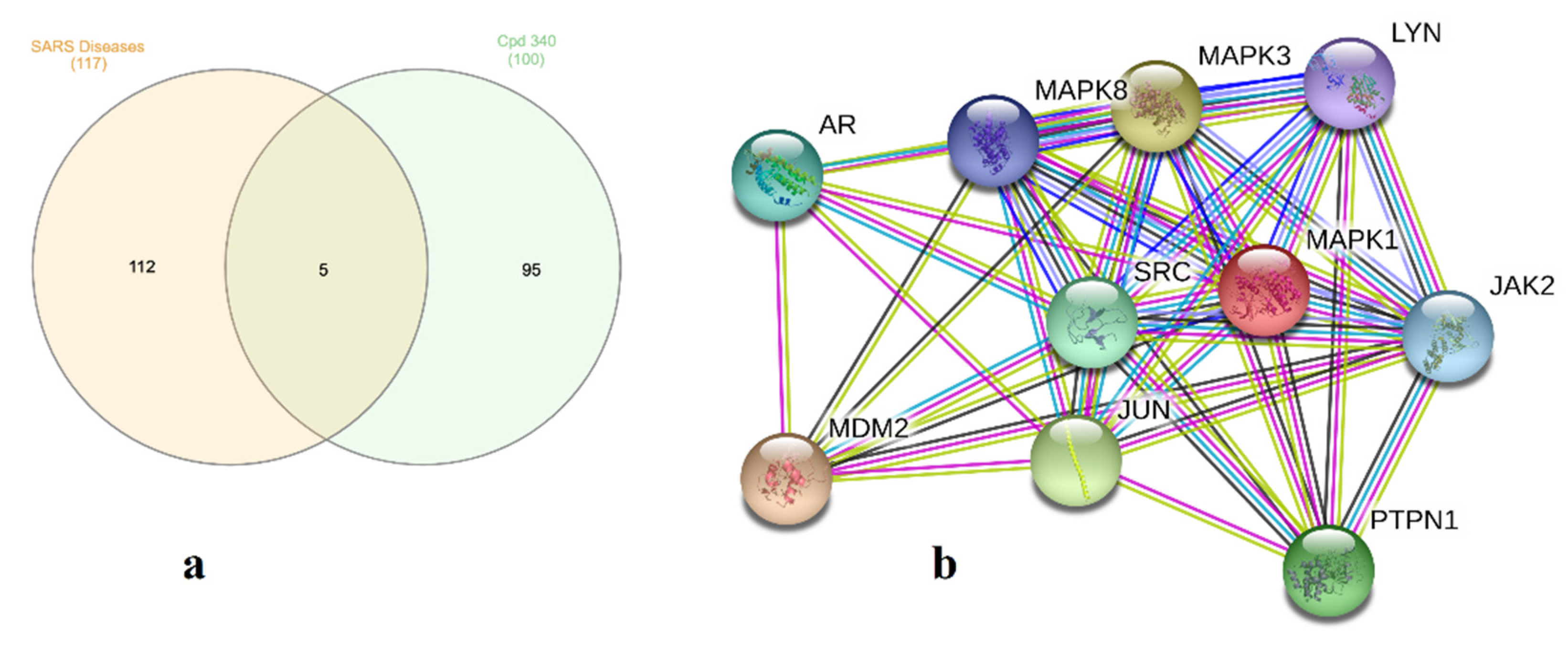
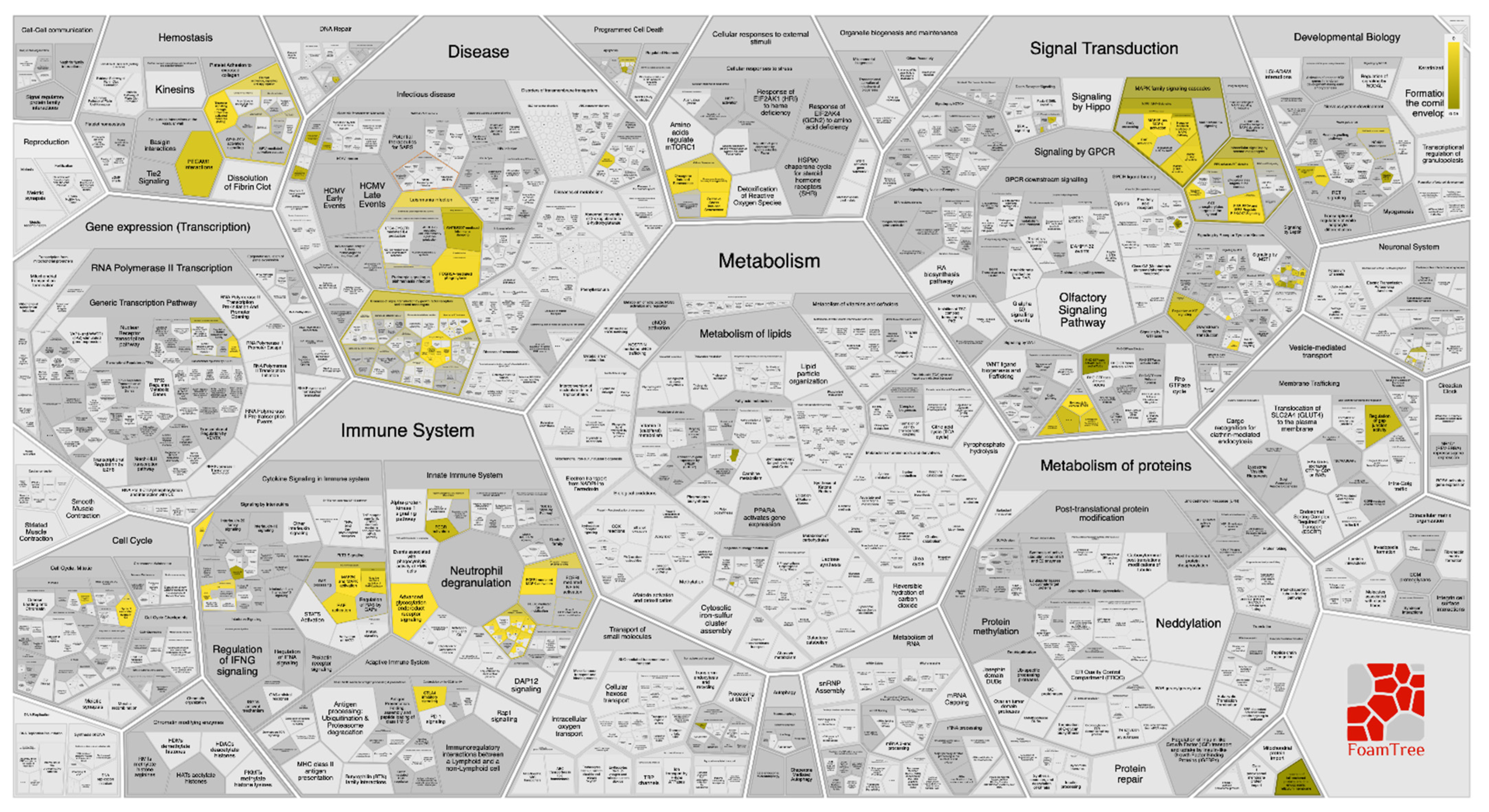
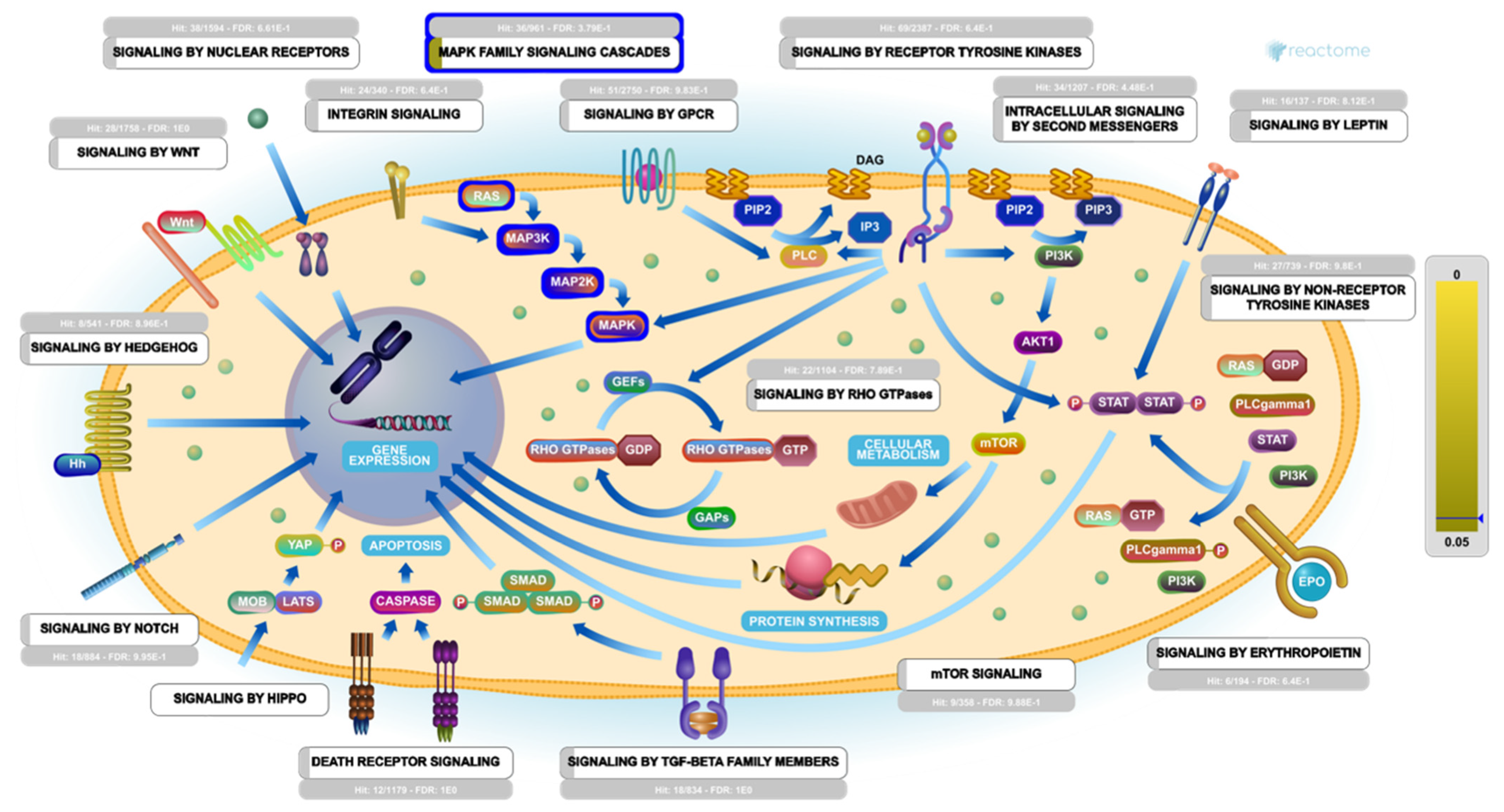
2.5. Molecular Target Prediction and Network Analysis
2.6. Pathway Enrichment Analysis (PEA)
3. Materials and Methods
3.1. Mpro Preparation
3.2. Inhibitor Preparation
3.3. Molecular Docking
3.4. Molecular Dynamics Simulations
3.5. MM-GBSA Binding Energy Calculations
3.6. Drug-Likeness Properties
3.7. Protein Interactions Network and Pathway Enrichment Analysis (PEA)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Wunderink, R.G. MERS, SARS and other coronaviruses as causes of pneumonia. Respirology 2017, 23, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Parrish, R.M.; Hohenstein, E.G.; Sherrill, D. Tractability gains in symmetry-adapted perturbation theory including coupled double excitations: CCD+ST(CCD) dispersion with natural orbital truncations. J. Chem. Phys. 2013, 139, 174102. [Google Scholar] [CrossRef] [PubMed]
- Dhand, R.; Li, J. Coughs and Sneezes: Their Role in Transmission of Respiratory Viral Infections, Including SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020, 202, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study. J. Chem. Inf. Model. 2020, 60, 3277–3286. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.; Mohamed, E.A.; Abdelrahman, A.H.; Allemailem, K.S.; Moustafa, M.F.; Shawky, A.M.; Mahzari, A.; Hakami, A.R.; Abdeljawaad, K.A.; Atia, M.A. Rutin and flavone analogs as prospective SARS-CoV-2 main protease inhibitors: In silico drug discovery study. J. Mol. Graph. Model. 2021, 105, 107904. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.; Abdelrahman, A.H.; Mohamed, T.A.; Atia, M.A.; Al-Hammady, M.A.; Abdeljawaad, K.A.; Elkady, E.M.; Moustafa, M.F.; Alrumaihi, F.; Allemailem, K.S.; et al. In Silico Mining of Terpenes from Red-Sea Invertebrates for SARS-CoV-2 Main Protease (M(pro)) Inhibitors. Molecules 2021, 26, 2082. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Abdeljawaad, K.A.; Abdelrahman, A.H.; Hegazy, M.E.F. Natural-like products as potential SARS-CoV-2 M(pro) inhibitors: In-silico drug discovery. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Abdelrahman, A.H.; Hegazy, M.E.F. In-silico drug repurposing and molecular dynamics puzzled out potential SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Abdelrahman, A.H.; Hussien, T.A.; Badr, E.A.; Mohamed, T.A.; El-Seedi, H.R.; Pare, P.W.; Efferth, T.; Hegazy, M.E.F. In silico drug discovery of major metabolites from spices as SARS-CoV-2 main protease inhibitors. Comput. Biol. Med. 2020, 126, 104046. [Google Scholar] [CrossRef]
- Sencanski, M.; Perovic, V.; Pajovic, S.B.; Adzic, M.; Paessler, S.; Glisic, S. Drug Repurposing for Candidate SARS-CoV-2 Main Protease Inhibitors by a Novel In Silico Method. Molecules 2020, 25, 3830. [Google Scholar] [CrossRef]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci. Rep. 2020, 10, 17716. [Google Scholar] [CrossRef]
- Zakaryan, H.; Arabyan, E.; Oo, A.; Zandi, K. Flavonoids: Promising natural compounds against viral infections. Arch. Virol. 2017, 162, 2539–2551. [Google Scholar] [CrossRef]
- Cherrak, S.A.; Merzouk, H.; Mokhtari-Soulimane, N. Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: A molecular docking and simulation studies. PLoS ONE 2020, 15, e0240653. [Google Scholar] [CrossRef]
- Jo, S.; Kim, S.; Kim, D.Y.; Kim, M.-S.; Shin, D.H. Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. J. Enzym. Inhib. Med. Chem. 2020, 35, 1539–1544. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Elkhawas, Y.A.; Elissawy, A.M.; El-Naggar, M.; Mostafa, N.M.; Al-Sayed, E.; Bishr, M.M.; Singab, A.N.B.; Salama, O.M. Chemical Diversity in Species Belonging to Soft Coral Genus Sacrophyton and Its Impact on Biological Activity: A Review. Mar. Drugs 2020, 18, 41. [Google Scholar] [CrossRef] [Green Version]
- Carté, B.K. Biomedical Potential of Marine Natural Products: Marine organisms are yielding novel molecules for use in basic research and medical applications. BioScience 1996, 46, 271–286. [Google Scholar] [CrossRef] [Green Version]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed Marine Natural Products in the Pharmaceutical and Cosmeceutical Industries: Tips for Success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef] [Green Version]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; the International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Hegazy, M.-E.F.; Mohamed, T.; Abdel-Latif, F.F.; Alsaid, M.S.; Shahat, A.; Paré, P.W. Trochelioid A and B, new cembranoid diterpenes from the Red Sea soft coral Sarcophyton trocheliophorum. Phytochem. Lett. 2013, 6, 383–386. [Google Scholar] [CrossRef]
- Hegazy, M.-E.F.; El-Beih, A.A.; Moustafa, A.Y.; Hamdy, A.A.; Alhammady, M.A.; Selim, R.M.; Abdel-Rehim, M.; Paré, P.W. Cytotoxic Cembranoids from the Red Sea Soft Coral Sarcophyton glaucum. Nat. Prod. Commun. 2011, 6, 1809–1812. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C. Coronavirus puts drug repurposing on the fast track. Nat. Biotechnol. 2020, 38, 379–381. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.A.; Abdelrahman, A.H.; Allemailem, K.S.; Almatroudi, A.; Moustafa, M.F.; Hegazy, M.E.F. In Silico Evaluation of Prospective Anti-COVID-19 Drug Candidates as Potential SARS-CoV-2 Main Protease Inhibitors. Protein J. 2021, 40, 296–309. [Google Scholar] [CrossRef]
- Chen, J.; Xia, L.; Liu, L.; Xu, Q.; Ling, Y.; Huang, D.; Huang, W.; Song, S.; Xu, S.; Shen, Y.; et al. Antiviral Activity and Safety of Darunavir/Cobicistat for the Treatment of COVID-19. In Open Forum Infectious Diseases; Oxford University Press: New York, NY, USA, 2020; Volume 7, p. ofaa241. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Kerrigan, J.E. Molecular dynamics simulations in drug design. In In Silico Models for Drug Discovery; Kortagere, S., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 95–113. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Shen, M.; Tian, S.; Li, Y.; Li, Q.; Xu, X.; Wang, J.; Hou, T. Drug-likeness analysis of traditional Chinese medicines: 1. property distributions of drug-like compounds, non-drug-like compounds and natural compounds from traditional Chinese medicines. J. Cheminform. 2012, 4, 31. [Google Scholar] [CrossRef] [Green Version]
- Hemmat, N.; Asadzadeh, Z.; Ahangar, N.K.; Alemohammad, H.; Najafzadeh, B.; Derakhshani, A.; Baghbanzadeh, A.; Baghi, H.B.; Javadrashid, D.; Najafi, S.; et al. The roles of signaling pathways in SARS-CoV-2 infection; lessons learned from SARS-CoV and MERS-CoV. Arch. Virol. 2021, 166, 675–696. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e1039. [Google Scholar] [CrossRef]
- Yosimichi, G.; Nakanishi, T.; Nishida, T.; Hattori, T.; Takano-Yamamoto, T.; Takigawa, M. CTGF/Hcs24 induces chondrocyte differentiation through a p38 mitogen-activated protein kinase (p38MAPK), and proliferation through a p44/42 MAPK/extracellular-signal regulated kinase (ERK). JBIC J. Biol. Inorg. Chem. 2001, 268, 6058–6065. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Sakagami, H.; Miwa, N. ACE2: The key Molecule for Understanding the Pathophysiology of Severe and Critical Conditions of COVID-19: Demon or Angel? Viruses 2020, 12, 491. [Google Scholar] [CrossRef] [PubMed]
- Grimes, J.M.; Grimes, K.V. p38 MAPK inhibition: A promising therapeutic approach for COVID-19. J. Mol. Cell. Cardiol. 2020, 144, 63–65. [Google Scholar] [CrossRef]
- Kono, M.; Tatsumi, K.; Imai, A.M.; Saito, K.; Kuriyama, T.; Shirasawa, H. Inhibition of human coronavirus 229E infection in human epithelial lung cells (L132) by chloroquine: Involvement of p38 MAPK and ERK. Antivir. Res. 2008, 77, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-H.; Gavagnin, M.; Cimino, G.; Guo, Y.-W. Two new biscembranes with unprecedented carbon skeleton and their probable biogenetic precursor from the Hainan soft coral Sarcophyton latum. Tetrahedron Lett. 2007, 48, 5313–5316. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- OMEGA 2.5.1.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2013.
- SZYBKI, 1.9.0.3; OpenEye Scientific Software: Santa Fe, NM, USA, 2016.
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- QUACPAC, 1.7.0.2; OpenEye Scientific Software: Santa Fe, NM, USA, 2016.
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gervasoni, S.; Vistoli, G.; Talarico, C.; Manelfi, C.; Beccari, A.R.; Studer, G.; Tauriello, G.; Waterhouse, A.M.; Schwede, T.; Pedretti, A. A Comprehensive Mapping of the Druggable Cavities within the SARS-CoV-2 Therapeutically Relevant Proteins by Combining Pocket and Docking Searches as Implemented in Pockets 2.0. Int. J. Mol. Sci. 2020, 21, 5152. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER16; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Roux, B.; Simonson, T. Implicit solvent models. Biophys. Chem. 1999, 78, 1–20. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision E01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA, B.D.S.V.; Version 2019; Dassault Systèmes BIOVIA: San Diego, CA, USA, 2019.
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heberle, H.; Meirelles, G.V.; Da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ma, X.; Song, Y.; Zhang, Y.; Xiong, W.; Li, L.; Zhou, L. Anti-colorectal cancer targets of resveratrol and biological molecular mechanism: Analyses of network pharmacology, human and experimental data. J. Cell. Biochem. 2019, 120, 11265–11273. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.K.; Saez-Rodriguez, J.; Clarke, D.; Sorger, P.K.; Lauffenburger, U.A. Training Signaling Pathway Maps to Biochemical Data with Constrained Fuzzy Logic: Quantitative Analysis of Liver Cell Responses to Inflammatory Stimuli. PLoS Comput. Biol. 2011, 7, e1001099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blucher, A.; McWeeney, S.K.; Stein, L.; Wu, G. Visualization of drug target interactions in the contexts of pathways and networks with ReactomeFIViz. F1000Research 2019, 8, 908. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name (Number) | Genus | 2D Chemical Structure | Docking Score (kcal/mol) | Binding Features b |
|---|---|---|---|---|
| Darunavir | --- |  | −8.2 | LEU167 (1.96 Å), GLU166 (1.94, 2.88 Å) |
| Sarelengan B (363) | S. elegans | 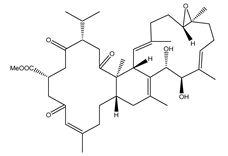 | −9.8 | GLY143 (2.39 Å), GLU166 (1.94 Å), GLN189 (2.58 Å), THR190 (2.33 Å) |
| Bislatumlide A (340) | S. latum |  | −9.6 | GLY143 (1.88 Å), GLU166 (2.68 Å) |
| Dioxanyalolide (347) | S. elegans | 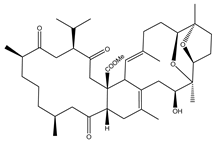 | −9.5 | GLU166 (2.07 Å) |
| Desacetylnyalolide (345) | S. elegans | 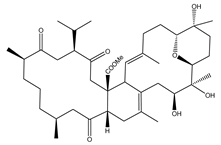 | −9.1 | GLU166 (1.66, 2.12 Å), THR190 (2.42 Å) |
| Lobophytone W (357) | S. elegans |  | −8.7 | HIS41 (2.01 Å), CYS145 (2.34 Å), GLU166 (2.35, 2.86 Å) |
| Compound Name | Estimated MM-GBSA Binding Energy (kcal/mol) | ||||||
|---|---|---|---|---|---|---|---|
| ΔEvdw | ΔEele | ΔEGB | ΔESUR | ΔGgas | ΔGSolv | ΔGbinding | |
| Bislatumlide A (340) | −56.1 | −27.7 | 45.6 | −6.6 | −83.8 | 39.0 | −44.8 |
| Darunavir | −47.4 | −15.1 | 33.8 | −6.2 | −62.5 | 27.7 | −34.8 |
| Compound Name | Acceptor | Donor | Angle (Degree) a | Distance (Å) a | Occupied (%) b |
|---|---|---|---|---|---|
| Bislatumlide A (340) | HIS41@ND1 | Bislatumlide A@O-H16 | 164 | 2.9 | 67.9 |
| GLU166@O | Bislatumlide A@O2-H25 | 142 | 2.8 | 90.3 | |
| GLN189@O | Bislatumlide A@O3-H47 | 145 | 2.6 | 88.9 | |
| Darunavir | GLU166@O | Darunavir @O5-H36 | 151 | 2.8 | 85.7 |
| Compound Name | mLogP | TPSA | nON | nOHNH | Nrotb | MWt | %ABS |
|---|---|---|---|---|---|---|---|
| Bislatumlide A (340) | 4.3 | 119.4 | 8 | 2 | 3 | 694.9 | 67.8% |
| Darunavir | 1.2 | 148.8 | 8 | 3 | 13 | 547.7 | 57.7% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Atia, M.A.M.; Mohamed, T.A.; Moustafa, M.F.; Hakami, A.R.; Khalifa, S.A.M.; Alhumaydhi, F.A.; Alrumaihi, F.; Abidi, S.H.; et al. Blue Biotechnology: Computational Screening of Sarcophyton Cembranoid Diterpenes for SARS-CoV-2 Main Protease Inhibition. Mar. Drugs 2021, 19, 391. https://doi.org/10.3390/md19070391
Ibrahim MAA, Abdelrahman AHM, Atia MAM, Mohamed TA, Moustafa MF, Hakami AR, Khalifa SAM, Alhumaydhi FA, Alrumaihi F, Abidi SH, et al. Blue Biotechnology: Computational Screening of Sarcophyton Cembranoid Diterpenes for SARS-CoV-2 Main Protease Inhibition. Marine Drugs. 2021; 19(7):391. https://doi.org/10.3390/md19070391
Chicago/Turabian StyleIbrahim, Mahmoud A. A., Alaa H. M. Abdelrahman, Mohamed A. M. Atia, Tarik A. Mohamed, Mahmoud F. Moustafa, Abdulrahim R. Hakami, Shaden A. M. Khalifa, Fahad A. Alhumaydhi, Faris Alrumaihi, Syed Hani Abidi, and et al. 2021. "Blue Biotechnology: Computational Screening of Sarcophyton Cembranoid Diterpenes for SARS-CoV-2 Main Protease Inhibition" Marine Drugs 19, no. 7: 391. https://doi.org/10.3390/md19070391
APA StyleIbrahim, M. A. A., Abdelrahman, A. H. M., Atia, M. A. M., Mohamed, T. A., Moustafa, M. F., Hakami, A. R., Khalifa, S. A. M., Alhumaydhi, F. A., Alrumaihi, F., Abidi, S. H., Allemailem, K. S., Efferth, T., Soliman, M. E., Paré, P. W., El-Seedi, H. R., & Hegazy, M.-E. F. (2021). Blue Biotechnology: Computational Screening of Sarcophyton Cembranoid Diterpenes for SARS-CoV-2 Main Protease Inhibition. Marine Drugs, 19(7), 391. https://doi.org/10.3390/md19070391