1. Introduction
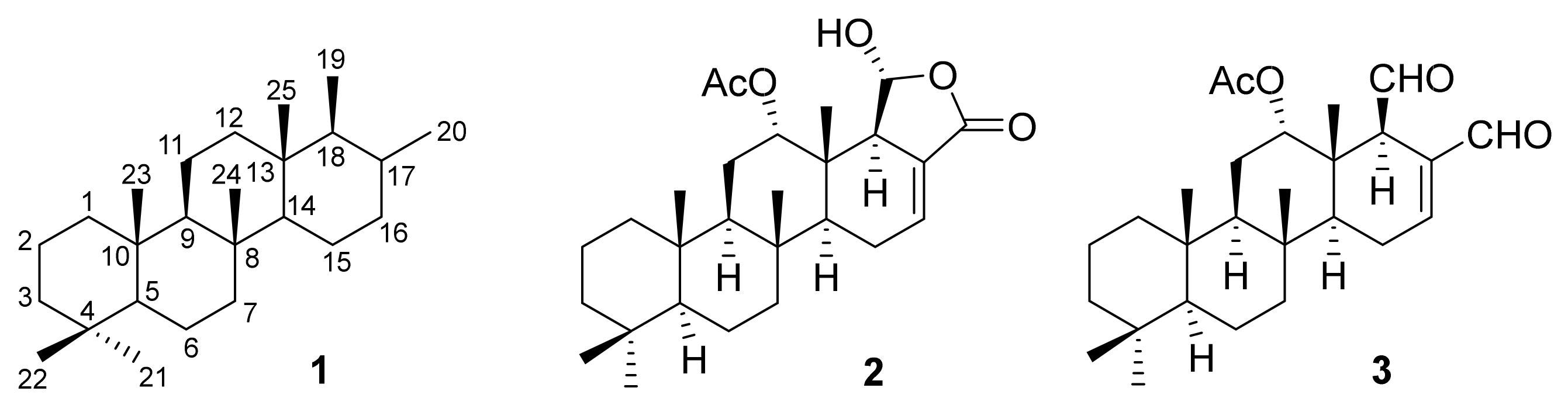
Scalaranic sesterterpenoids are natural products with a tetracyclic carbon skeleton
1 (
Figure 1). The first representatives of this terpenoids subclass were isolated in the beginning of the 1970s. In particular, scalarine (
2) was isolated by Ernesto Fattorusso and collaborators from the see sponges
Cacospongia scalaris [
1], collected in the Mediteranian Sea. Soon after, Guido Cimino and collaborators identified the bioactive sesterterpenoid (-)-scalaradial (
3) in the extract of another sea sponge,
Cacospongia mollior [
2].
Marine organisms such as sponges or mollusks represent the main source for the scalaranic sesterterpenoids’ isolation [
3,
4,
5,
6]. Some recent works report their isolation also from terrestrial plants [
7] and fungi [
8], thus keeping the focus and the scientific interest towards such compounds. The last 5 years witnessed more than 30 high impact publications connected to scalaranes [
9]. This is mainly due to the wide range of their biological activities, including antifeedant, antimicrobial, antifungal, anti-HIV properties, cytotoxicity and anti-inflammatory activity, etc. [
3,
8]. However, a broader investigation of scalaranes in medicinal chemistry studies is still hampered by their relative scarcity in natural sources and a lot of efforts have been put on the elaboration of pathways for their target synthesis [
10]. The structural complexity of the scalarane architecture is connected to their polycyclic backbone, stereochemical issues and specific oxygenations. While the first two challenges have been addressed successfully in several synthetic strategies, the introduction of oxygenated functional groups, especially in the C-12 position of the tetracyclic system still represents a relevant synthetic hurdle. Only few works on the synthesis of cycle B- [
11] and C-functionalized [
12,
13,
14,
15,
16] scalaranes have been reported since 2004. In particular, previous successful reports on the synthesis of C-12—functionalized scalaranes make use of
ent-isocopalic compounds and assemble the D-cycle via a Diels-Alder cycloaddition approach [
12,
13,
14,
15] or employing an intramolecular Heck reaction of tricyclic cheilanthanes [
16]. The most successful example [
15] demonstrates the synthesis of the C-12 functionalized scalaranic framework over 18 synthetic steps with an 4.5% overall yield. We present in the current paper an alternative synthetic rout towards the scalaranes functionalized at the C-12 position.
2. Results and Discussion
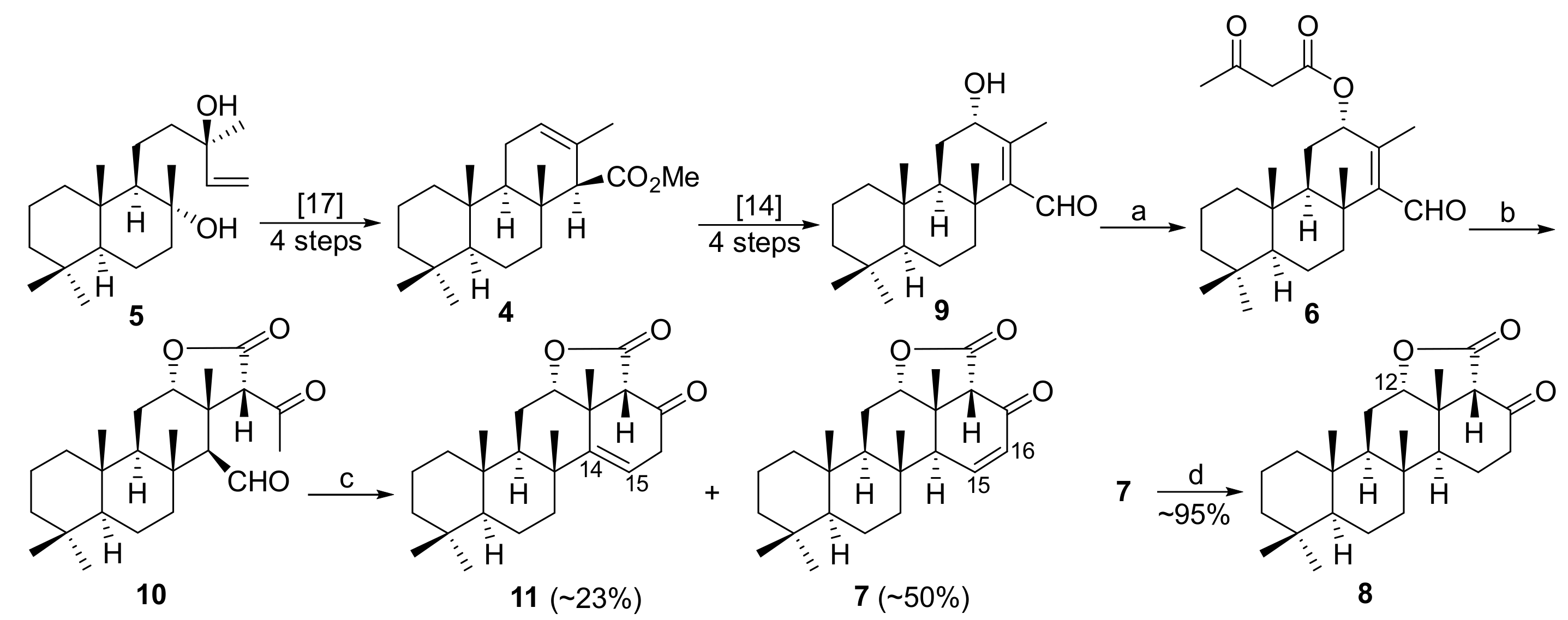
In order to elaborate an alternative strategic approach for the synthesis of a C-12-functionalized tetracyclic framework, we addressed a synthetic pathway basing on the readily available methyl
ent-isocopalate (
4) as a convenient chiral building block which can be prepared easily from the commercial (-)-sclareol (
5). It can be further oxygenated at the C-12 position and homologated with a C-4 fragment in the form of the acetoacetate ester
6 as a pre-requisite of an intramolecular sequence of a Michael–aldol reactions, leading to the closure of the D-cycle in lactone
7 with the required
trans-stereochemistry (
Scheme 1).
The lactone 7 is a valuable intermediate to access highly functionalized scalaranes on flexible manipulation of its functional groups. In our hands, the hydrogenation of the double bond delivered the 17-oxo-20-norscalaran-12α,19-O-lactone (8).
Implementation of the planned synthetic strategy was straightforward (
Scheme 2). The isocopalic hydroxyaldehyde (
9) obtained by a known sequence of transformations from
5 via
4 [
14,
17] was esterified with diketene under mild conditions in dichloromethane, according to the method [
18].
The ester
6 resulted in a good yield, and due to its instability was submitted to the next step without purification. The Michael reaction was initiated on immediate treatment of crude ester
6 with caesium carbonate in acetonitrile [
19]. The desired lactone
10 was obtained with a good yield (~61% over two steps) and its structure was demonstrated basing on spectral data.
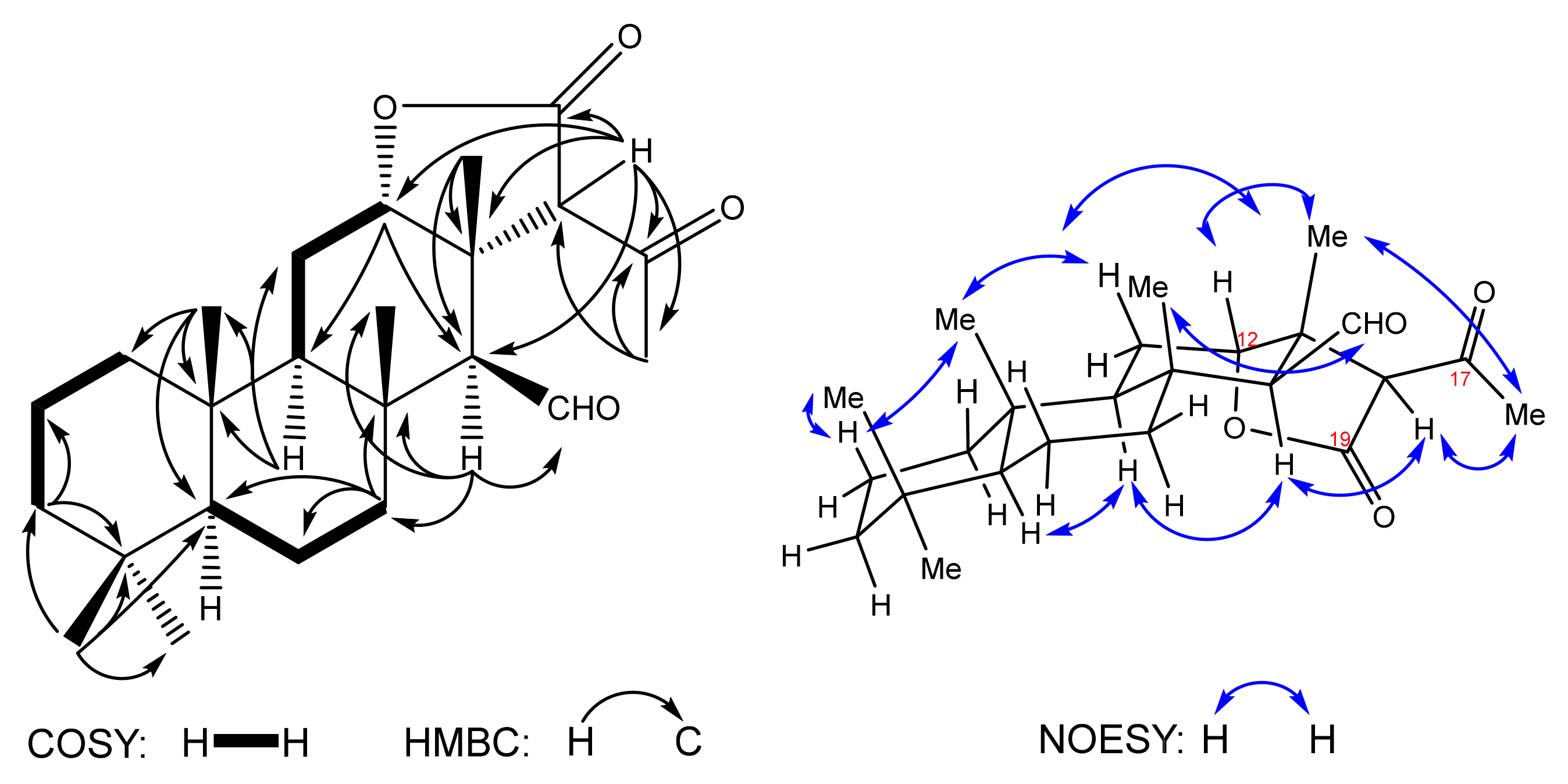
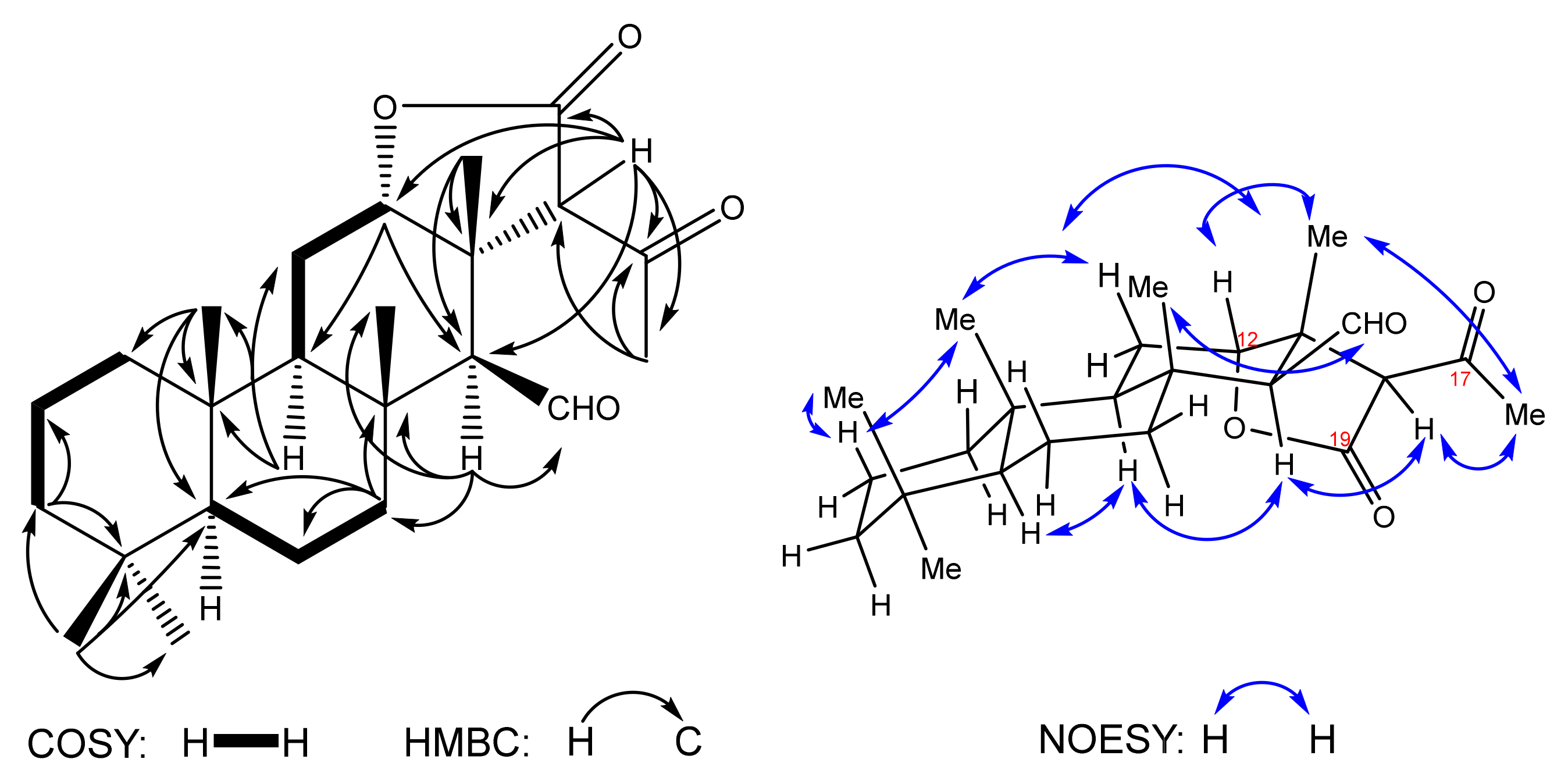
The IR spectrum of compound
10 shows the presence of the aliphatic C-H bonds (2922 cm
−1) and carbonyl groups (1774, 1711 cm
−1). The
13C spectrum shows peaks of 24 carbons: 6 methyl and 6 methine carbons, 6 methylenic carbons, an oxymethine (δ
C 84.4), aldehyde (δ
C 204.6) and 6 quaternary carbons, including two carbonyls (δ
C 172.8, 203.2). Attribution of
13C peaks and assignment of all protons chemical shifts was performed on the basis of 2D HSQC, HMBC and
1H-
1H COSY correlations. In particular,
1H and
13C NMR signals of six methyl groups at δ
H 0.86 (3H-21)/δ
C 33.2 (C-21), 0.82 (3H-22)/21.3 (C-22), 0.86 (3H-23)/15.9 (C-23), 1.21 (3H-24)/19.0 (C-24), 1.22 (3H-25)/15.5 (C-25) have been attributed basing on HMBC correlations, along with the methyl adjacent to the keto group found at δ
H 2.34 (H-16)/δ
C 33.3 (C-16) (
Figure 2). The triplet of the oxymethine proton is detected at δ
H 4.63 (t, 2.9, H-12)/δ
C 84.4 (C-12) and the doublet of the aldehyde proton at δ
H 10.02 (d, 1.4, C-15)/δ
C 204.6 [C-15(CHO)]. The methine protons are confirmed at δ
H 0.93 (m, H-5), 1.28 (m, H-9), 1.86 (bs, H-14) and 3.93 (s, H-18) by HSQC cross peaks with carbons at δ
C 56.2 (C-5), 49.7 (C-9), 65.2 (C-14) and 66.5 (C-18), respectively. HMBC correlations H-18→C-12, C-13, C-14 (
Figure 2) confirm the formation of the new bond after the Michael reaction leading to the α-lactone cycle and the pendant methyl ketone.
The relative stereochemistry was established on the basis of the NOESY spectrum (
Figure 2). The configuration of the 12β-H proton which corresponds to the starting substrate
6 was confirmed by H-12↔H3-25 correlations. The β-orientation of the aldehyde group is proven by correlations H-14↔H-9 and H-15(CHO)↔H3-24.
The intramolecular aldol reaction of ketoaldehyde 10 was triggered upon treatment with PTSA. The cyclization occurred with a good yield and selectivity; the desired unsaturated ketolactone 7 predominated over its isomer 11, which was formed as a result of double bond migration under acidic reaction conditions. Such isomerizations are known in aldol-related cyclizations; we did not make any attempts to optimize this transformation.
The IR spectrum of compound
7 shows the presence of the aliphatic C-H bonds (2920, 2865 cm
−1) and carbonyl group (1760 cm
−1). The structure of compound
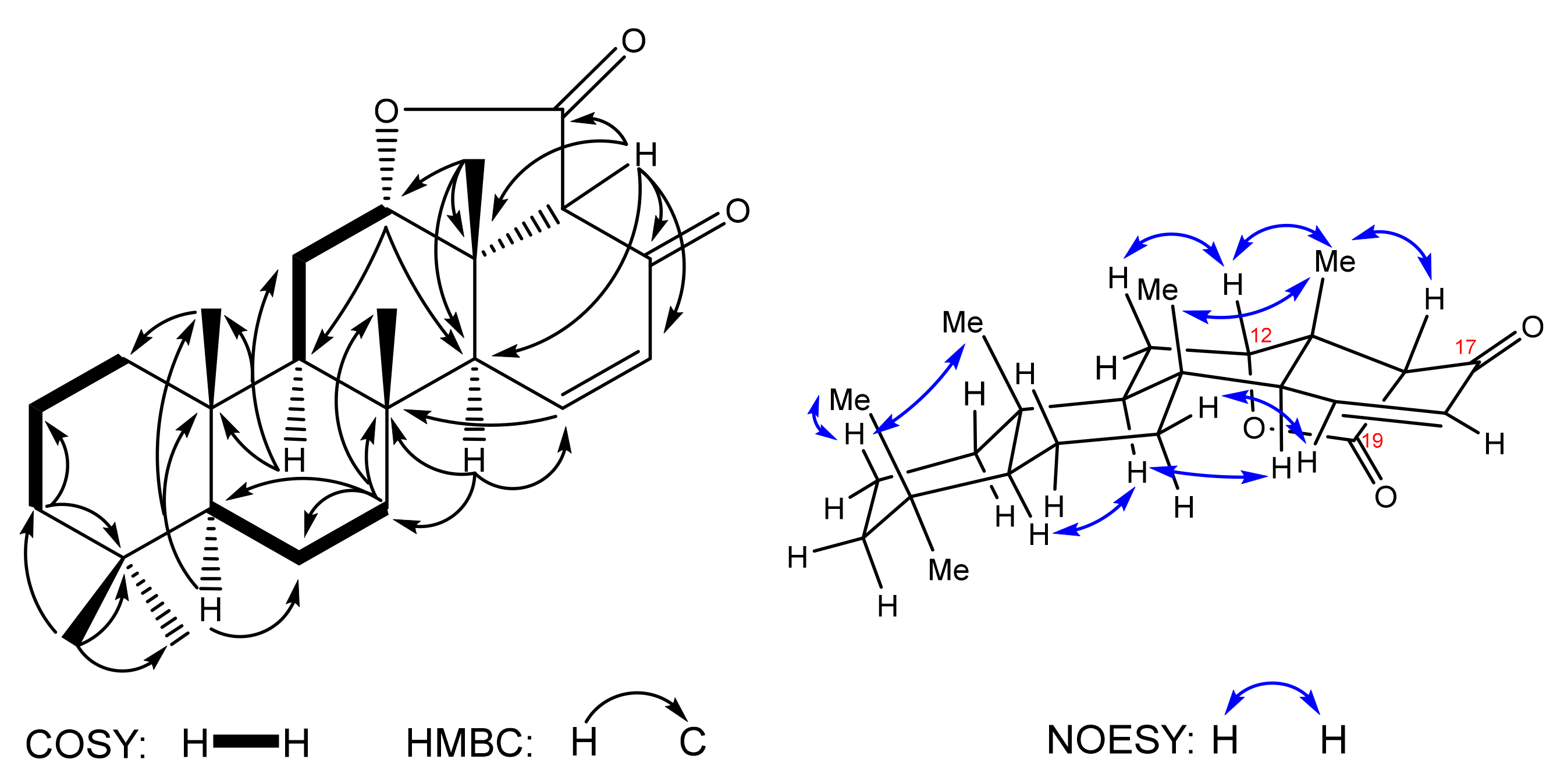
7 was elucidated on the basis of NMR spectral data, in particular of 2D HSQC, HMBC and
1H-
1H COSY correlations (
Figure 3).
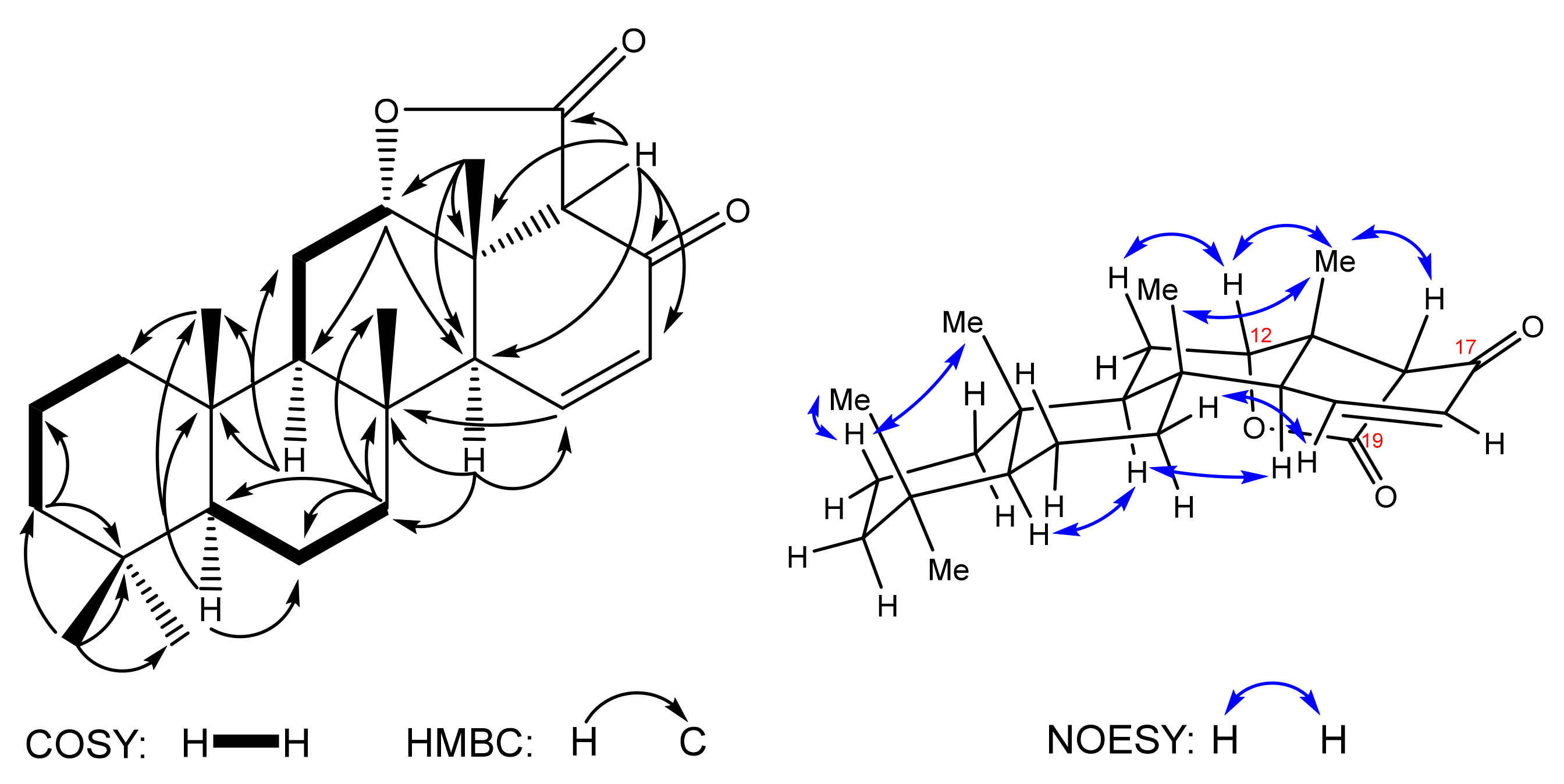
The 1H and 13C NMR show neither aldehyde group nor methyl ketone specific signals, whereas a double bond is clearly detected (δC 129.9, 149.5). In this line, the 1H-1H COSY cross peaks corresponding to H-15↔H-16↔H-17 correlations show convincingly the D-ring closure as a result of the intramolecular aldol reaction in the substrate 8. On the basis of 13C and HSQC spectra, the carbon backbone of compound 7 is revealed to include 24 carbon atoms: 5 methyl, 6 methylene groups and 7 methine groups, 6 quaternary carbons, including two carbonyls (δC 169.7, 188.6). Attribution of 13C peaks and assignment of all protons chemical shifts resulted in five methyls at δH 0.85 (3H-21)/δC 33.2 (C-21), 0.82 (3H-22)/21.3 (C-22), 0.86 (3H-23)/15.6 (C-23), 0.99 (3H-24)/18.0 (C-24) and 1.27 (3H-25)/18.4 (C-25). The methine protons are confirmed at δH 0.89 (m, H-5), 1.32 (m, H-9), 4.38 (t, 2.8, H-12), 2.39 (t, 3, H-14), 3.07 (s, H-18) by HSQC cross peaks with carbons at δC 56.6 (C-5), 52.2 (C-9), 82.8 (C-12), 50.4 (C-14) and 64.9 (C-18), respectively. The protons attached to sp2 carbons are detected at δH 7.09 (dd, 10, 3, H-15)/δC 149.5 (C-15) and 6.09 (dd, 10, 3, H-16)/129.9 (C-16).
The careful examination of 2D NMR confirmed assembling of the pentacyclic system including tetracyclic nor-scalaranic framework condensed with the C-12–C-18 lactone ring and oxygenated at C-17 with the keto group. The relative stereochemistry of lactone
7 was established on the basis of NOESY spectrum (
Figure 3). Correlation H-12↔H3-25↔H-18 clearly shows the α-orientation of the lactone ring, and H-14 α-orientation is proven by H-14↔H-9 correlation.
The spectral data of minor lactone 11 are very much similar to those of major compound 7. The only major difference represents the double bond position in cycle D, which is trisubstituted and placed at C-14–C-15 carbon atoms.
The major pentacyclic ketolactone
7 represents a very useful compound for a flexible generation of a whole array of molecular diversity. Direct short range functionalizations are feasible in cycles C and D, and, evidently, olefination of the C-17 keto group can provide the C-25—scalaranic backbone. In order to finally prove the relative stereochemistry of lactone
7, we performed X-ray analysis of its hydrogenation product
8, which turned out to provide suitable crystals for this investigation. The hydrogenation of
7 went smoothly (95%) after treatment with palladium under hydrogen gas atmosphere. The spectral data of saturated ketolactone
8 have shown a perfect match to its suggested stereochemistry. In particular, 2D NMR experiments confirmed the structural changes of the substrate
7, consisting of the modified chemical shift values for C-15 and C-16 positions to δ
H 1.96–1.63 (m, 2H-15)/δ
C 18.5 (C-15) and 2.58–2.36 (m, 2H-16)/48.8 (C-16). Compound
8 shows a total of 24 carbon atoms, including 6 methyl, 8 methylene and 5 methine groups, and 6 quaternary carbons. Attribution of
13C peaks and assignment of all protons chemical shifts show methyl groups at δ
H 0.85 (3H-21)/δ
C 33.2 (C-21), 0.82 (3H-22)/21.3 (C-22), 0.85 (3H-23)/15.8 (C-23), 0.90 (3H-24)/17.0 (C-24) and 1.25 (3H-25)/18.4 (C-25). The attribution of C-H groups included signals at δ
H 0.89 (m, H-5), 1.34 (m, H-9), 4.29 (t, 2.8, H-12), 1.54 (m, H-14), 3.05 (s, H-18), which correspond to carbon atoms at δ
C 56.4 (C-5), 52.4 (C-9), 84.2 (C-12), 50.4 (C-14) and 67.5 (C-18). NOESY correlations for compound
8 confirm the desired
trans-stereochemistry between newly built cycles of the tetracyclic scalaranic framework (
Figure 4).
The chemical composition and crystal structure of compound
8 were confirmed by single crystal X-ray diffraction. A single crystal of ketolactone
8 was obtained on its crystallization from ethyl acetate-diethyl ether solvent mixture (1:1). According to X-ray crystallography, compound
8 exhibits a molecular crystal structure crystallizing the P212121 Shohnke space group of the orthorhombic system with one neutral entity in the asymmetric part, as shown in
Figure 4. In the crystal, the neutral molecules are interacting through C-H
···O hydrogen bonding to form infinite supramolecular ribbons running along an axis. A detailed report on the X-ray experiment, including one-dimensional architecture and crystal packing, is available as
Supplementary Materials.
3. Materials and Methods
3.1. General Experimental Procedures
Melting points were measured with a Boethius heating stage. Optical rotations: Jasco-DIP-370 polarimeter; 5 cm cell; in CHCl3. IR Spectra: Spectrum-100 FT-IR spectrophotometer (PerkinElmer), with the universal ATR sampling accessory; ν in cm−1. 1H- and 13C-NMR Spectra: Bruker-Avance-III spectrometer (400.13 and 100.61 MHz); in CDCl3; δ in ppm rel. to CHCl3 as internal standard (δH 7.26 and δC 77.0), J in Hz. The carbon and hydrogen content of compounds were determined by standard microanalysis on Vario-EL-III-CHNOS Elemental Analyzer. Commercial Merck silica gel 60 (70–230 mesh ASTM) was used for flash chromatography and Merck pre-coated silica gel plates were used for TLC. The chromatograms were sprayed with 0.1% solution of cerium (IV) sulfate in 2N sulfuric acid, and heated at 80 °C for 5 min to detect the spots. Treatment of reaction mixtures in organic solvents included the extraction by diethyl ether, washing of the extract with water up to neutral reaction, drying over anhydrous Na2SO4, filtering and solvent removal in vacuum.
3.2. Single Crystal X-Ray Diffraction
X-ray diffraction measurements were carried out with a Rigaku Oxford-Diffraction XCALIBUR E CCD diffractometer equipped with graphite-monochromated MoKα radiation. A single crystal was positioned at 40 mm from the detector and 201 frames were measured each for 125 s over 1° scan width. The unit cell determination and data integration were carried out using the CrysAlis package of Oxford Diffraction [
20]. The structures were solved by Intrinsic Phasing using Olex2 [
21] software with the SHELXT [
22] structure solution program, and refined by full-matrix least-squares on
F2 with SHELXL-2015 [
23] using an anisotropic model for non-hydrogen atoms. In the absence of significant anomalous scattering, the absolute configuration of the structures could not be reliably determined. Friedel pairs were merged and any references to the Flack parameter were removed. The H atoms were placed geometrically and constrained to ride on their parent atoms with d
CH = 0.96 Å and Uiso values of 1.2 Ueq of the parent atoms. The crystallographic data and refinement details are quoted in
Table S1, whereas bond lengths and angles are given in
Table S2 (
Supplementary Materials available).
3.3. 12α-Hydroxy-ent-isocopal-13,14-en-15-al (9)
Compound
9 was obtained according to the described method [
14]. 12α-Hydroxy-ent-isocopal-13,14-en-15-al (
9) was obtained as a white crystalline solid. Mp: 123–125 °C; (Lit. [
13] Mp: 134–135 °C);
–69.8 (c 0.31, CHCl
3). IR (υ, cm
−1): 3388, 2869, 1678, 1456, 1379, 1042, 733.
1H (400.13 MHz, CDCl
3) δ
H: 0.81 (3H, s, H-19), 0.83 (3H, s, H-18), 0.87 (3H, s, H-20), 1.16 (3H, s, H-17), 2.12 (3H, s, H-16), 4.04 (1H, dd,
J = 4.5, 1.2 Hz, H-12), 10.08 (1H, s, CHO).
13C (100.61 MHz, CDCl
3) δ
C: 16.5 (q, C-20), 16.8 (q, C-16), 18.4 (t, C-2), 18.5 (t, C-6), 19.8 (q, C-17), 21.2 (q, C-19), 27.1 (t, C-11), 33.2 (q, C-18), 33.2 (s, C-4), 37.0 (s, C-10), 37.6 (t, C-7), 38.9 (s, C-8), 39.6 (t, C-1), 42.0 (t, C-3), 50.3 (d, C-9), 56.5 (d, C-5), 70.8 (d, C-12), 145.1 (d, C-14), 148.1 (s, C-13), 194.3 (s, C-15).
3.4. Synthesis of Compound 6
Et3N (80 μL, 0.57 mmol) and diketene (45 μL, 0.57 mmol) were added to a solution of hydroxyaldehyde 9 (117 mg, 0.38 mmol) and benzene (2 mL) in the inert atmosphere. The reaction mixture was stirred for 30 min at 0 °C and 2 h at room temperature. After the usual work-up, the extract was dried and filtered. The solvent was removed under reduced pressure and the residue (~156 mg) of compound 6 was obtained, pale yellow viscous oil. Because the substance 6 is unstable, it was used in the next step without any purification. 1H (400.13 MHz, CDCl3) δH: 1.17 (3H, s, Me-24), 1.97 (3H, s, Me-25), 2.27 (3H, s, Me-16), 3.51 (2H, s, H-18), 5.31 (1H, bd, J = 3.9 Hz, H-12), 10.08 [1H, s, C-15(CHO)]; 13C (100.61 MHz, CDCl3) δC: 16.2 (q, C-23), 16.2 (q, C-25), 18.4 (t, C-2), 18.4 (t, C-6), 19.9 (q, C-24), 21.2 (q, C-22), 33.2 (s, C-4), 36.9 (s, C-10), 37.5 (t, C-7), 38.5 (s, C-8), 39.4 (t, C-1), 41.9 (t, C-3), 50.3 (t, C-18), 50.5 (d, C-9), 56.2 (d, C-5), 73.8 (d, C-12), 143.0 (s, C-14), 147.39 (s, C-13), 166.7 (s, C-19), 193.9 (s, C-15), 200.0 (s, C-17).
3.5. Synthesis of Compound 10
To a solution of compound 6 (140 mg, 0.36 mmol) and anhydrous MeCN, and inert atmosphere, was added anhydrous Cs2CO3 (122 mg, 0.37 mmol). The reaction mixture was stirred for 15 min at room temperature and 2 h at reflux. After the usual work-up, the extract was dried and filtered. The solvent was removed, and the residue (~146 mg) was purified on a silica gel (5 g) column (petroleum ether–ethyl acetate, gradient elution), resulting in compound 10 (85 mg, ~61% over two steps from 9), pale yellow viscous oil. –19.7 (c 0.21, CHCl3). IR (ν, cm−1): 2922, 2870, 1774, 1711, 1390, 1207. 1H (400.13 MHz, CDCl3) δH: 0.82 (3H, s, Me-22), 0.86 (6H, s, Me-21, 23), 1.21 (3H, s, Me-24), 1.24 (3H, s, Me-25), 1.86 (1H, bs, H-14) 2.34 (3H, s, Me-16), 3.93 (1H, s, H-18), 4.63 (1H, t, J = 2.9 Hz, H-12), 10.02 (1H, d, J = 1.4 Hz, CHO). 13C (100.61 MHz, CDCl3) δC: 15.5 (q, C-25), 15.9 (q, C-23), 17.8 (t, C-6), 18.3 (t, C-2), 19.0 (q, C-24), 20.3 (t, C-11), 21.3 (q, C-22), 33.2 (q, C-21), 33.3 (s, C-4), 33.3 (q, C-16), 37.1 (s, C-10), 37.5 (s, C-8), 39.1 (t, C-1), 41.5 (t, C-7), 41.8 (t, C-3), 44.9 (s, C-13), 49.7 (d, C-9), 56.2 (d, C-5), 65.2 (d, C-14), 66.5 (d, C-18), 84.4 (d, C-12), 172.8 (s, C-19), 203.2 (s, C-17), 204.6 (s, C-15). Anal. Calc. for C24H36O4: C 74.19, H 9.49; found: C 74.24, H 9.41.
3.6. Intramolecular Aldol Condensation of Compound 10
To a solution of compound 10 (78 mg, 0.20 mmol) and anhydrous benzene (3 mL), p-TsOH (11 mg, 0.06 mmol) was added, and the reaction mixture was refluxed for 4 h. After the usual work-up, the extract was dried and filtered. The solvent was removed, and the residue (~76 mg) was purified on a silica gel (3.5 g) column (petroleum ether—ethyl acetate, gradient elution), resulting in compound 11 (18 mg, 23%) and compound (7) (39.1 mg, ~50%).
3.6.1. Compound 11
Pale yellow amorphous gum. IR (ν, cm−1): 2922, 2870, 1774, 1711, 1390, 1207. IR (ν, cm−1): 2916, 2872, 1756, 1677, 1142, 1070. 1H (400.13 MHz, CDCl3) δH: 0.83 (3H, s, Me-22), 0.87 (3H, s, Me-21), 0.90 (3H, s, Me-23), 1.14 (3H, s, Me-24), 1.40 (3H, s, Me-25), 3.19 (1H, s, H-18), 4.42 (1H, t, J = 2.8 Hz, H-12), 2.88–2.93 (1H, dd, J = 22.7, 2.7 Hz, H-16), 3.10–3.16 (1H, dd, J = 22.7, 5.2 Hz, H-16), 5.66 (1H, dd, J = 5.1, 2.7 Hz, H-15). 13C (100.61 MHz, CDCl3) δC: 16.1 (q, C-23), 18.5 (t, C-2), 18.5 (t, C-6), 20.7 (t, C-11), 21.5 (q, C-22), 23.2 (q, C-24), 23.6 (q, C-25), 33.3 (s, C-4), 33.3 (q, C-21), 37.5 (s, C-10), 38.2 (t, C-16), 39.4 (t, C-1), 39.6 (s, C-8), 39.9 (t, C-7), 41.8 (t, C-3), 47.6 (s, C-13), 48.4 (d, C-9), 56.2 (d, C-5), 65.1 (d, C-18), 85.1 (d, C-12), 117.6 (d, C-15), 148.2 (s, C-14), 170.3 (s, C-19), 201.7 (s, C-17). Anal. Calc. for C24H34O3: C 77.80, H 9.25; found: C 77.49, H 9.31.
3.6.2. Compound 7
White crystalline solid. Mp: 260–262 °C; –14.6 (c 0.19, CHCl3). IR (ν, cm−1): 2920, 2865, 1760, 1681, 1149, 1077. 1H (400.13 MHz, CDCl3) δH: 0.82 (3H, s, Me-22), 0.85 (3H, s, Me-21), 0.86 (3H, s, Me-23), 0.99 (3H, s, Me-24), 1.27 (3H, s, Me-25), 2.39 (1H, t, J = 3 Hz, H-15), 3.07 (1H, s, H-18), 4.38 (1H, t, J = 2.8 Hz, H-12), 6.09 (1H, dd, J = 10, 3 Hz, H-16), 7.09 (1H, dd, J = 10, 3 Hz, H-15). 13C (100.61 MHz, CDCl3) δC: 15.6 (q, C-23), 17.7 (t, C-6), 18.0 (q, C-24), 18.4 (t, C-2), 18.4 (q, C-25), 20.1 (t, C-11), 21.3 (q, C-22), 33.2 (q, C-21), 33.3 (s, C-4), 35.6 (s, C-8), 37.0 (s, C-10), 39.1 (t, C-1), 40.1 (t, C-7), 41.8 (t, C-3), 48.0 (s, C-13), 50.4 (d, C-14), 52.2 (d, C-9), 56.6 (d, C-5), 64.9 (d, C-18), 82.8 (d, C-12), 129.9 (d, C-16), 149.5 (d, C-15), 169.7 (s, C-19), 188.6 (s, C-17). Anal. Calc. for C24H34O3: C 77.80, H 9.25; found: C 77.58, H 9.38.
3.7. Hydrogenation of Unsaturated Ketone 7
To a solution of compound 7 (30 mg, 0.08 mmol) and EtOAc (4 mL), 10% Pd/C (9.4 mg, 0.009 mmol) was added and was stirred for 15 min. Afterwards, a stream of hydrogen was added to the reaction mixture and was stirred for 24 h at room temperature. The mixture was filtered, and the solvent was removed in vacuum. The residue (29 mg) was purified on a silica gel (1.0 g) column (petroleum ether—ethyl acetate, gradient elution), resulting in compound 8 (28,6 mg, ~95%) as a white crystalline solid. Mp: 279–281 °C; 32.2 (c 0.9, CHCl3). IR (ν, cm−1): 2957, 2923, 2845, 1776, 1712, 1190, 1037. 1H (400.13 MHz, CDCl3) δH: 0.82 (3H, s, Me-22), 0.85 (3H, s, Me-23), 0.86 (3H, s, Me-22), 0.90 (3H, s, Me-24), 1.25 (3H, s, Me-25), 3.05 (1H, s, H-18), 4.29 (1H, t, J = 2.8 Hz, H-12). 13C (100.61 MHz, CDCl3) δC: 15.8 (q, C-23), 17.0 (q, C-24), 18.1 (t, C-6), 18.4 (t, C-2), 18.4 (q, C-25), 18.5 (t, C-15), 20.5 (t, C-11), 21.3 (q, C-22), 33.2 (q, C-21), 33.3 (s, C-4), 36.9 (s, C-8), 37.1 (s, C-10), 39.3 (t, C-1), 40.8 (t, C-7), 40.8 (t, C-16), 41.9 (t, C-3), 44.9 (s, C-13), 50.4 (d, C-14), 52.5 (d, C-9), 56.4 (d, C-5), 67.5 (d, C-18), 84.2 (d, C-12), 171.0 (s, C-19), 202.4 (s, C-17). Anal. Calc. for C24H36O3: C 77.38, H 9.74; found: C 77.46, H 9.68.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}