
New Andrastin-Type Meroterpenoids from the Marine-Derived Fungus Penicillium sp.


Abstract
1. Introduction
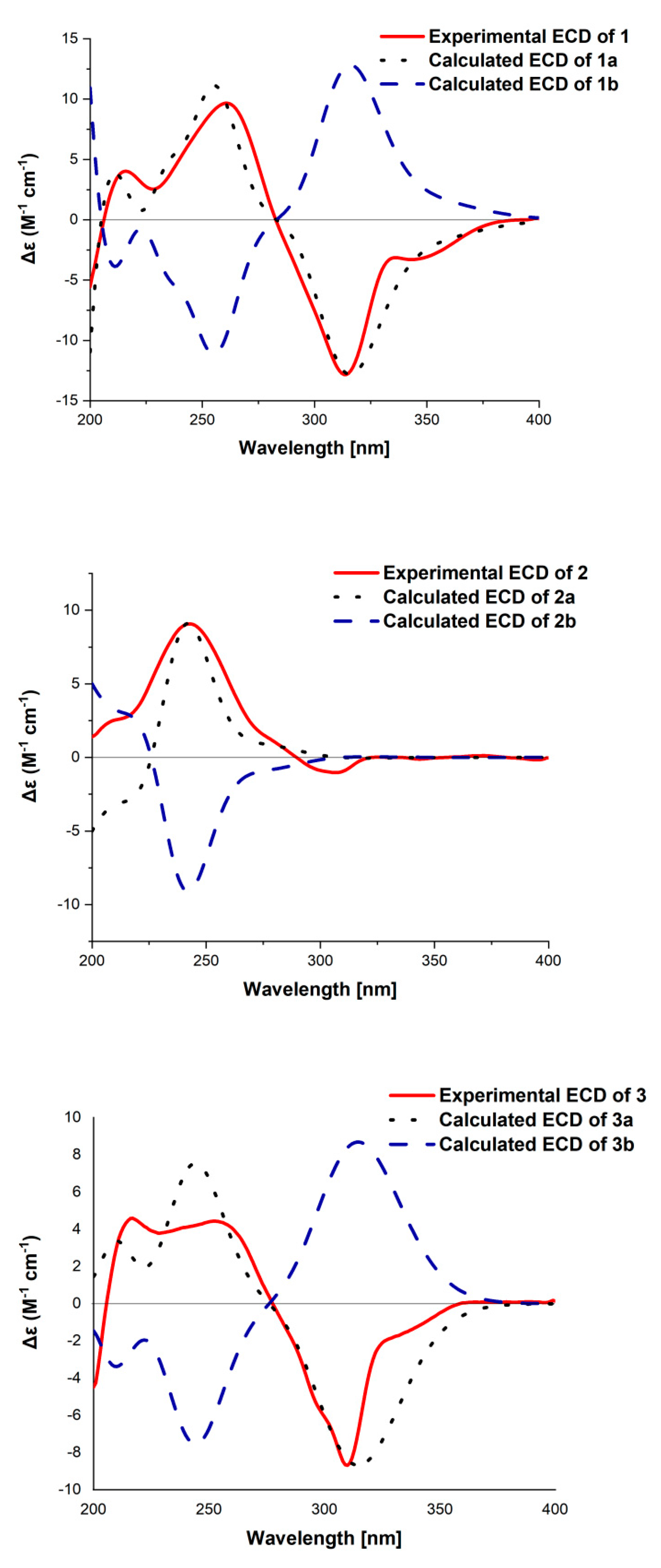
2. Results
3. Experimental Section
3.1. General Experimental Procedure
3.2. Strain and Fermentation
3.3. Extraction and Isolation
3.4. Bioassays for Cytotoxic Activity
3.5. ECD Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Geris, R.; Simpson, T.J. Meroterpenoids produced by fungi. Nat. Prod. Rep. 2009, 26, 1063–1094. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Abe, I. Biosynthesis of fungal meroterpenoids. Nat. Prod. Rep. 2016, 33, 26–53. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Awakawa, T.; Abe, I. Reconstituted biosynthesis of fungal meroterpenoid andrastin A. Tetrahedron 2013, 69, 8199–8204. [Google Scholar] [CrossRef]
- Matsuda, Y.; Quan, Z.; Mitsuhashi, T.; Li, C.; Abe, I. Cytochrome P450 for citreohybridonol synthesis: Oxidative derivatization of the andrastin scaffold. Org. Lett. 2016, 18, 296–299. [Google Scholar] [CrossRef]
- Uchida, R.; Shiomi, K.; Inokoshi, J.; Tanaka, H.; Iwai, Y.; Omura, S. Andrastins A–C, new protein farnesyltransferase inhibitors, produced by Penicillium sp. FO-3929. J. Antibiot. 1996, 49, 1278–1280. [Google Scholar] [CrossRef]
- Kosemura, S.; Matsunaga, K.; Yamamura, S.; Kubota, M.; Ohba, S. The structures of citreohybridone A and B novel sesterterpenoid-type metabolites of a hybrid strain KO 0031 derived from Penicillium citreo-viride B. IFO 6200 and 4692. Tetrahedron Lett. 1991, 32, 3543–3546. [Google Scholar] [CrossRef]
- Kosemura, S.; Matsuo, S.; Yamamura, S.; Citreohybriddione, C. A meroterpenoid of a hybrid strain KO 0031 derived from Penicillium citreo-viride B. IFO 6200 and 4692. Phytochemistry 1996, 43, 1231–1234. [Google Scholar] [CrossRef]
- Cheng, Z.B.; Wei, X.B.; Wang, Y.Y.; Bai, S.Y.; Liu, L.J.; Luo, Z.H.; Yuan, W.J.; Li, Q. Two new meroterpenoids and two new monoterpenoids from the deep seaderived fungus Penicillium sp. YPGA11. Fitoterapia 2019, 133, 120–124. [Google Scholar] [CrossRef]
- Gao, S.S.; Shang, Z.; Li, X.M.; Li, C.S.; Cui, C.M.; Wang, B.G. Secondary metabolites produced by solid fermentation of the marine-derived fungus Penicillium commune QSD-17. Biosci. Biotechnol. Biochem. 2012, 76, 358–360. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.L.; Xia, J.M.; Lin, T.; Lin, Y.J.; Lin, Y.K.; Xia, M.L.; Chen, H.F.; Luo, Z.H.; Shao, Z.Z.; Yang, X.W. Andrastone A from the deep-sea-derived fungus Penicillium allii-sativi acts as an inducer of caspase and RXR α-dependent apoptosis. Front. Chem. 2019, 7, 692. [Google Scholar] [CrossRef]
- Özkaya, F.C.; Ebrahim, W.; Klopotowski, M.; Liu, Z.; Janiak, C.; Proksch, P. Isolation and X-ray structure analysis of citreohybridonol from marine-derived Penicillium atrovenetum. Nat. Pro. Res. 2018, 32, 840–843. [Google Scholar] [CrossRef]
- Kosemura, S.; Meroterpenoids from Penicillium citreo-viride B. IFO 4692 and 6200 hybrid. Tetrahedron 2003, 59, 5055–5072. [Google Scholar] [CrossRef]
- Wang, X.R.; Sena, J.G.; Hoover, A.R.; King, J.B.; Ellis, T.K.; Powell, D.R.; Cichewicz, R.H. Chemical epigenetics alters the secondary metabolite composition of guttate excreted by an Atlantic forest soil-derived Penicillium citreonigrum. J. Nat. Prod. 2010, 73, 942–948. [Google Scholar] [CrossRef]
- Cheng, X.; Liang, X.; Zheng, Z.H.; Zhang, X.X.; Lu, X.H.; Yao, F.H.; Qi, S.H. Penicimeroterpenoids A−C, meroterpenoids with rearrangement skeletons from the marine-derived fungus Penicillium sp. SCSIO 41512. Org. Lett. 2020, 62, 6330–6333. [Google Scholar] [CrossRef] [PubMed]
- Powers, Z.; Scharf, A.; Cheng, A.; Yang, F.; Himmelbauer, M.; Mitsuhashi, T.; Barra, L.; Taniguchi, Y.; Kikuchi, T.; Fujita, M.; et al. Biomimetic synthesis of meroterpenoids by dearomatization-driven polycyclization. Angew. Chem. Int. Ed. 2019, 58, 16141–16146. [Google Scholar] [CrossRef]
- Zong, Y.; Wang, W.J.; Xu, T. Total synthesis of bioactive marine meroterpenoids: The cases of liphagal and frondosin B. Mar. Drugs. 2018, 16, 115. [Google Scholar] [CrossRef]
- Kuan, K.K.W.; Markwell-Heys, A.W.; Cruickshank, M.C.; Tran, D.P.; Adlington, R.M.; Baldwin, J.E.; George, J.H. Biomimetic synthetic studies on meroterpenoids from the marine sponge aka coralliphaga: Divergent total syntheses of siphonodictyal B, liphagal and corallidictyals A–D. Bioorgan. Med. Chem. 2019, 27, 2449–2465. [Google Scholar] [CrossRef]
- Matsuda, Y.; Awakawa, T.; Wakimoto, T.; Abe, I. Spiro-Ring formation is catalyzed by a multifunctional dioxygenase in austinol biosynthesis. J. Am. Chem. Soc. 2013, 135, 10962–10965. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Iwabuchi, T.; Wakimoto, T.; Awakawa, T.; Abe, I. Spiro-Ring Formation is catalyzed by a multifunctional dioxygenase in austinol biosynthesis. J. Am. Chem. Soc. 2015, 137, 3393–3401. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2020, 37, 175–223. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.W.; Xie, C.L.; Xia, J.M.; He, Z.H. Andrastone compound and its preparation method and application in preparation of antiallergic drug. Patent CN 111217878 A; China, 2020. [Google Scholar]
- Liu, L.; Chen, X.X.; Li, D.; Zhang, Y.; Li, L.; Guo, L.D.; Cao, Y.; Che, Y.S. Bisabolane sesquiterpenoids from the plant endophytic fungus Paraconiothyrium brasiliense. J. Nat. Prod. 2015, 78, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; revision C 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1a | 2.32, m | 27.8, CH2 | 2.31, dt (12.9 5.5) | 26.6 CH2 | 2.75, m | 23.1, CH2 |
| 1b | 1.01, dt (12.0 5.5) | 1.01, m | 2.03, m | |||
| 2 | 1.59, m | 23.4, CH2 | 1.71, m | 23.6, CH2 | 1.82, m | 26.3, CH2 |
| 3 | 4.65, m | 77.1, CH | 4.65, t (2.5) | 76.9, CH | 4.73, t (2.5) | 77.7, CH |
| 4 | 37.0, C | 37.4, C | 38.7, C | |||
| 5 | 1.78, m | 47.7, CH | 1.80, m | 46.0, CH | 2.45, dd (11.1, 6.8) | 41.8, CH |
| 6a | 2.02, m | 16.9, CH2 | 2.16, m | 16.9, CH2 | 1.62, m | 18.2, CH2 |
| 6b | 1.81, m | 1.81, m | 1.62, m | |||
| 7a | 2.81, td (13.5, 3.5) | 30.8, CH2 | 2.84, td (13.1, 4.4) | 32.6, CH2 | 2.72, m | 29.7, CH2 |
| 7b | 2.36, m | 2.09, m | 1.95, m | |||
| 8 | 38.6, C | 39.7, C | 39.3, C | |||
| 9 | 2.19, s | 53.5, CH | 2.19, s | 147.9, C | 132.7, C | |
| 10 | 52.3, C | 55.1, C | 141.9, C | |||
| 11 | 5.82, s | 126.4, CH | 5.55, s | 125.8, CH | 4.75, s | 70.8, CH |
| 12 | 132.9, C | 76.0, C | 79.4, C | |||
| 13 | 60.9, C | 53.1, C | 55.0, C | |||
| 14 | 70.6, C | 71.8, C | 72.7, C | |||
| 15 | 210.6, C | 202.0, C | 197.7, C | |||
| 16 | 72.1, C | 75.6, C | 77.0, C | |||
| 17 | 206.8, C | 202.3, C | 204.4, C | |||
| 18 | 1.38, s | 19.6, CH3 | 1.31, s | 7.6, CH3 | 1.26, s | 7.4, CH3 |
| 19 | 1.29, s | 16.4, CH3 | 1.25, s | 10.4, CH3 | 1.42, s | 10.8, CH3 |
| 20 | 1.68, s | 18.9, CH3 | 1.26, s | 24.4, CH3 | 1.36, s | 22.2, CH3 |
| 21 | 10.1, s | 204.5, CH | 10.1, s | 202.1, CH | 170.8, CH | |
| 22 | 170.7, C | 170.9, C | 2.12, s | 21.3, CH3 | ||
| 23 | 2.10, s | 21.3, CH3 | 2.10, s | 21.6, CH3 | 0.87, s | 21.3, CH3 |
| 24 | 0.88, s | 21.4, CH3 | 0.93, s | 21.4, CH3 | 1.00, s | 24.8, CH3 |
| 25 | 0.94, s | 26.5, CH3 | 0.96, s | 26.8, CH3 | 1.75, s | 24.8, C |
| 26 | 1.15, s | 19.9, CH3 | 1.44, s | 26.2, CH3 | 168.0, C | |
| 27 | 167.3, C | 167.5, C | 3.61, s | 52.0, CH3 | ||
| 28 | 3.61, s | 52.0, CH3 | 3.62, s | 52.3, CH3 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, J.; Huo, R.; Liu, G.; Liu, L. New Andrastin-Type Meroterpenoids from the Marine-Derived Fungus Penicillium sp. Mar. Drugs 2021, 19, 189. https://doi.org/10.3390/md19040189
Ren J, Huo R, Liu G, Liu L. New Andrastin-Type Meroterpenoids from the Marine-Derived Fungus Penicillium sp. Marine Drugs. 2021; 19(4):189. https://doi.org/10.3390/md19040189
Chicago/Turabian StyleRen, Jinwei, Ruiyun Huo, Gaoran Liu, and Ling Liu. 2021. "New Andrastin-Type Meroterpenoids from the Marine-Derived Fungus Penicillium sp." Marine Drugs 19, no. 4: 189. https://doi.org/10.3390/md19040189
APA StyleRen, J., Huo, R., Liu, G., & Liu, L. (2021). New Andrastin-Type Meroterpenoids from the Marine-Derived Fungus Penicillium sp. Marine Drugs, 19(4), 189. https://doi.org/10.3390/md19040189