
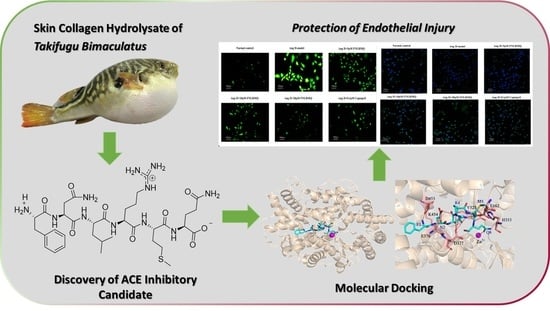
ACE Inhibitory Peptide from Skin Collagen Hydrolysate of Takifugu bimaculatus as Potential for Protecting HUVECs Injury

 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
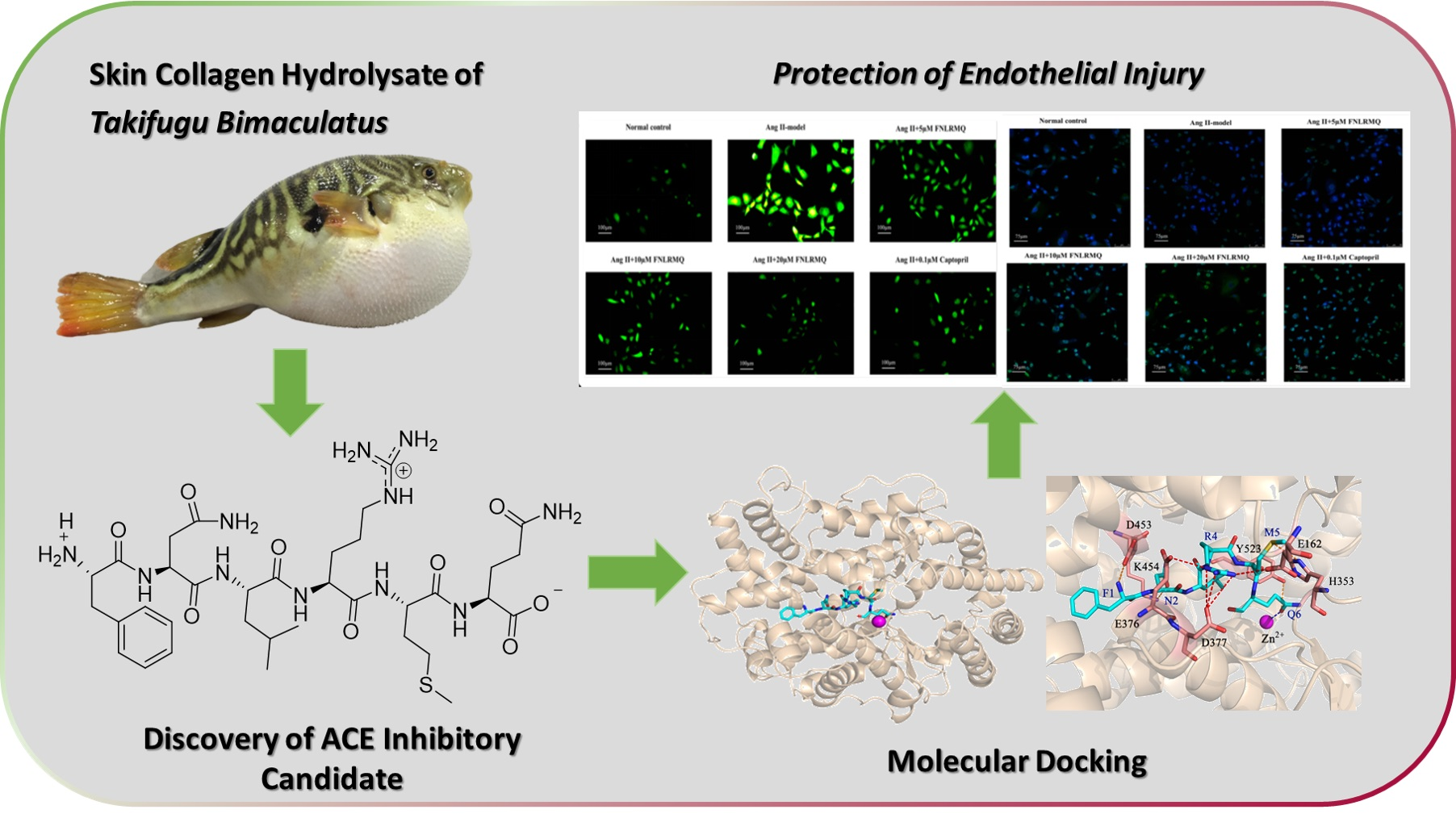
2.1. Screening and Identification of ACE Inhibitory Peptides
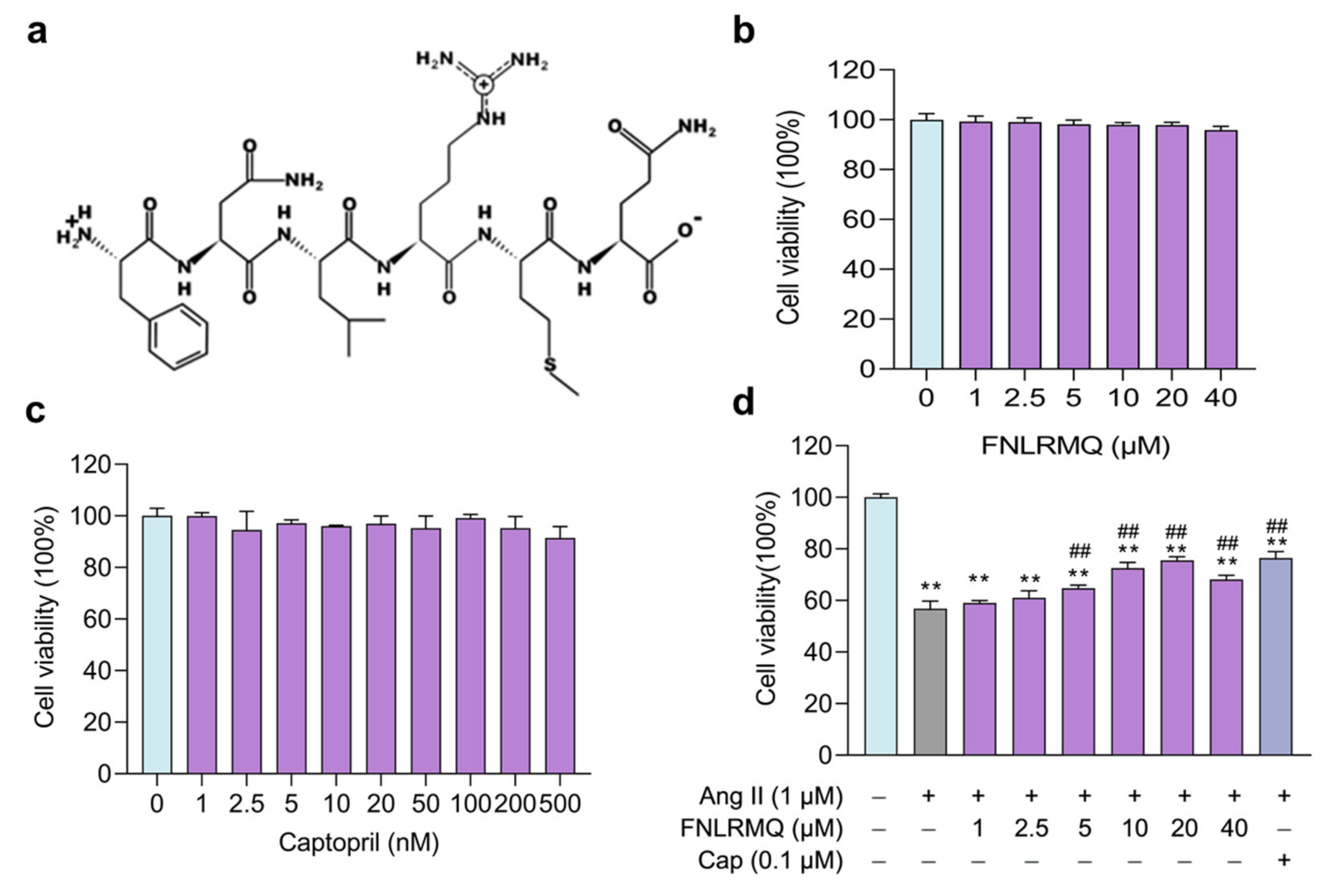
2.2. FNLRMQ Enhanced Cell Viability in Ang-II-induced HUVECs
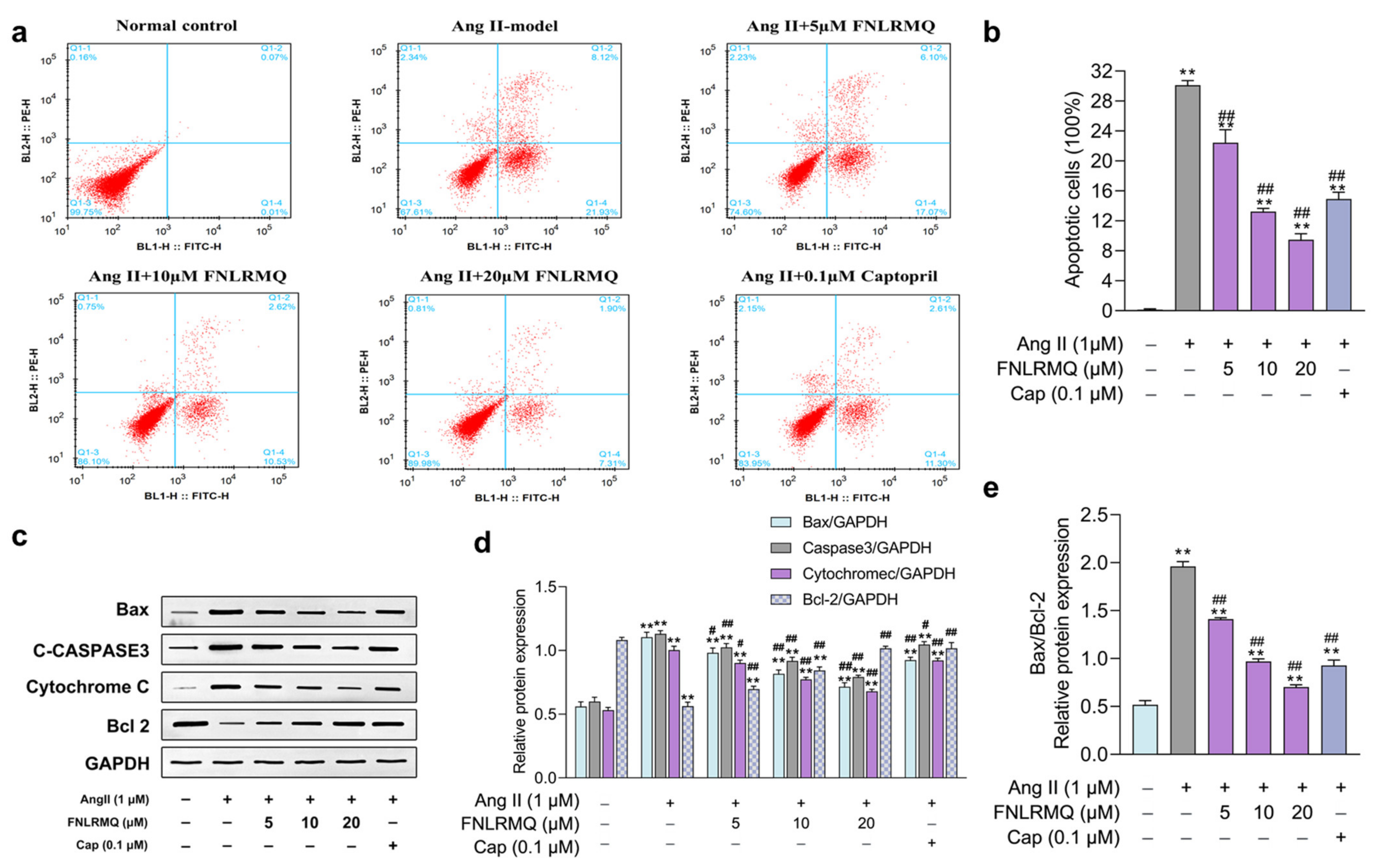
2.3. FNLRMQ Ameliorated Ang-II-Induced HUVEC Apoptosis
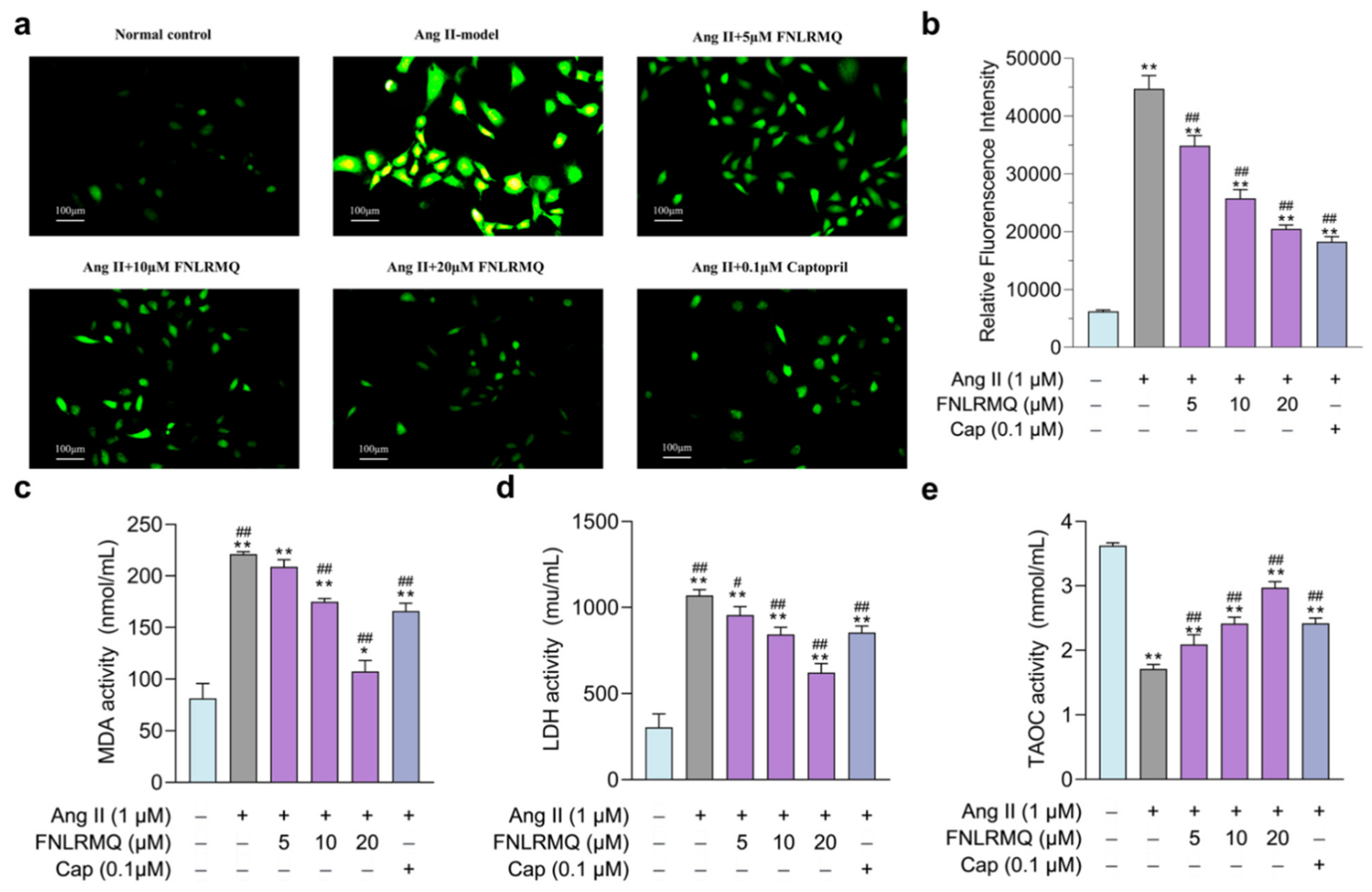
2.4. Protective Effects of FNLRMQ on Oxidative Stress in Ang-II-Induced HUVECs
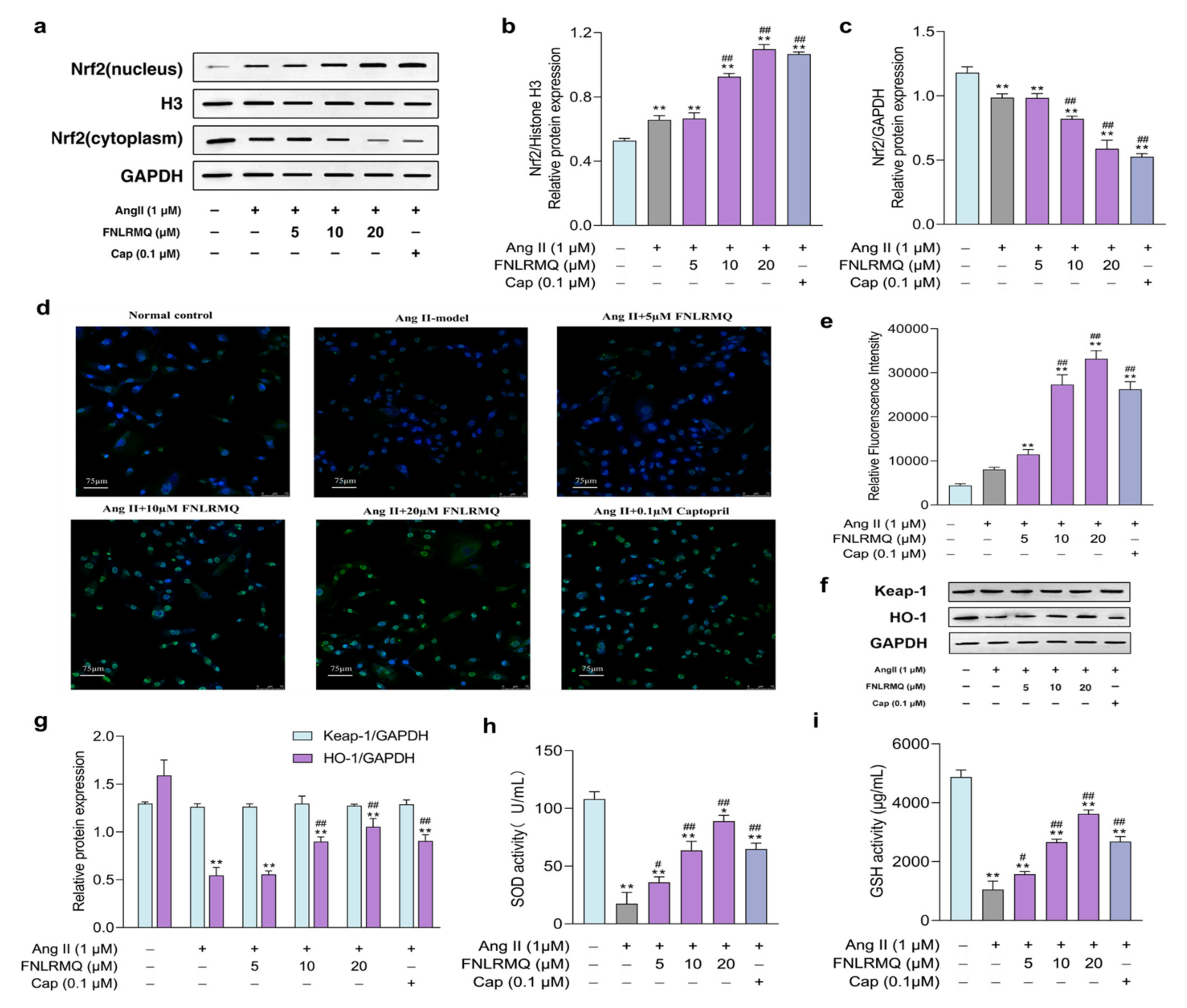
2.5. FNLRMQ Facilitated the Translocation of Nrf2 and Activated the Nrf2/HO-1 Pathway
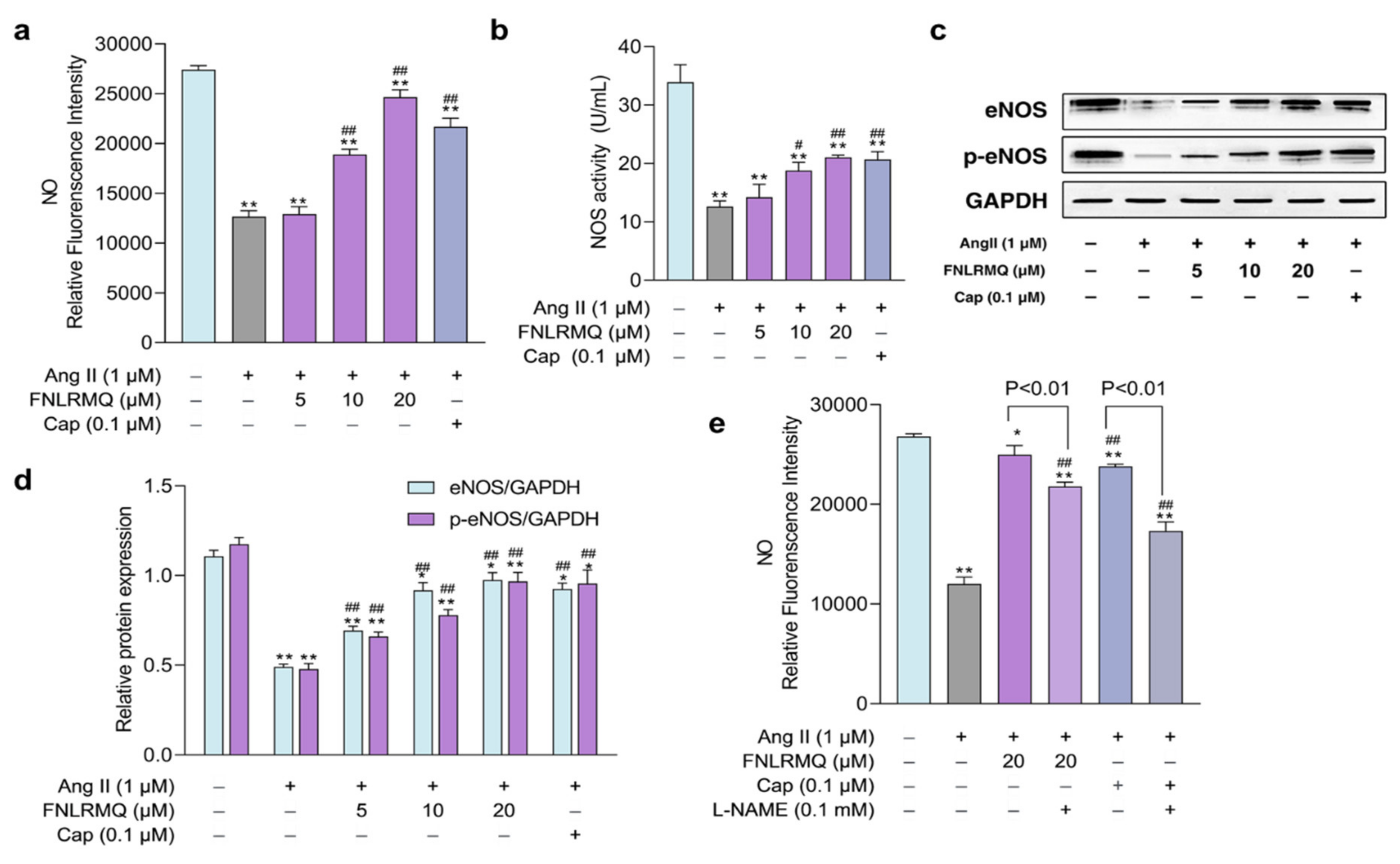
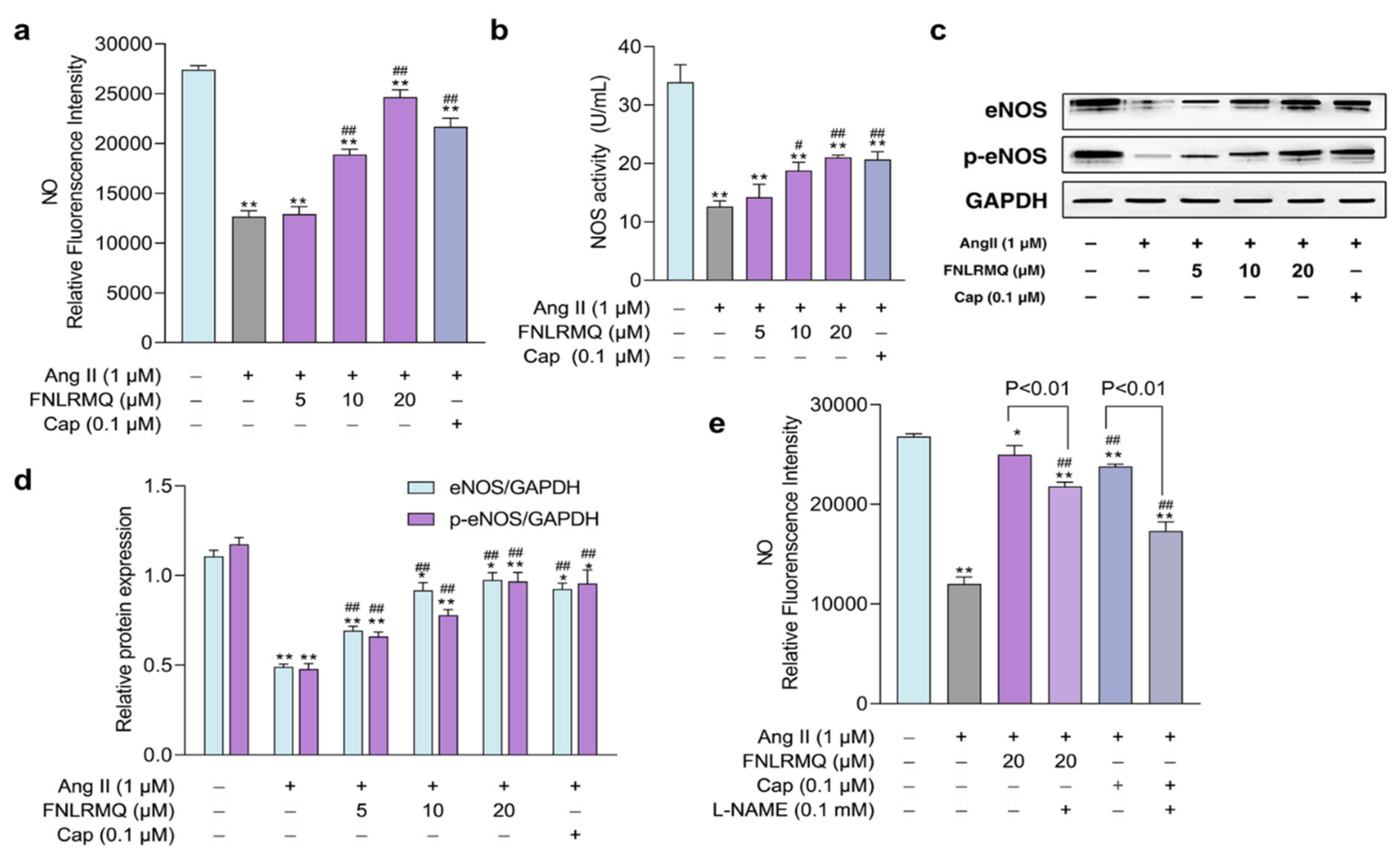
2.6. The Effects of FNLRMQ on the Production of NO and NOS
2.7. The Role of the eNOS in Response to Ang-II-Induced Oxidative Stress
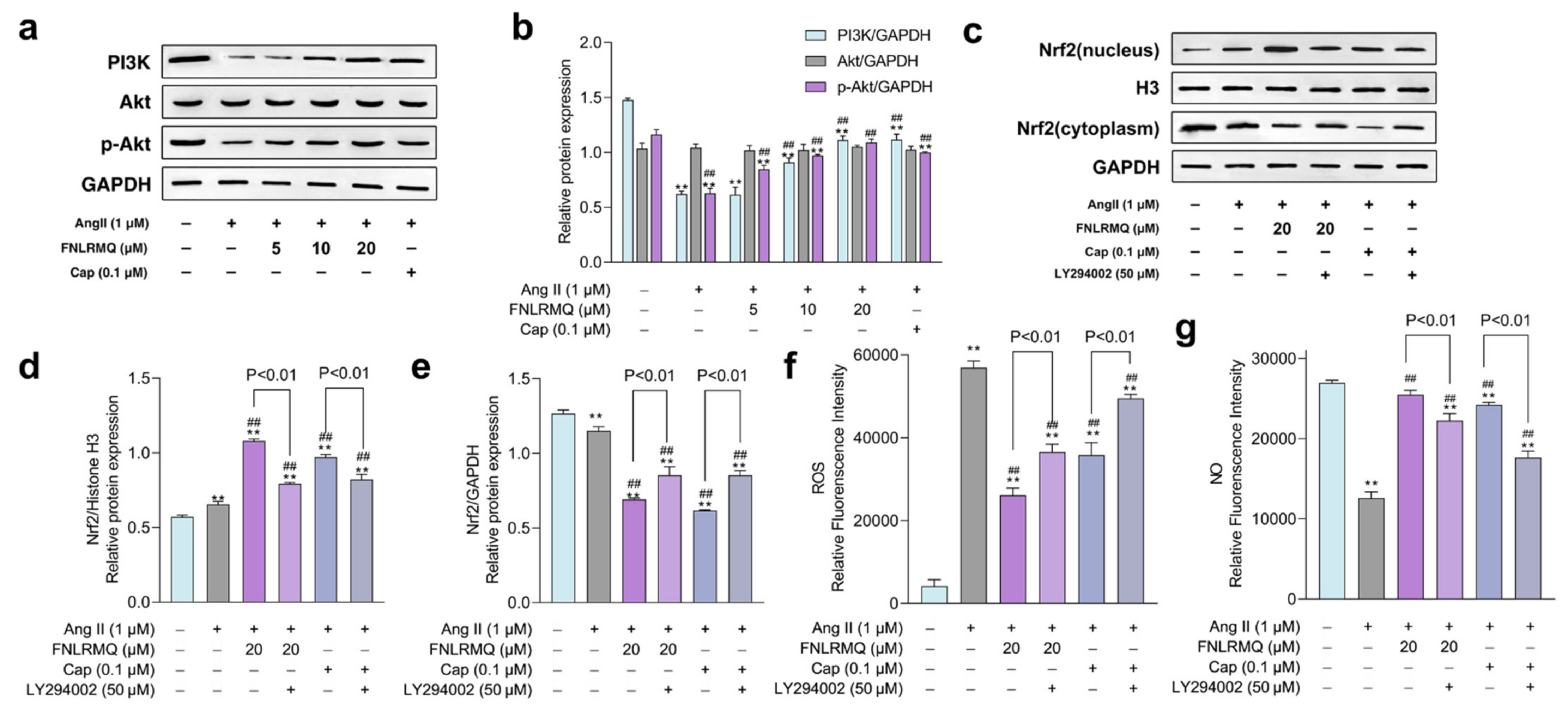
2.8. FNLRMQ Attenuated Ang-II-Induced HUVECs Dysfunction via PI3K/Akt Pathway
3. Materials and Methods
3.1. Materials and Reagents
3.2. Production of Collagen Hydrolysates
3.3. Fractionation by Ultrafiltration
3.4. Screening and Identification of the Potential Bioactive Peptides
3.5. Determination of ACE Inhibitory activity
3.6. Cell Culture
3.7. Cell Viability Assessment
3.8. Apoptosis Analysis
3.9. Detection of ROS and NO
3.10. Biochemical Indicator Detection
3.11. Subcellular Fractionation
3.12. Western Blotting Analysis
3.13. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Dharmashankar, K.; Widlansky, M.E. Vascular endothelial function and hypertension: Insights and directions. Curr. Hypertens. Rep. 2010, 12, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Schulz, E.; Gori, T.; Münzel, T. Oxidative stress and endothelial dysfunction in hypertension. Hypertens. Res. 2011, 34, 665–673. [Google Scholar] [CrossRef]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fekete, A.A.; Givens, D.I.; Lovegrove, J.A. The impact of milk proteins and peptides on blood pressure and vascular function: A review of evidence from human intervention studies. Nutr. Res. Rev. 2013, 26, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Munzel, T.; Sinning, C.; Post, F.; Warnholtz, A.; Schulz, E. Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann. Med. 2008, 40, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Thuillez, C.; Richard, V. Targeting endothelial dysfunction in hypertensive subjects. J. Hum. Hypertens. 2005, 19, S21–S25. [Google Scholar] [CrossRef] [PubMed]
- Versari, D.; Daghini, E.; Virdis, A.; Ghiadoni, L.; Taddei, S. Endothelial Dysfunction as a Target for Prevention of Cardiovascular Disease. Diabetes Care 2009, 32, S314–S321. [Google Scholar] [CrossRef] [Green Version]
- Park, K.H.; Park, W.J. Endothelial dysfunction: Clinical implications in cardiovascular disease and therapeutic approaches. J. Korean Med. Sci. 2015, 30, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Lu, Y.R.; Chen, Y.N.; Cheng, J.Q. The role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 2015, 225, R83–R99. [Google Scholar] [CrossRef] [Green Version]
- Dimmeler, S.; Rippmann, V.; Weiland, U.; Haendeler, J.; Zeiher, A.M. Angiotensin II induces apoptosis of human endothelial cells—Protective effect of nitric oxide. Circ. Res. 1997, 81, 970–976. [Google Scholar] [CrossRef]
- Filippatos, G.; Tilak, M.; Pinillos, H.; Uhal, B.D. Regulation of apoptosis by angiotensin II in the heart and lungs (review). Int. J. Mol. Med. 2001, 7, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Q.; Wang, J.J.; Liang, J.Q.; Feng, D.; Deng, F.; Yang, Y.; Lu, Y.; Hu, Z. Propofol prevents human umbilical vein endothelial cell injury from Ang II-induced apoptosis by activating the ACE2-(1-7)-Mas axis and eNOS phosphorylation. PLoS ONE 2018, 13, e0199373. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.X.; Yang, J.J.; Yang, T.; Xue, Y.L.; Liu, J.; Li, Y.J.; Zhang, D.D.; Xu, J.W.; Bian, K. Alpha-asarone protects endothelial cells from injury by angiotensin II. Evid. Based Complement. Altern. Med. 2014, 2014, 682041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Xu, Y.; Jiang, L. Sulforaphane attenuates angiotensin II-induced human umbilical vein endothelial cell injury by modulating ROS-mediated mitochondrial signaling. Hum. Exp. Toxicol. 2020, 39, 734–747. [Google Scholar] [CrossRef]
- Enseleit, F.; Luscher, T.F.; Ruschitzka, F. Angiotensin-converting enzyme inhibition and endothelial dysfunction: Focus on ramipril. Eur. Heart J. 2003, 5, A31–A36. [Google Scholar] [CrossRef]
- Bots, M.L.; Remme, W.J.; Luscher, T.F.; Fox, K.M.; Bertrand, M.; Ferrari, R.; Simoons, M.L.; Grobbee, D.E.; Investigators, E.-P. ACE inhibition and endothelial function: Main findings of PERFECT, a sub-study of the EUROPA trial. Cardiovasc. Drugs Ther. 2007, 21, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Tiefenbacher, C.P.; Friedrich, S.; Bleeke, T.; Vahl, C.; Chen, X.B.; Niroomand, F. ACE inhibitors and statins acutely improve endothelial dysfunction of human coronary arterioles. Am. J. Physiol. Heart Circ. 2004, 286, H1425–H1432. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kim, H.; Kim, Y.H.; Chung, W.-S.; Suh, J.K.; Kim, S.J. Antioxidant effect of captopril and enalapril on reactive oxygen species-induced endothelial dysfunction in the rabbit abdominal aorta. Korean J. Thorac. Cardiovasc. Surg. 2013, 46, 14–21. [Google Scholar] [CrossRef]
- Sica, D.A. Angiotensin-converting enzyme inhibitors side effects--physiologic and non-physiologic considerations. J. Clin. Hypertens. 2004, 6, 410–416. [Google Scholar] [CrossRef]
- Ishida, Y.; Shibata, Y.; Fukuhara, I.; Yano, Y.; Takehara, I.; Kaneko, K. Effect of an excess intake of casein hydrolysate containing Val-Pro-Pro and Ile-Pro-Pro in subjects with normal blood pressure, high-normal blood pressure, or mild hypertension. Biosci. Biotechnol. Biochem. 2011, 75, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Gogineni, V.; Hamann, M.T. Marine natural product peptides with therapeutic potential: Chemistry, biosynthesis, and pharmacology. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 81–196. [Google Scholar] [CrossRef]
- Ovchinnikova, T.V. Structure, Function, and therapeutic potential of marine bioactive peptides. Mar. Drugs 2019, 17, 505. [Google Scholar] [CrossRef] [Green Version]
- Pujiastuti, D.Y.; Amin, M.N.G.; Alamsjah, M.A.; Hsu, J.L. Marine organisms as potential sources of bioactive peptides that inhibit the activity of angiotensin i-converting enzyme: A review. Molecules 2019, 24, 2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuine, R.; Rathnayake, A.U.; Byun, H.-G. Biological activity of peptides purified from fish skin hydrolysates. Fish. Aquat. Sci. 2019, 22, 10. [Google Scholar] [CrossRef] [Green Version]
- Suleria, H.A.R.; Gobe, G.; Masci, P.; Osborne, S.A. Marine bioactive compounds and health promoting perspectives; innovation pathways for drug discovery. Trends Food Sci. Technol. 2016, 50, 44–55. [Google Scholar] [CrossRef]
- Li, N.; Shen, Q.; Wang, J.H.; Han, C.H.; Ji, R.; Li, F.Q.; Jiang, T. Tetrodotoxin detection and species identification of pufferfish in retail roasted fish fillet by DNA barcoding in China. Food Addit. Contam. Part A 2015, 32, 2148–2153. [Google Scholar] [CrossRef]
- Ahmed, M.; Verma, A.K.; Patel, R. Collagen extraction and recent biological activities of collagen peptides derived from sea-food waste: A review. Sustain. Chem. Pharm. 2020, 18, 100315. [Google Scholar] [CrossRef]
- Ji, Y.; Liu, Y.; Gong, Q.L.; Zhou, L.; Wang, Z.P. Toxicity of cultured puffer fish and seasonal variations in China. Aquac. Res. 2011, 42, 1186–1195. [Google Scholar] [CrossRef]
- Shi, Y.; Zhou, Z.X.; Liu, B.; Kong, S.N.; Chen, B.H.; Bai, H.Q.; Li, L.B.; Pu, F.; Xu, P. Construction of a high-density genetic linkage map and qtl mapping for growth-related traits in Takifugu bimaculatus. Mar. Biotechnol. 2020, 22, 130–144. [Google Scholar] [CrossRef]
- Sheng, Y.Z.; Sun, Y.L.; Zhang, X.; Wan, H.F.; Yao, C.J.; Liang, K.Y.; Li, L.B.; Liu, B.; Zhong, J.X.; Zhang, Z.P.; et al. Characterization of two myostatin genes in pufferfish Takifugu bimaculatus: Sequence, genomic structure, and expression. PeerJ 2020, 8, e9655. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Ngo, D.-H.; Vo, T.-S. Marine fish-derived bioactive peptides as potential antihypertensive agents. Adv. Food Nutr. Res. 2012, 65, 249–260. [Google Scholar] [PubMed]
- Aluko, R.E. Antihypertensive peptides from food proteins. Annu. Rev. Food Sci. Technol. 2015, 6, 235–262. [Google Scholar] [CrossRef]
- Momose, N.; Fukuo, K.; Morimoto, S.; Ogihara, T. Captopril inhibits endothelin-1 secretion from endothelial cells through bradykinin. Hypertension 1993, 21, 921–924. [Google Scholar] [CrossRef] [Green Version]
- Barton, M.; Haudenschild, C.C. Endothelium and atherogenesis: Endothelial therapy revisited. J. Cardiovasc. Pharmacol. 2001, 38, S23–S25. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Barker, T.A.; Berk, B.C. Angiotensin II and the endothelium-diverse signals and effects. Hypertension 2005, 45, 163–169. [Google Scholar] [CrossRef]
- Wen, H.; Gwathmey, J.K.; Xie, L.-H. Oxidative stress-mediated effects of angiotensin II in the cardiovascular system. World J. Hypertens. 2012, 2, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, D.; Nihei, J.; Cardillo, F.; Singh, R. The ACE inhibitors enalapril and captopril modulate cytokine responses in Balb/c and C57Bl/6 normal mice and increase CD4(+)CD103(+)CD25(negative) splenic T cell numbers. Cell. Immunol. 2010, 260, 92–97. [Google Scholar] [CrossRef]
- Shi, X.K.; Guan, Y.H.; Jiang, S.Y.; Li, T.D.; Sun, B.; Cheng, H. Renin-angiotensin system inhibitor attenuates oxidative stress induced human coronary artery endothelial cell dysfunction via the PI3K/AKT/mTOR pathway. Arch. Med. Sci. 2019, 15, 152–164. [Google Scholar] [CrossRef]
- Yu, W.; Akishita, M.; Xi, H.; Nagai, K.; Sudoh, N.; Hasegawa, H.; Kozaki, K.; Toba, K. Angiotensin converting enzyme inhibitor attenuates oxidative stress-induced endothelial cell apoptosis via p38 MAP kinase inhibition. Clin. Chim. Acta 2006, 364, 328–334. [Google Scholar] [CrossRef]
- Chen, J.L.; Tan, L.; Li, C.Y.; Zhou, C.X.; Hong, P.Z.; Sun, S.L.; Qian, Z.J. Mechanism analysis of a novel angiotensin-I-converting enzyme inhibitory peptide from isochrysis zhanjiangensis microalgae for suppressing vascular injury in human umbilical vein endothelial cells. J. Agric. Food Chem. 2020, 68, 4411–4423. [Google Scholar] [CrossRef]
- Khodapasand, E.; Jafarzadeh, N.; Farrokhi, F.; Kamalidehghan, B.; Houshmand, M. Is Bax/Bcl-2 ratio considered as a prognostic marker with age and tumor location in colorectal cancer? Iran. Biomed. J. 2015, 19, 69–75. [Google Scholar] [PubMed]
- Shao, W.J.; Yu, Z.W.; Chiang, Y.T.; Yang, Y.; Chai, T.Y.; Foltz, W.; Lu, H.G.; Fantus, I.G.; Jin, T.R. Curcumin prevents high fat diet induced insulin resistance and obesity via attenuating lipogenesis in liver and inflammatory pathway in adipocytes. PLoS ONE 2012, 7, e28784. [Google Scholar] [CrossRef]
- Sena, C.M.; Leandro, A.; Azul, L.; Seica, R.; Perry, G. Vascular oxidative stress: Impact and therapeutic approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayem, A.A.; Hossain, M.K.; Lee, S.B.; Kim, K.; Saha, S.K.; Yang, G.M.; Choi, H.Y.; Cho, S.G. The role of reactive oxygen species (ROS) in the biological activities of metallic nanoparticles. Int. J. Mol. Sci. 2017, 18, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nita, M.; Grzybowski, A. The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxidative Med. Cell. 2016, 2016, 3164734. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.S.; Ren, J.H.; Lu, J.P.; Fan, Y. Atorvastatin protects against angiotensin II-induced injury and dysfunction in human umbilical vein endothelial cells through Bradykinin 2 receptors. J. Cardiovasc. Pharmacol. 2010, 56, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gong, F.; Chen, M.F.; Li, C.; Hong, P.; Sun, S.; Zhou, C.; Qian, Z.J. In vitro vascular-protective effects of a tilapia by-product oligopeptide on angiotensin II-induced hypertensive endothelial injury in HUVEC by Nrf2/NF-κB pathways. Mar. Drugs 2019, 17, 431. [Google Scholar] [CrossRef] [Green Version]
- Li, H.G.; Horke, S.; Forstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef]
- Forstermann, U.; Xia, N.; Li, H.G. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.L.; Liang, L.W.; Zhao, Y.; Zhang, H. Epigallocatechin-3-gallate ameliorates angiotensin II-induced oxidative stress and apoptosis in human umbilical vein endothelial cells through the activation of Nrf2/Caspase-3 signaling. J. Vasc. Res. 2017, 54, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Tian, T.; Wang, Y.P.; Li, Z.; Xing, K.; Tian, G. SIRT6 protects vascular endothelial cells from angiotensin II-induced apoptosis and oxidative stress by promoting the activation of Nrf2/ARE signaling. Eur. J. Pharmacol. 2019, 859, 172516. [Google Scholar] [CrossRef] [PubMed]
- da Costa, R.M.; Rodrigues, D.; Pereira, C.A.; Silva, J.F.; Alves, J.V.; Lobato, N.S.; Tostes, R.C. Nrf2 as a potential mediator of cardiovascular risk in metabolic diseases. Front. Pharmacol. 2019, 10, 382. [Google Scholar] [CrossRef] [Green Version]
- Donato, A.J.; Gano, L.B.; Eskurza, I.; Silver, A.E.; Gates, P.E.; Jablonski, K.; Seals, D.R. Vascular endothelial dysfunction with aging: Endothelin-1 and endothelial nitric oxide synthase. Am. J. Physiol. Heart Circ. 2009, 297, H425–H432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourque, S.L.; Davidge, S.T.; Adams, M.A. The interaction between endothelin-1 and nitric oxide in the vasculature: New perspectives. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1288–R1295. [Google Scholar] [CrossRef] [Green Version]
- Tassone, E.J.; Sciacqua, A.; Andreozzi, F.; Presta, I.; Perticone, M.; Carnevale, D.; Casaburo, M.; Hribal, M.L.; Sesti, G.; Perticone, F. Angiotensin (17) counteracts the negative effect of angiotensin II on insulin signalling in HUVECs. Cardiovasc. Res. 2013, 99, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Oak, J.H.; Cai, H. Attenuation of angiotensin II signaling recouples eNOS and inhibits nonendothelial NOX activity in diabetic mice. Diabetes 2007, 56, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Fulton, D.; Gratton, J.P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [CrossRef]
- Lee, J.H.; Parveen, A.; Do, M.H.; Lim, Y.; Shim, S.H.; Kim, S.Y. Lespedeza cuneata protects the endothelial dysfunction via eNOS phosphorylation of PI3K/Akt signaling pathway in HUVECs. Phytomedicine 2018, 48, 1–9. [Google Scholar] [CrossRef]
- Cui, J.S.; Zhuang, S.J.; Qi, S.H.; Li, L.; Zhou, J.W.; Zhang, W.; Zhao, Y.; Qi, N.; Yin, Y.J.; Huang, L. Hydrogen sulfide facilities production of nitric oxide via the Akt/endothelial nitric oxide synthases signaling pathway to protect human umbilical vein endothelial cells from injury by angiotensin II. Mol. Med. Rep. 2017, 16, 6255–6261. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.T.; Stephens, L.R. PI3K signalling in inflammation. Biochim.-Mol. Cell Biol. Lipids 2015, 1851, 882–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Huang, X.; Luo, Z.H.; He, J.J.; Haider, F.; Son, C.; Peng, L.; Chen, T.; Wu, B.L. Hypoxia-activated PI3K/Akt inhibits oxidative stress via the regulation of reactive oxygen species in human dental pulp cells. Oxidative Med. Cell. Longev. 2019, 2019, 6595189. [Google Scholar] [CrossRef]
- Wang, D.Z.; Jin, M.Y.; Zhao, X.Y.; Zhao, T.Y.; Lin, W.; He, Z.L.; Fan, M.J.; Jin, W.; Zhou, J.; Jin, L.W.; et al. FGF1(Delta HBS) ameliorates chronic kidney disease via PI3K/AKT mediated suppression of oxidative stress and inflammation. Cell Death Dis. 2019, 10, 464. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sherratt, P.J.; Huang, H.C.; Yang, C.S.; Pickett, C.B. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element—Degradation of Nrf2 by the 26 S proteasome. J. Biol. Chem. 2003, 278, 4536–4541. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, S.J.; Han, Z.H.; Li, Y.Q.; Xue, J.H.; Gao, D.F.; Wu, X.S.; Wang, C.X. PI3K/AKT signaling pathway plays a role in enhancement of eNOS activity by recombinant human angiotensin converting enzyme 2 in human umbilical vein endothelial cells. Int. J. Clin. Exp. Pathol. 2014, 7, 8112–8117. [Google Scholar]
- Attique, S.A.; Hassan, M.; Usman, M.; Atif, R.M.; Mahboob, S.; Al-Ghanim, K.A.; Bilal, M.; Nawaz, M.Z. A Molecular docking approach to evaluate the pharmacological properties of natural and synthetic treatment candidates for use against hypertension. Int. J. Environ. Res. Public Health 2019, 16, 523. [Google Scholar] [CrossRef] [Green Version]
- Ma, F.F.; Wang, H.; Wei, C.K.; Thakur, K.; Wei, Z.J.; Jiang, L. Three novel ACE inhibitory peptides isolated from ginkgo biloba seeds: Purification, inhibitory kinetic and mechanism. Front. Pharmacol. 2019, 9, 1579. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, S.; Pan, N.; Xu, M.; Su, Y.; Qiao, K.; Chen, B.; Zheng, B.; Xiao, M.; Liu, Z. ACE Inhibitory Peptide from Skin Collagen Hydrolysate of Takifugu bimaculatus as Potential for Protecting HUVECs Injury. Mar. Drugs 2021, 19, 655. https://doi.org/10.3390/md19120655
Cai S, Pan N, Xu M, Su Y, Qiao K, Chen B, Zheng B, Xiao M, Liu Z. ACE Inhibitory Peptide from Skin Collagen Hydrolysate of Takifugu bimaculatus as Potential for Protecting HUVECs Injury. Marine Drugs. 2021; 19(12):655. https://doi.org/10.3390/md19120655
Chicago/Turabian StyleCai, Shuilin, Nan Pan, Min Xu, Yongchang Su, Kun Qiao, Bei Chen, Bingde Zheng, Meitian Xiao, and Zhiyu Liu. 2021. "ACE Inhibitory Peptide from Skin Collagen Hydrolysate of Takifugu bimaculatus as Potential for Protecting HUVECs Injury" Marine Drugs 19, no. 12: 655. https://doi.org/10.3390/md19120655
APA StyleCai, S., Pan, N., Xu, M., Su, Y., Qiao, K., Chen, B., Zheng, B., Xiao, M., & Liu, Z. (2021). ACE Inhibitory Peptide from Skin Collagen Hydrolysate of Takifugu bimaculatus as Potential for Protecting HUVECs Injury. Marine Drugs, 19(12), 655. https://doi.org/10.3390/md19120655