
Isomalabaricane Triterpenes from the Marine Sponge Rhabdastrella sp.

 ,
, 
 ,
,
Abstract
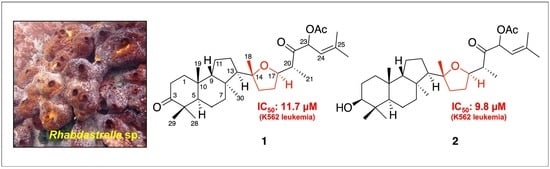
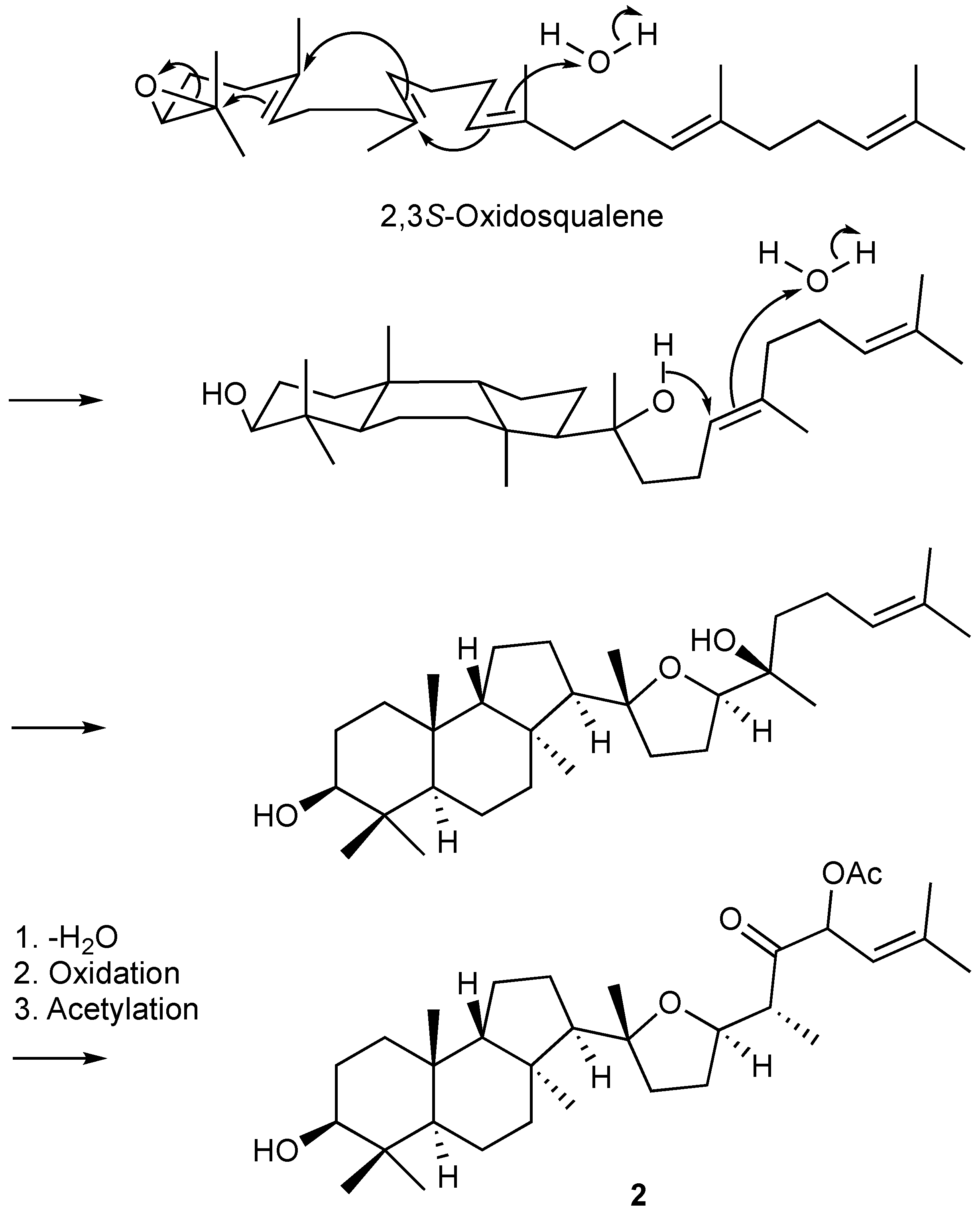
1. Introduction
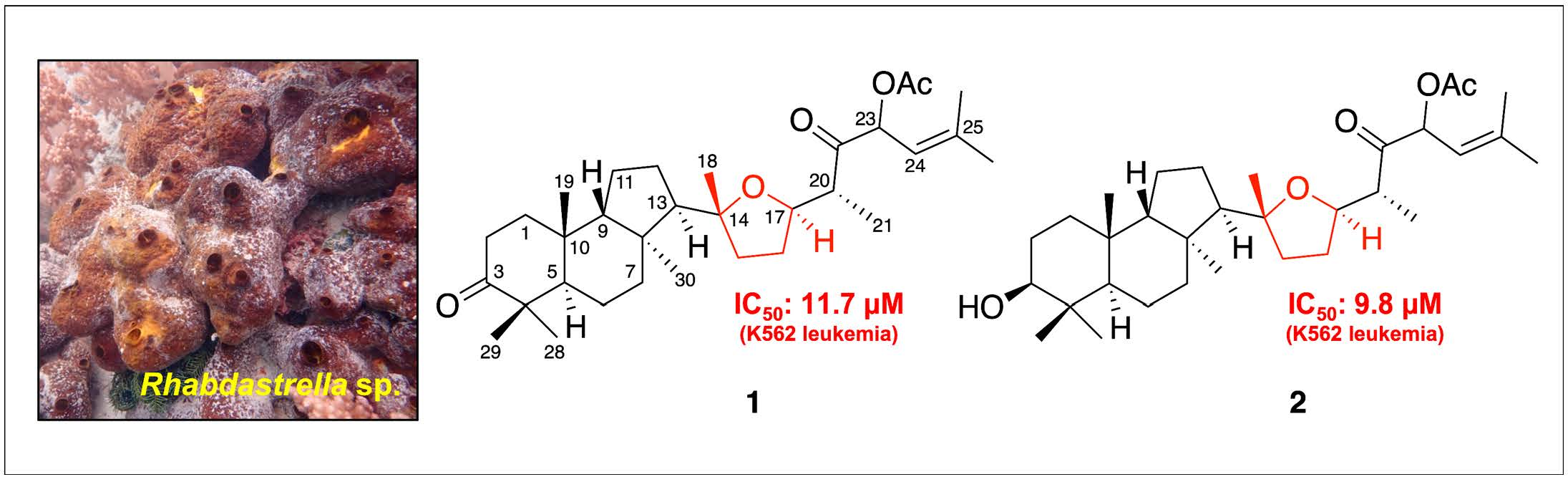
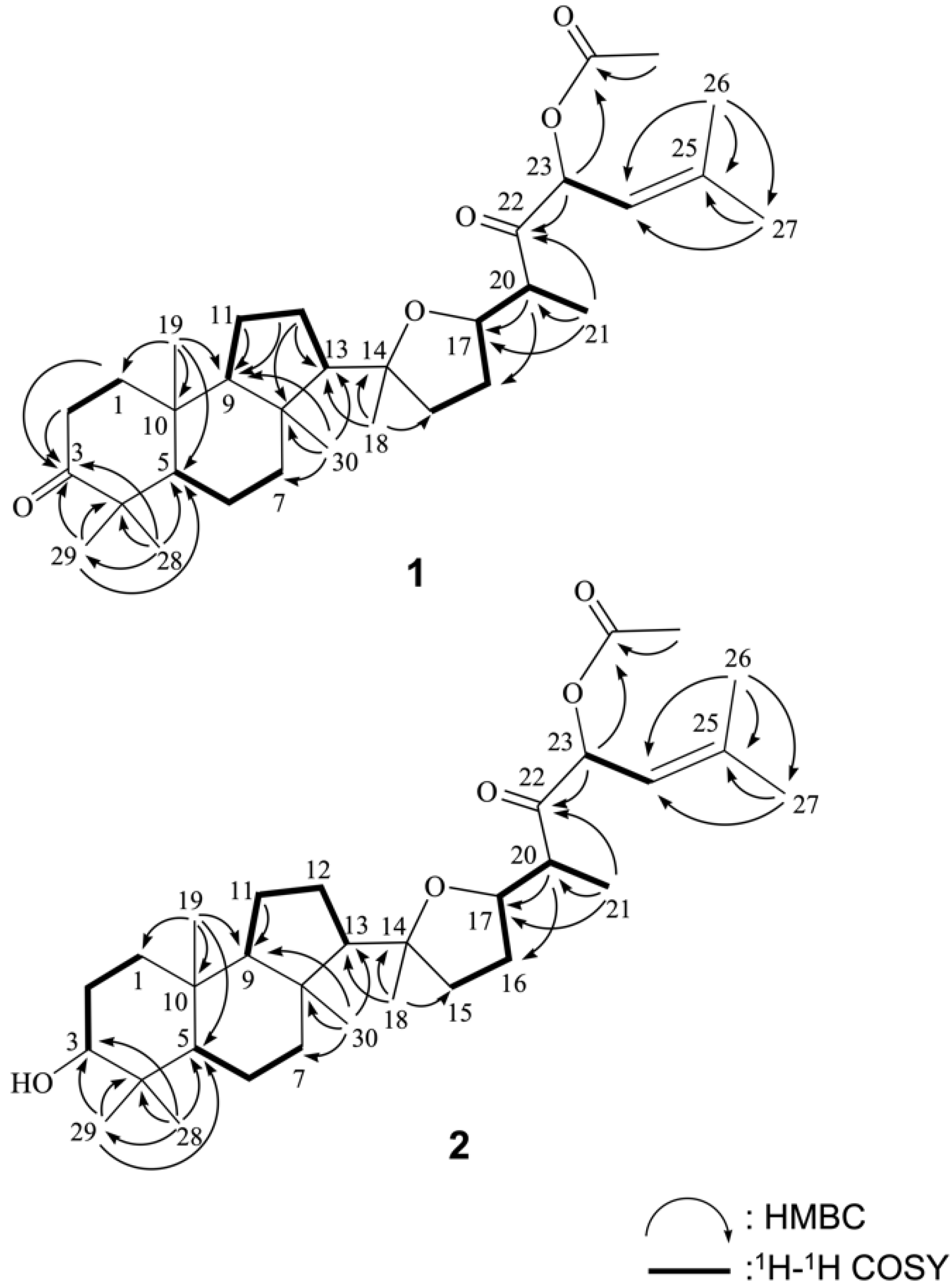
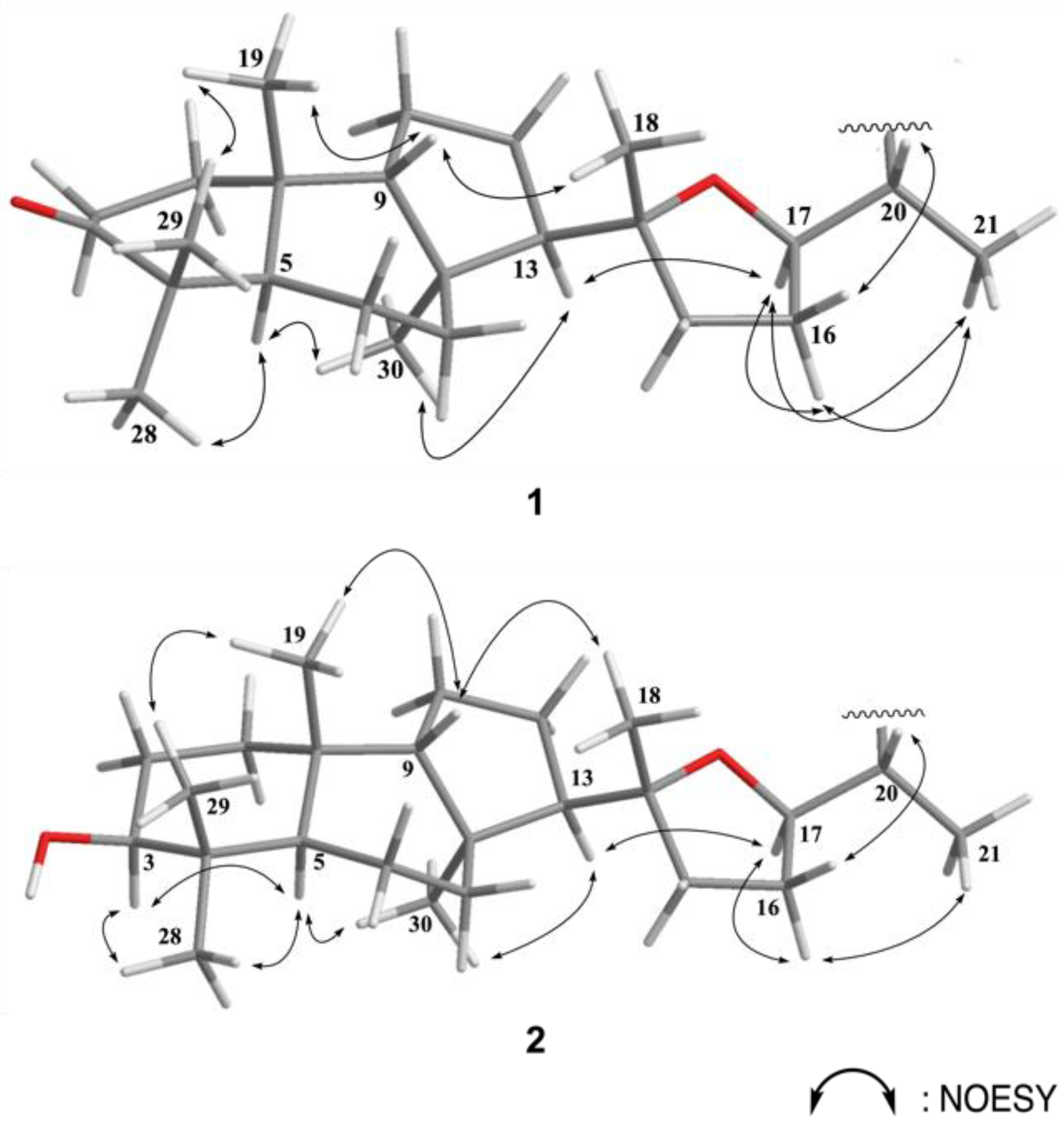
2. Results
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Animal Material
3.3. Extraction and Isolation
3.4. MTT Cell Proliferation Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Tasdemir, D.; Mangalindan, G.C.; Concepcion, G.P.; Verbitski, S.M.; Rabindran, S.; Miranda, M.; Greenstein, M.; Hooper, J.N.; Harper, M.K.; Ireland, C.M. Bioactive isomalabaricane triterpenes from the marine sponge Rhabdastrella globostellata. J. Nat. Prod. 2002, 65, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Clement, J.A.; Li, M.; Hecht, S.M.; Kingston, D.G. Bioactive isomalabaricane triterpenoids from Rhabdastrella globostellata that stabilize the binding of DNA polymerase beta to DNA. J. Nat. Prod. 2006, 69, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Lv, F.; Deng, Z.; Li, J.; Fu, H.; van Soest, R.W.; Proksch, P.; Lin, W. Isomalabaricane-type compounds from the marine sponge Rhabdastrella aff. distincta. J. Nat. Prod. 2004, 67, 2033–2036. [Google Scholar] [CrossRef] [PubMed]
- Meragelman, K.M.; McKee, T.C.; Boyd, M.R. New cytotoxic isomalabaricane triterpenes from the sponge Jaspis species. J. Nat. Prod. 2001, 64, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Sanagawa, M.; Watanabe, Y.; Setiawan, A.; Arai, M.; Kobayashi, M. Novel isomarabarican triterpenes, exhibiting selective anti-proliferative activity against vascular endothelial cells, from marine sponge Rhabdastrella globostellata. Bioorg. Med. Chem. 2007, 15, 4818–4828. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.L.; McKee, T.C.; Cardellina, J.H.; Leid, M.; Boyd, M.R. Cytotoxic triterpenes from a marine sponge, Stelletta sp. J. Nat. Prod. 1996, 59, 1047–1050. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, S.A.; Rajesh, K.G.; Adavi, R.V.B.; Raghuram, R.A.; Ravi, K.B.; Appa, R.V.N.A. New malabaricane triterpenes from the oleoresin of Ailanthus malabarica. Fitoterapia 2015, 100, 166–173. [Google Scholar]
- William, F.P.; Iain, C.P.; Ashok, G.B.; Sukh, D. The structure of malabricol. Tetrahedron Lett. 1979, 43, 4153–4154. [Google Scholar]
- Miyabi, H.; Kazuomi, T.; Toshiyuki, H.; Hiroaki, O.; Tatsuhiko, F.; Shin-ichi, A.; Yusuke, T.; Ryuji, I.; Masaharu, K.; Matsumi, D.; et al. Cytotoxic isomalabaricane derivatives and a monocyclic triterpene glycoside from the sponge Rhabdastrella globostellata. J. Nat. Prod. 2010, 73, 1512–1518. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH (J in Hz) a | δC (Mult.) b | 1H–1H COSY | HMBC |
|---|---|---|---|---|
| 1 | 2.08 m; 1.46 m | 31.9 (CH2) d | H-2 | C-3 |
| 2 | 2.66 m; 2.30 m | 33.7 (CH2) | C-3 | |
| 3 | 220.3 (C) | |||
| 4 | 46.8 (C) | |||
| 5 | 2.23 m | 45.7 (CH) | H-6 | |
| 6 | 1.45 m; 1.34 m | 20.0 (CH2) | H-5, H-7 | |
| 7 | 2.15 m; 1.35 m | 32.3 (CH2) | C-5, C-9 | |
| 8 | 42.2 (C) | |||
| 9 | 1.44 m | 52.9 (CH) | H-11 | |
| 10 | 35.0 (C) | |||
| 11 | 1.52 m; 1.30 m | 21.4 (CH2) | H-9, H-12 | C-9 |
| 12 | 1.85 m; 1.55 m | 25.4 (CH2) | H-11, H-13 | C-8, C-9, C-13 |
| 13 | 1.78 m | 59.8 (CH) | H-12 | |
| 14 | 86.2 (C) | |||
| 15 | 1.80 m | 38.6 (CH2) | H-16 | |
| 16 | 1.97 m; 1.58 m | 31.0 (CH2) | H-17 | |
| 17 | 3.76 ddd (14.0, 8.5, 4.5) c | 79.8 (CH) | H-20 | |
| 18 | 1.17 s | 24.5 (CH3) | C-13, C-14, C-15 | |
| 19 | 0.79 s | 24.0 (CH3) | C-1, C-5, C-9, C-10 | |
| 20 | 2.70 m | 47.6 (CH) | H-17, H-21 | C-16, C-17 |
| 21 | 0.95 d (7.0) | 12.1 (CH3) | H-20 | C-17, C-20, C-22 |
| 22 | 207.4 (C) | |||
| 23 | 6.09 d (10.0) | 77.0 (CH) | H-24 | OAc |
| 24 | 5.11 d (10.0) | 116.9 (CH) | H-23 | |
| 25 | 142.6 (C) | |||
| 26 | 1.81 s | 26.0 (CH3) | C-24, C-25, C-27 | |
| 27 | 1.89 s | 19.0 (CH3) | C-24, C-25, C-26 | |
| 28 | 1.02 s | 19.3 (CH3) | C-3, C-4, C-5, C-29 | |
| 29 | 1.06 s | 29.3 (CH3) | C-3, C-4, C-5, C-28 | |
| 30 | 1.09 s | 31.8 (CH3) | C-7, C-8, C-9, C-13 | |
| OAc | 2.12 s | 20.9 (CH3)169.8 (C) |
| Position | δH (J in Hz) a | δC (Mult.) b | 1H–1H COSY | HMBC |
|---|---|---|---|---|
| 1 | 1.48 m; 1.41 m | 33.9 (CH2) d | H-2 | |
| 2 | 1.73 m; 1.61 m | 29.2 (CH2) | H-3 | |
| 3 | 3.24 dd (11.5,6.5) | 79.5 (CH) | ||
| 4 | 39.0 (C) | |||
| 5 | 1.54 m | 46.6 (CH) | H-6 | |
| 6 | 1.64 m; 1.38 m | 18.5 (CH2) | H-5, H-7 | |
| 7 | 2.04 m; 1.35 m | 32.9 (CH2) | ||
| 8 | 42.0 (C) | |||
| 9 | 1.42 m | 55.0 (CH) | H-11 | |
| 10 | 35.6 (C) | |||
| 11 | 1.49 m; 1.30 m | 21.2 (CH2) | H-9. H-12 | C-9 |
| 12 | 1.82 m; 1.54 m | 25.0 (CH2) | H-11, H-13 | |
| 13 | 1.78 m | 59.9 (CH) | H-12 | |
| 14 | 86.2 (C) | |||
| 15 | 1.80 m; 1.75 m | 38.8 (CH2) | H-16 | |
| 16 | 1.98 m; 1.60 m | 31.1 (CH2) | H-17 | |
| 17 | 3.75 ddd (13.5, 8.5, 4.5) c | 79.5 (CH) | H-20 | |
| 18 | 1.16 s | 24.5 (CH3) | C-13, C-14, C-15 | |
| 19 | 0.95 s | 23.1 (CH3) | C-1, C-5, C-9, C-10 | |
| 20 | 2.69 ddd (13.5, 8.5, 1.5) | 47.5 (CH) | H-17, H-21 | C-16, C-17 |
| 21 | 0.94 d (6.0) | 12.1 (CH3) | H-20 | C-17, C-20, C-22 |
| 22 | 207.4 (C) | |||
| 23 | 6.10 d (10.0) | 78.7 (CH) | H-24 | C-22, C-24, OAc |
| 24 | 5.11 d (10.0) | 116.9 (CH) | H-23 | |
| 25 | 142.6 (C) | |||
| 26 | 1.81 s | 26.0 (CH3) | C-24, C-25, C-27 | |
| 27 | 1.89 s | 19.0 (CH3) | C-24, C-25, C-26 | |
| 28 | 0.98 s | 15.9 (CH3) | C-3, C-4, C-5, C-29 | |
| 29 | 0.78 s | 29.1 (CH3) | C-3, C-4, C-5, C-28 | |
| 30 | 1.04 s | 31.6 (CH3) | C-7, C-8, C-9, C-14 | |
| OAc | 2.10 s | 20.9 (CH3)169.8 (C) |
| Compounds | Cell Lines (IC50 μM) | |||
|---|---|---|---|---|
| DLD-1 | T-47D | Molt4 | K562 | |
| 1 | – a | – a | 16.54 | 11.71 |
| 2 | – a | 17.48 | 11.03 | 9.81 |
| Doxorubicin b | 0.42 | 0.18 | 0.02 | 0.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, K.-H.; Huang, Z.-H.; El-Shazly, M.; Peng, B.-R.; Wei, W.-C.; Su, J.-H. Isomalabaricane Triterpenes from the Marine Sponge Rhabdastrella sp. Mar. Drugs 2021, 19, 206. https://doi.org/10.3390/md19040206
Lai K-H, Huang Z-H, El-Shazly M, Peng B-R, Wei W-C, Su J-H. Isomalabaricane Triterpenes from the Marine Sponge Rhabdastrella sp. Marine Drugs. 2021; 19(4):206. https://doi.org/10.3390/md19040206
Chicago/Turabian StyleLai, Kuei-Hung, Zheng-Hao Huang, Mohamed El-Shazly, Bo-Rong Peng, Wen-Chi Wei, and Jui-Hsin Su. 2021. "Isomalabaricane Triterpenes from the Marine Sponge Rhabdastrella sp." Marine Drugs 19, no. 4: 206. https://doi.org/10.3390/md19040206
APA StyleLai, K.-H., Huang, Z.-H., El-Shazly, M., Peng, B.-R., Wei, W.-C., & Su, J.-H. (2021). Isomalabaricane Triterpenes from the Marine Sponge Rhabdastrella sp. Marine Drugs, 19(4), 206. https://doi.org/10.3390/md19040206