
Advances in Phenazines over the Past Decade: Review of Their Pharmacological Activities, Mechanisms of Action, Biosynthetic Pathways and Synthetic Strategies

Abstract
:1. Introduction
2. Natural Phenazine Derivatives
2.1. Biological Activity of Known Phenazines
2.2. Natural Phenazines
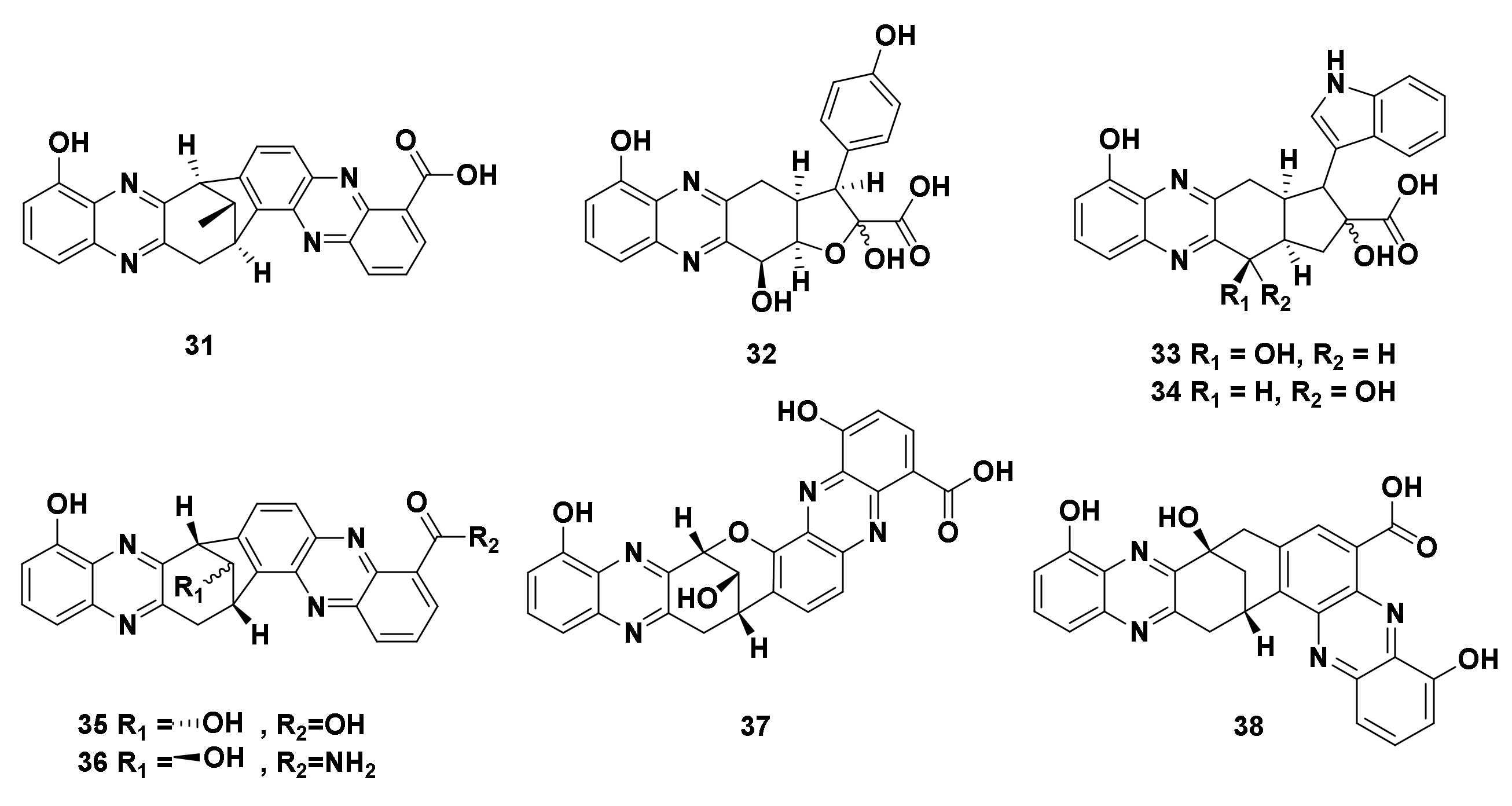
2.2.1. Terpenoid Phenazines
2.2.2. Glycosylated Phenazines
2.2.3. Divergent Fused Phenazines
2.2.4. Biological Activity of New Phenazines
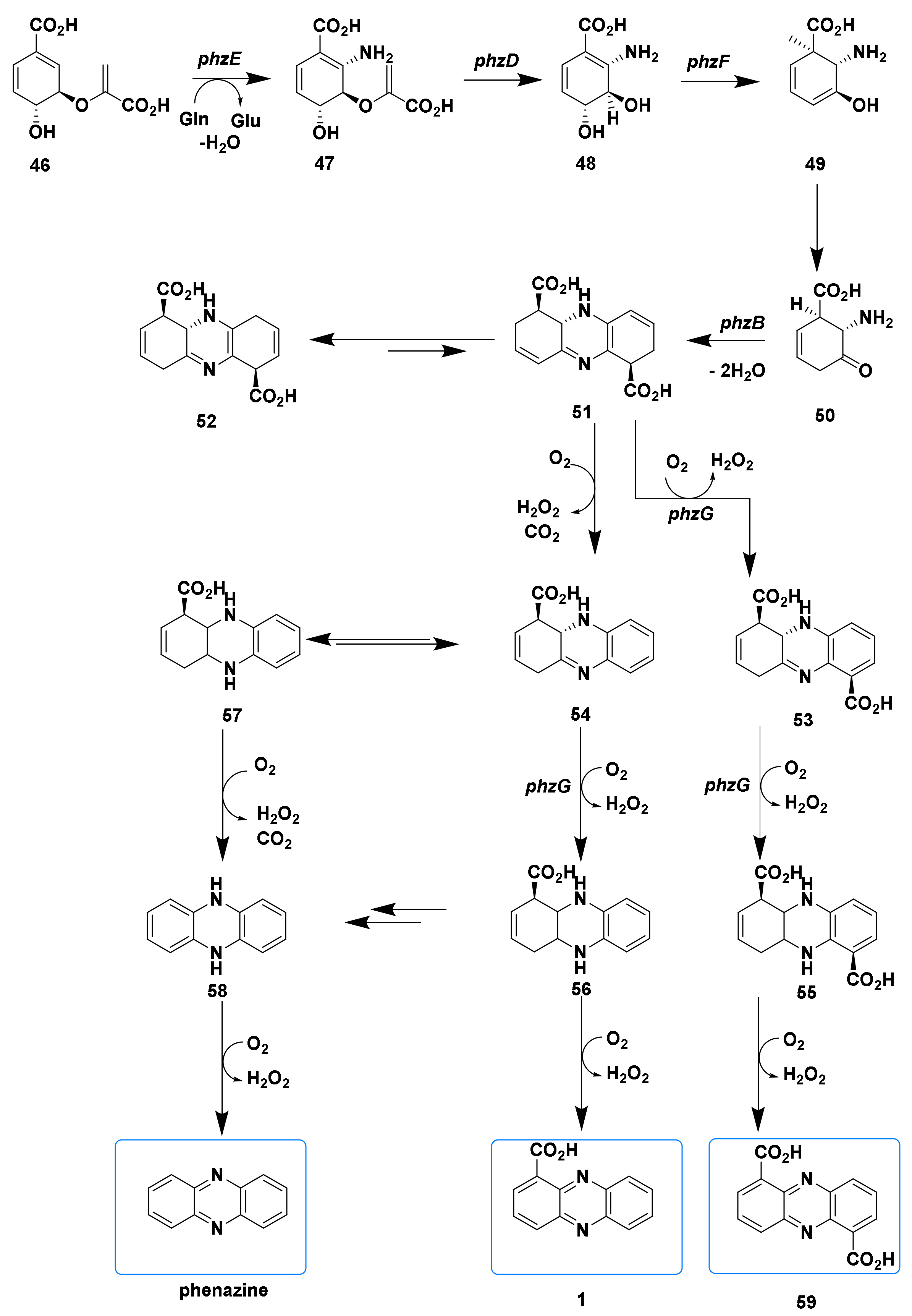
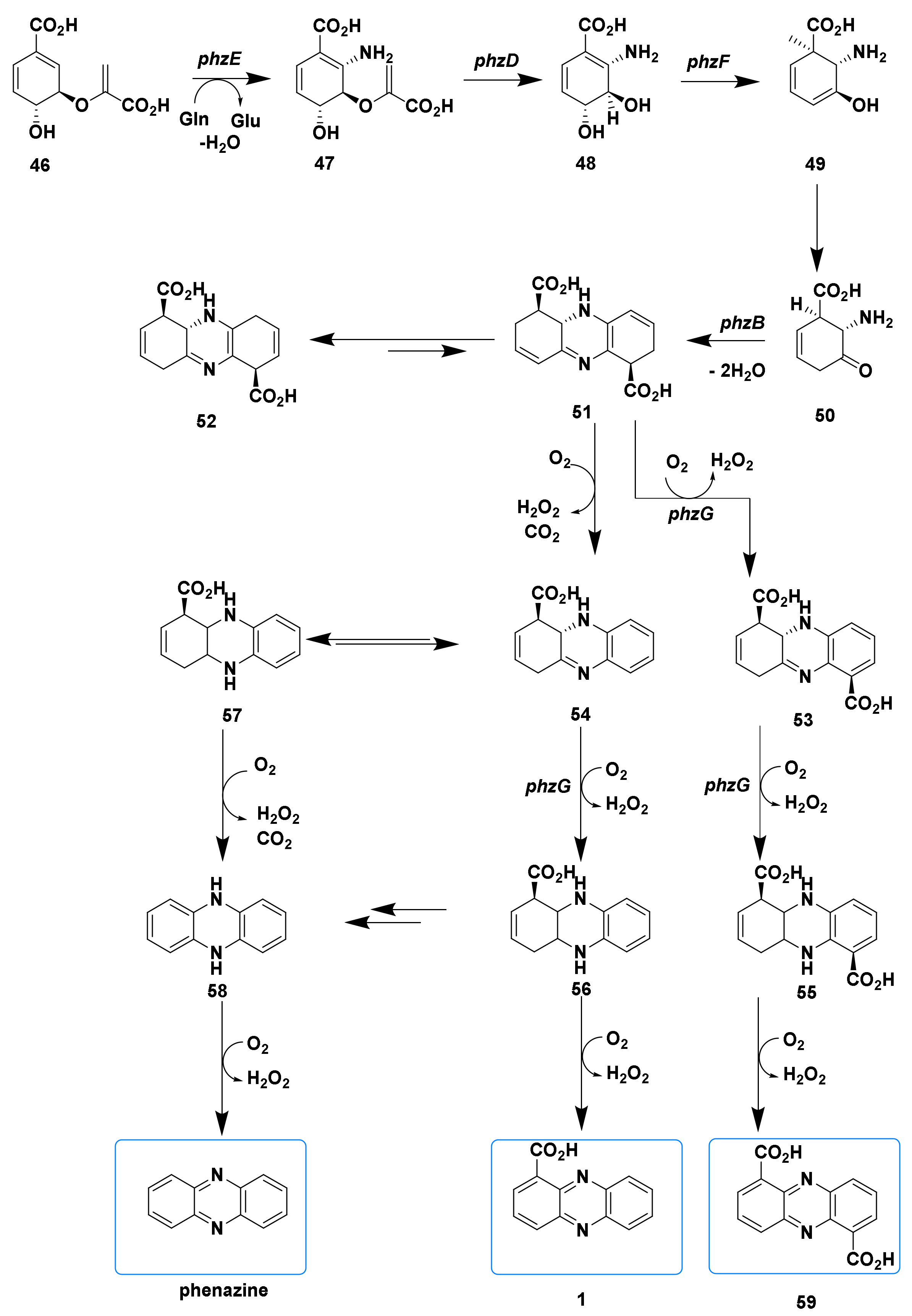
3. The Progresses of Biosynthetic Pathways of Phenazines
4. Synthetic Phenazine Derivatives
4.1. Antimicrobial Activity
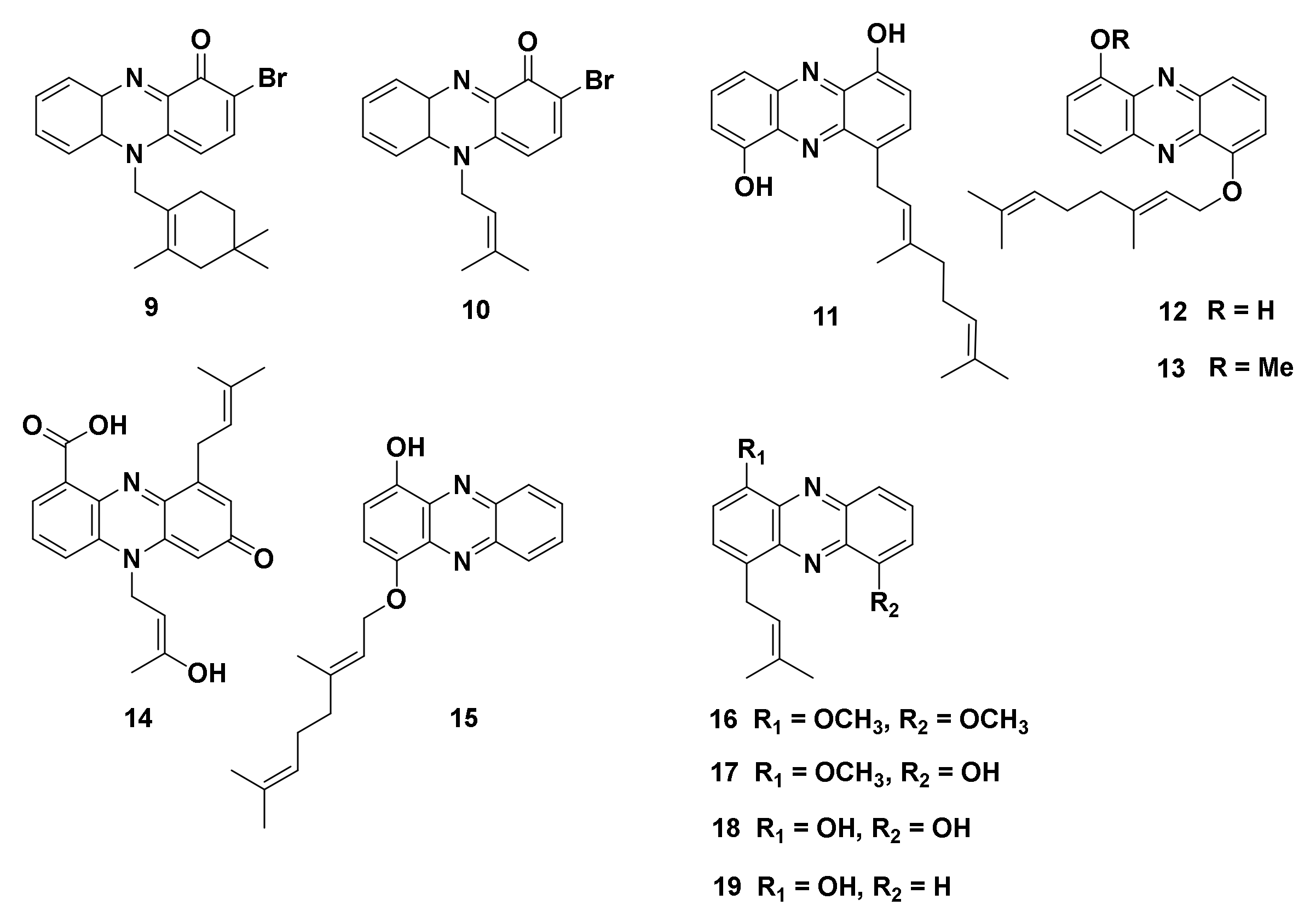
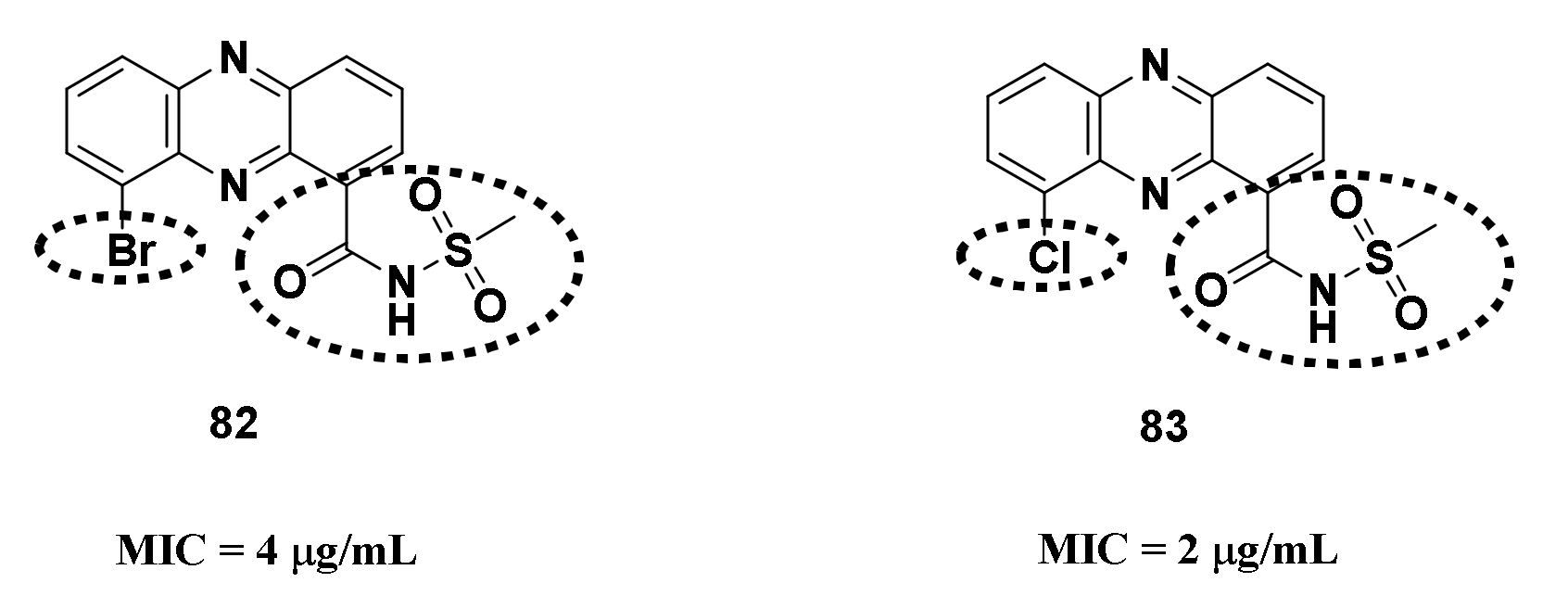
4.1.1. Halogenated Phenazine Derivatives
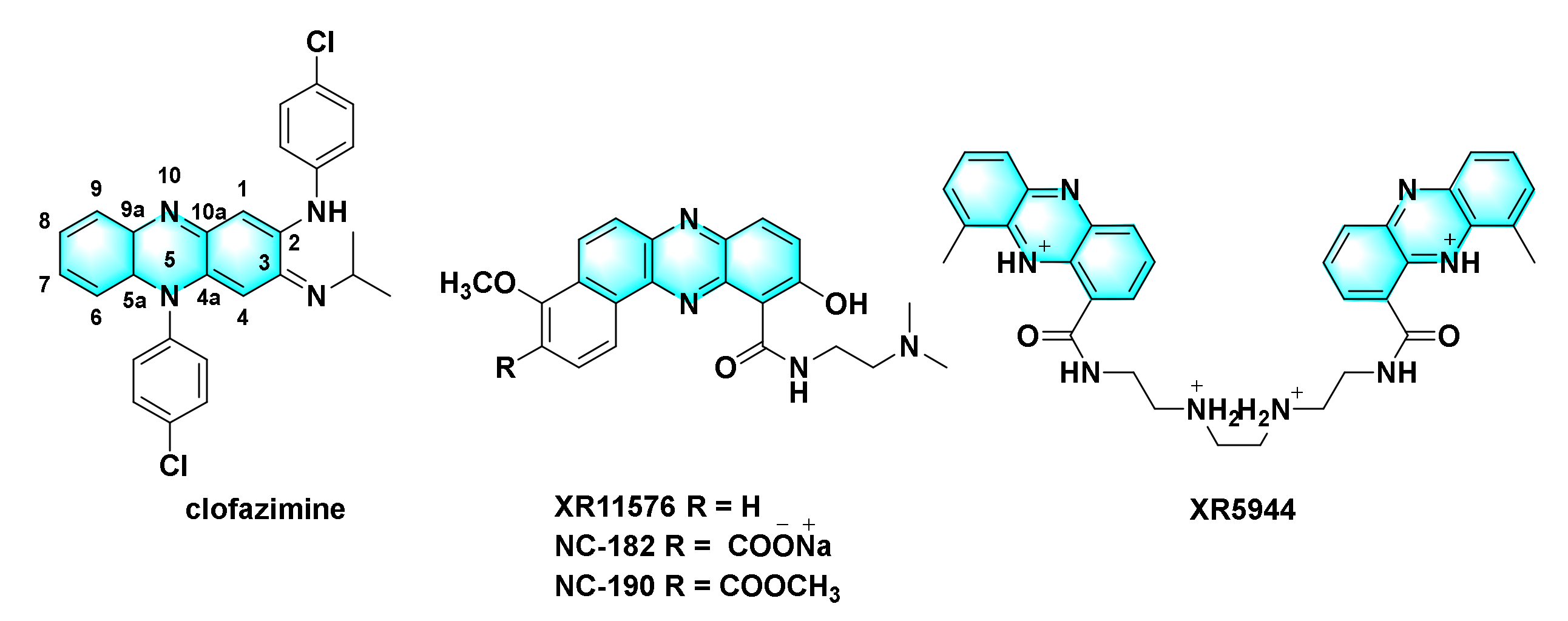
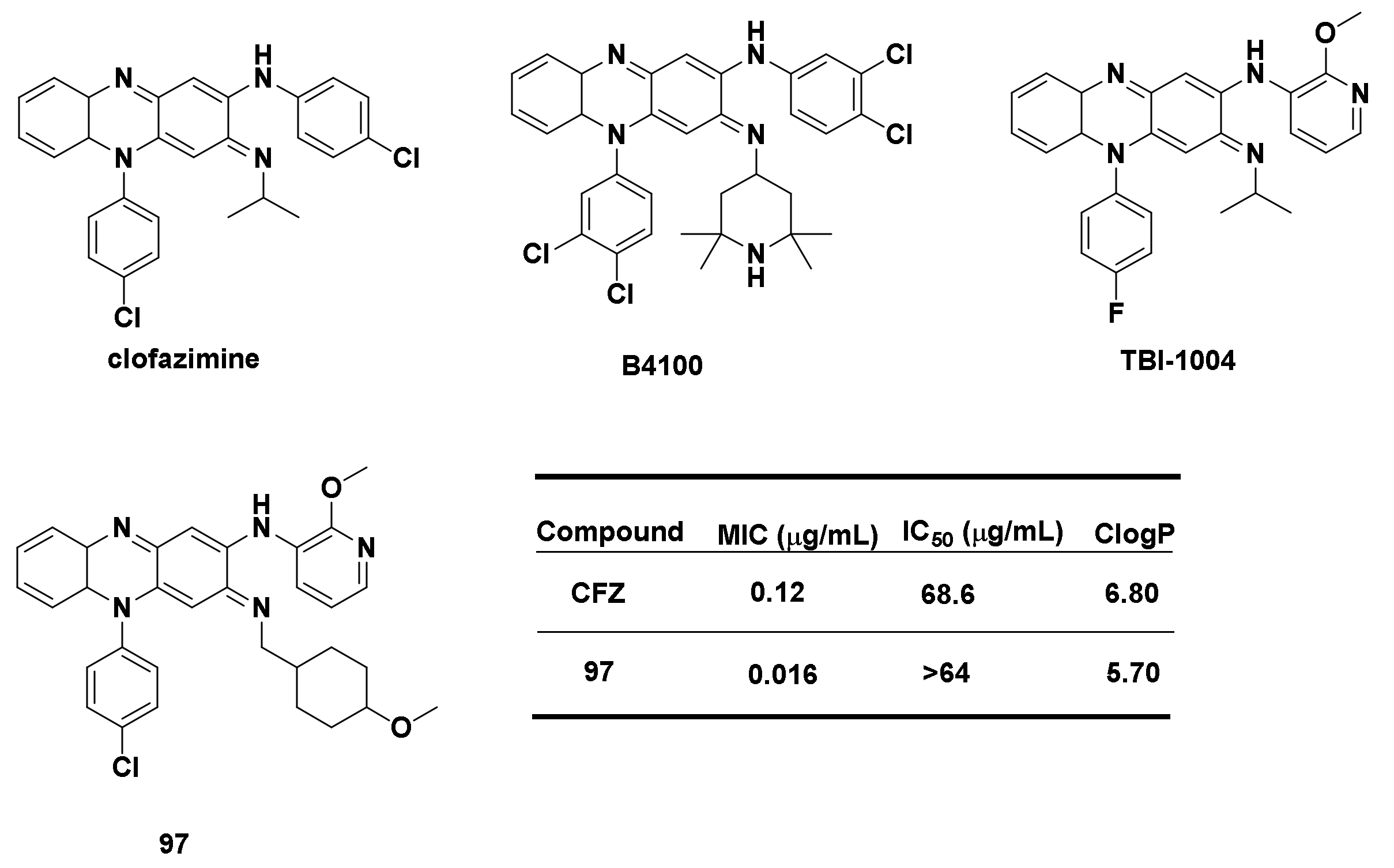
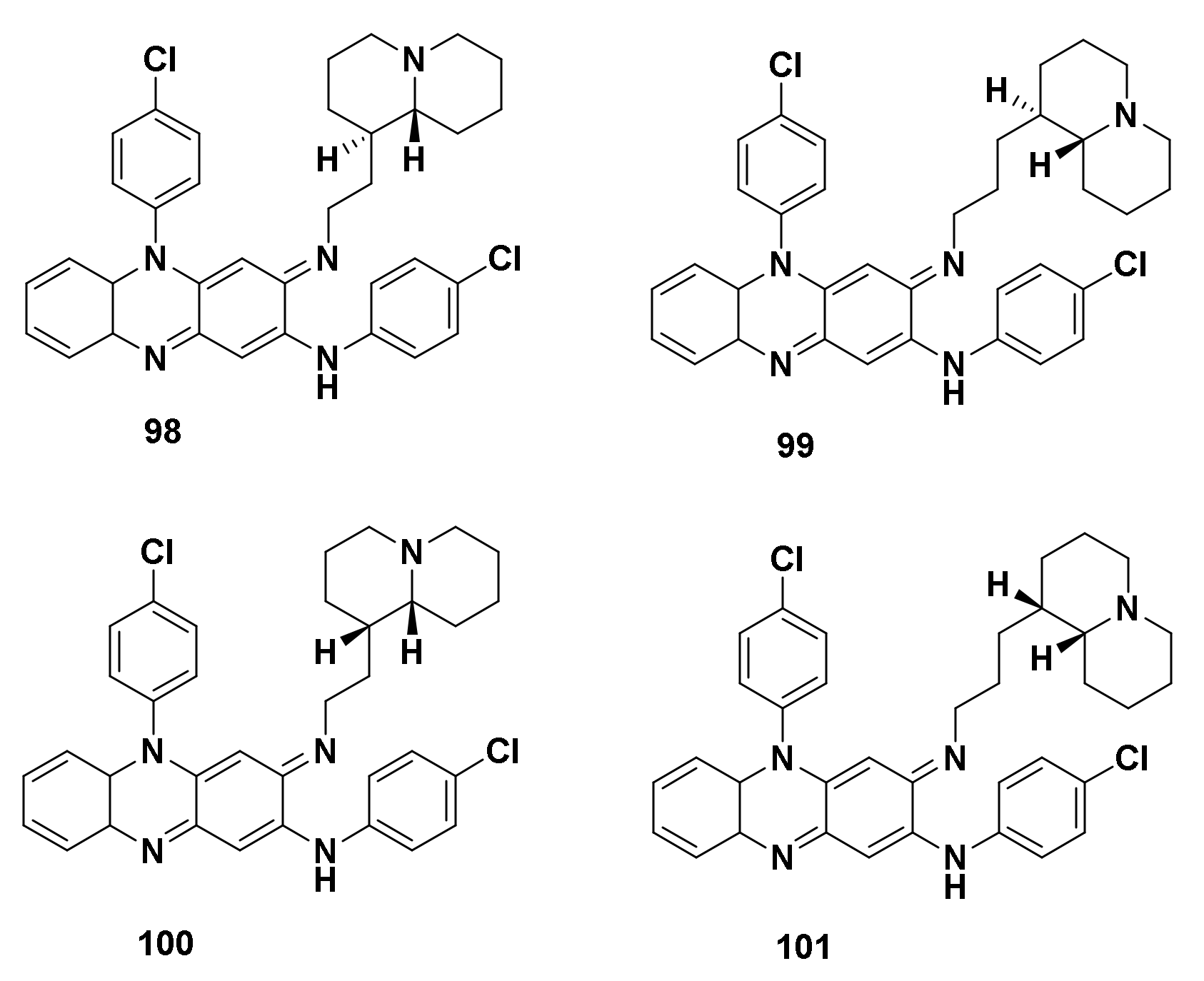
4.1.2. Derivatives of Clofazimine
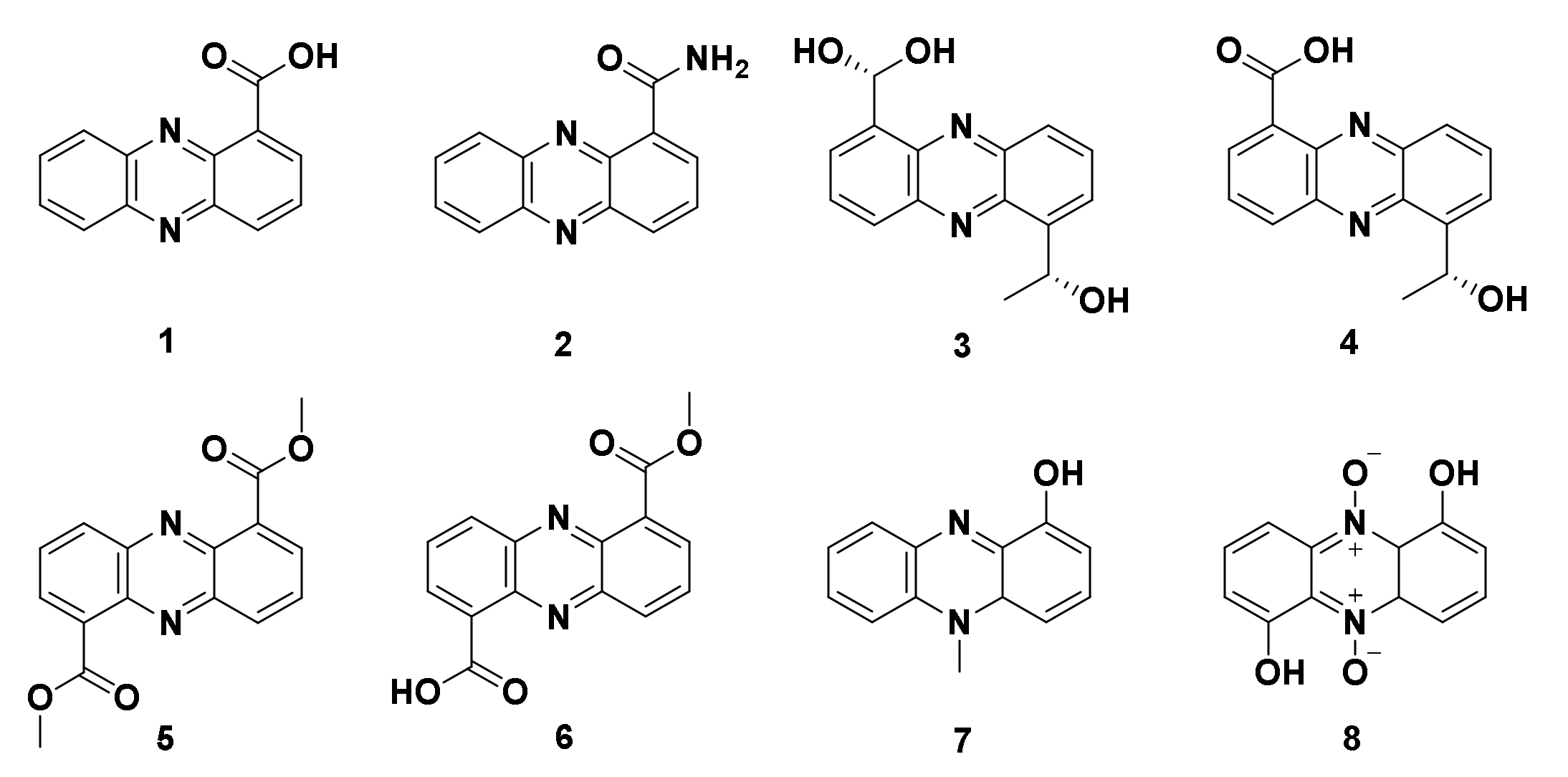
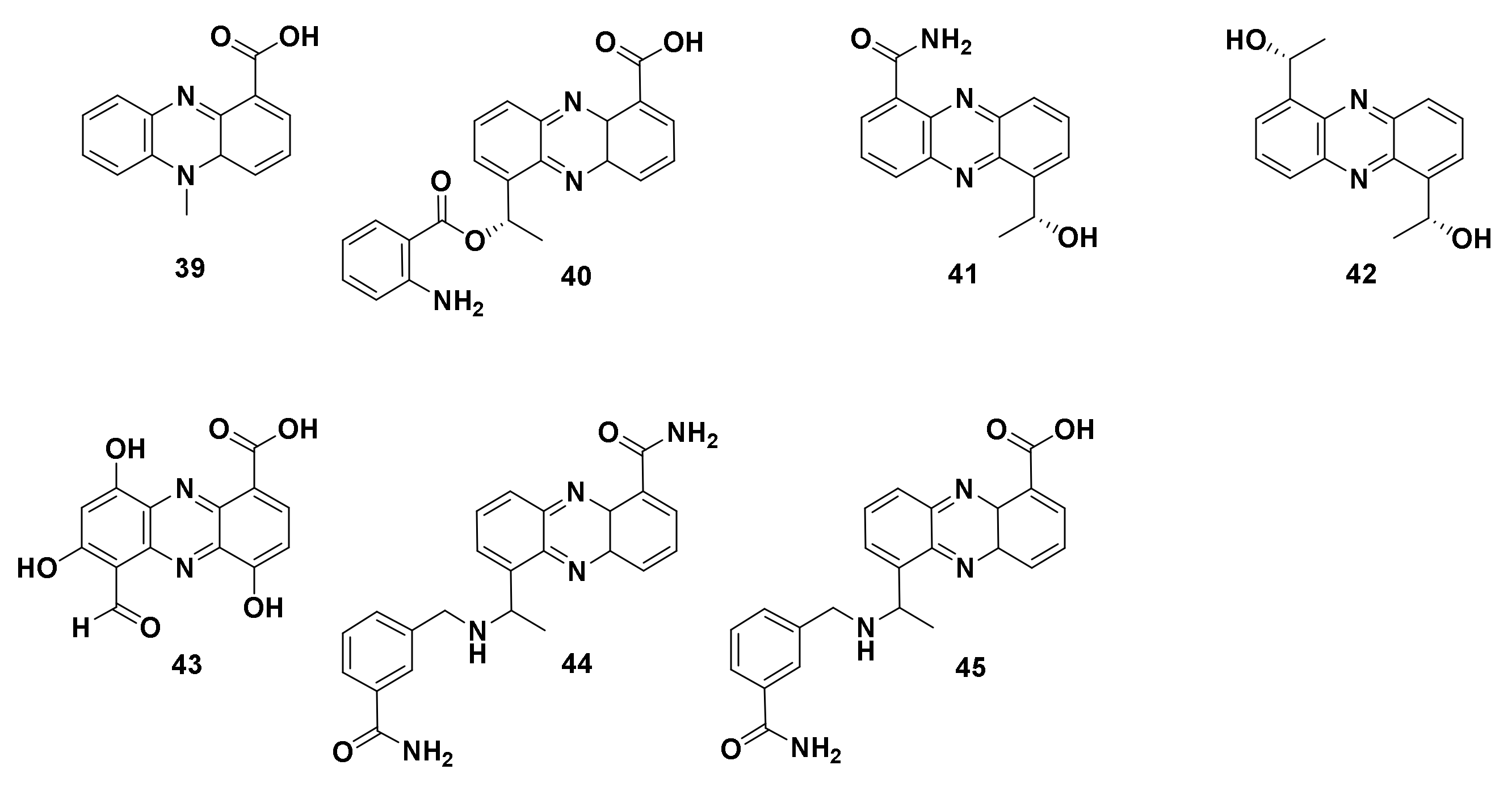
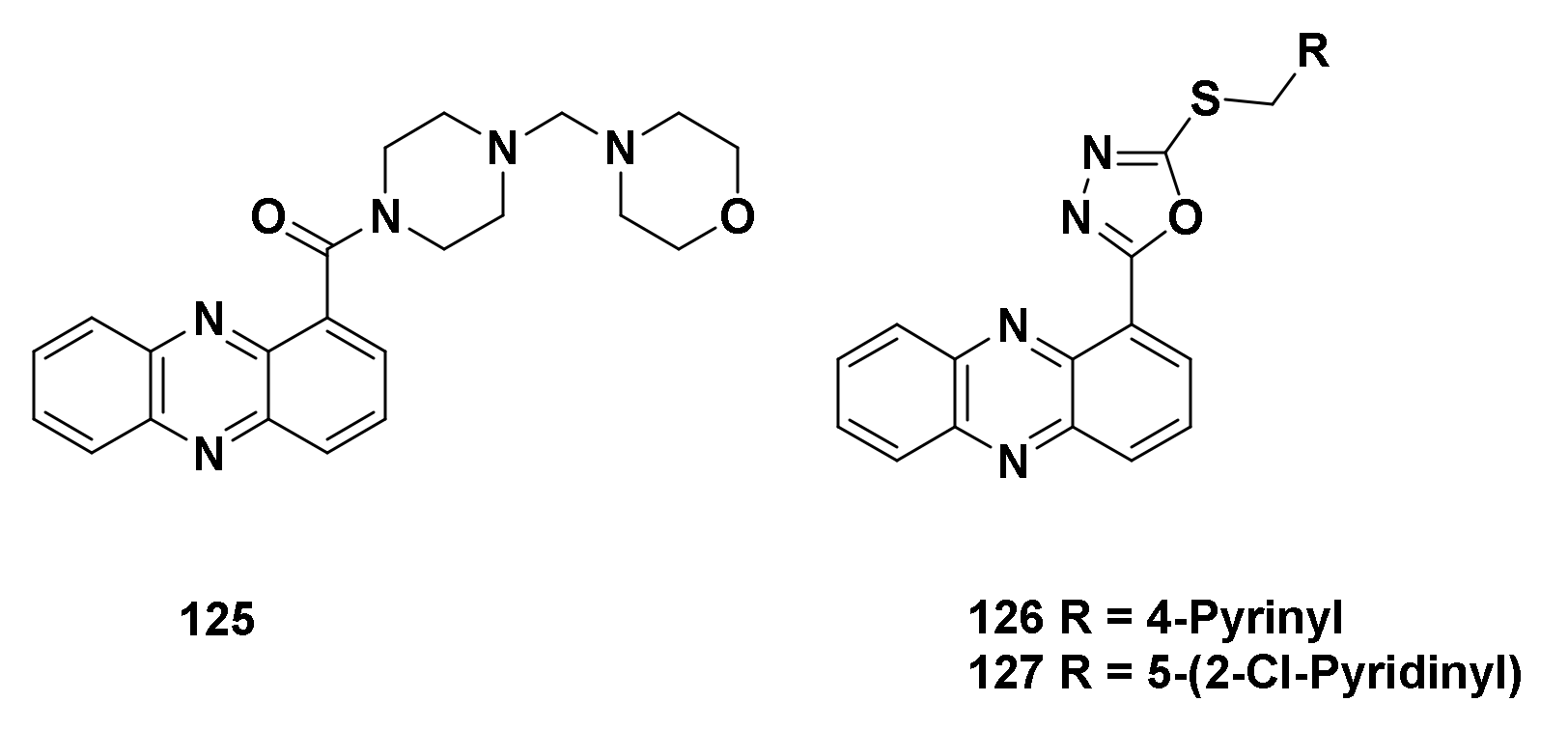
4.1.3. Derivatives of Phenazine-1-carboxylic acid
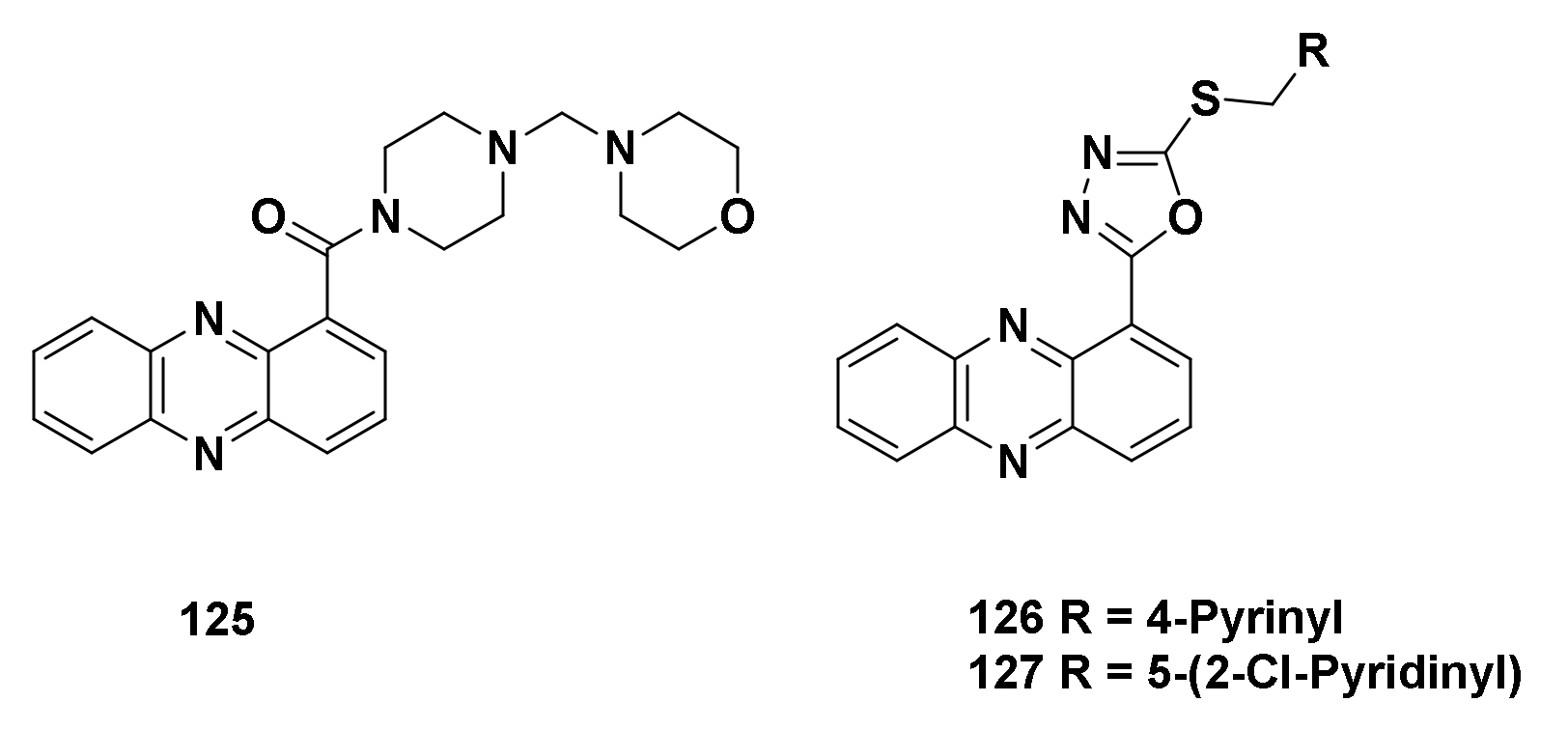
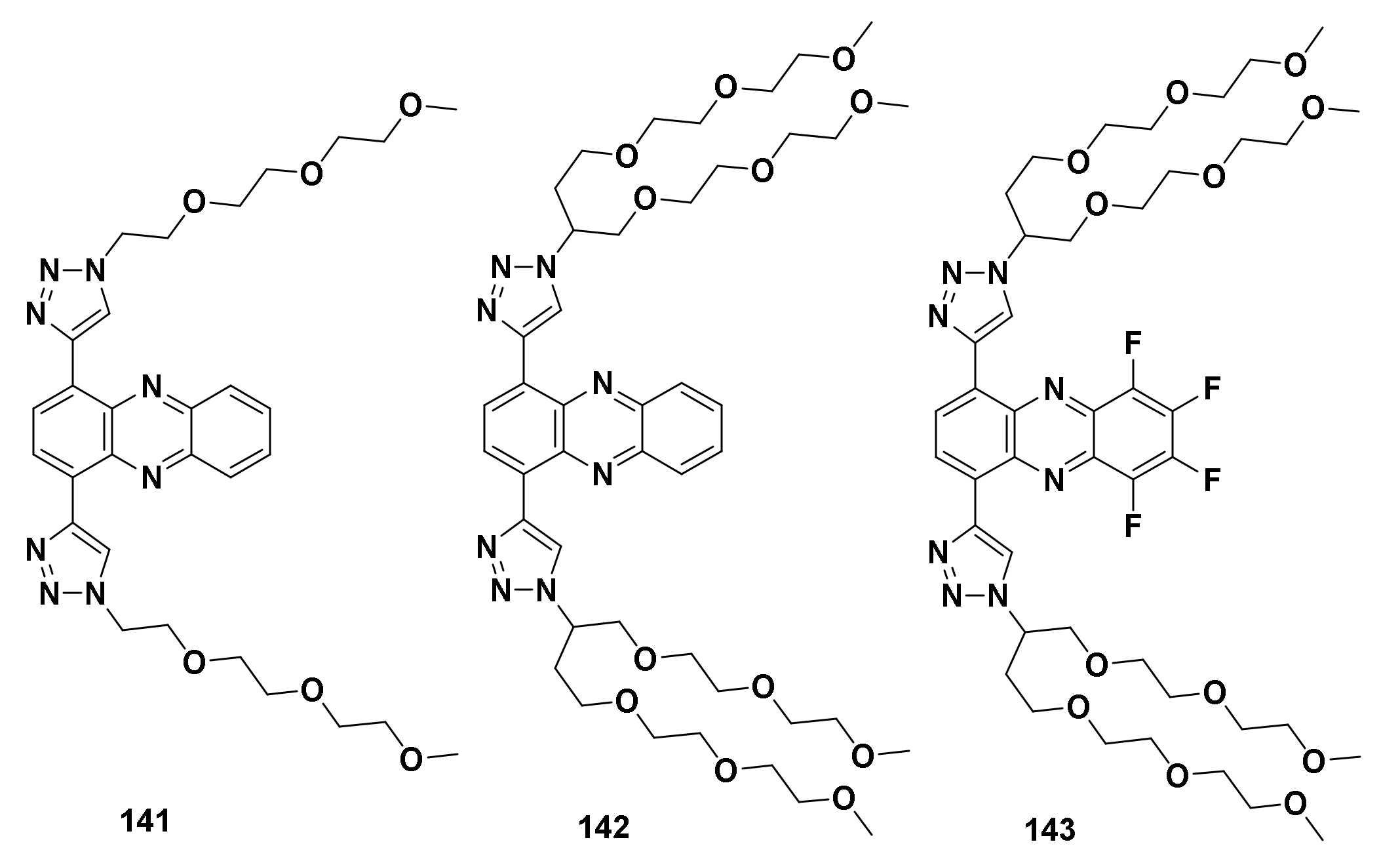
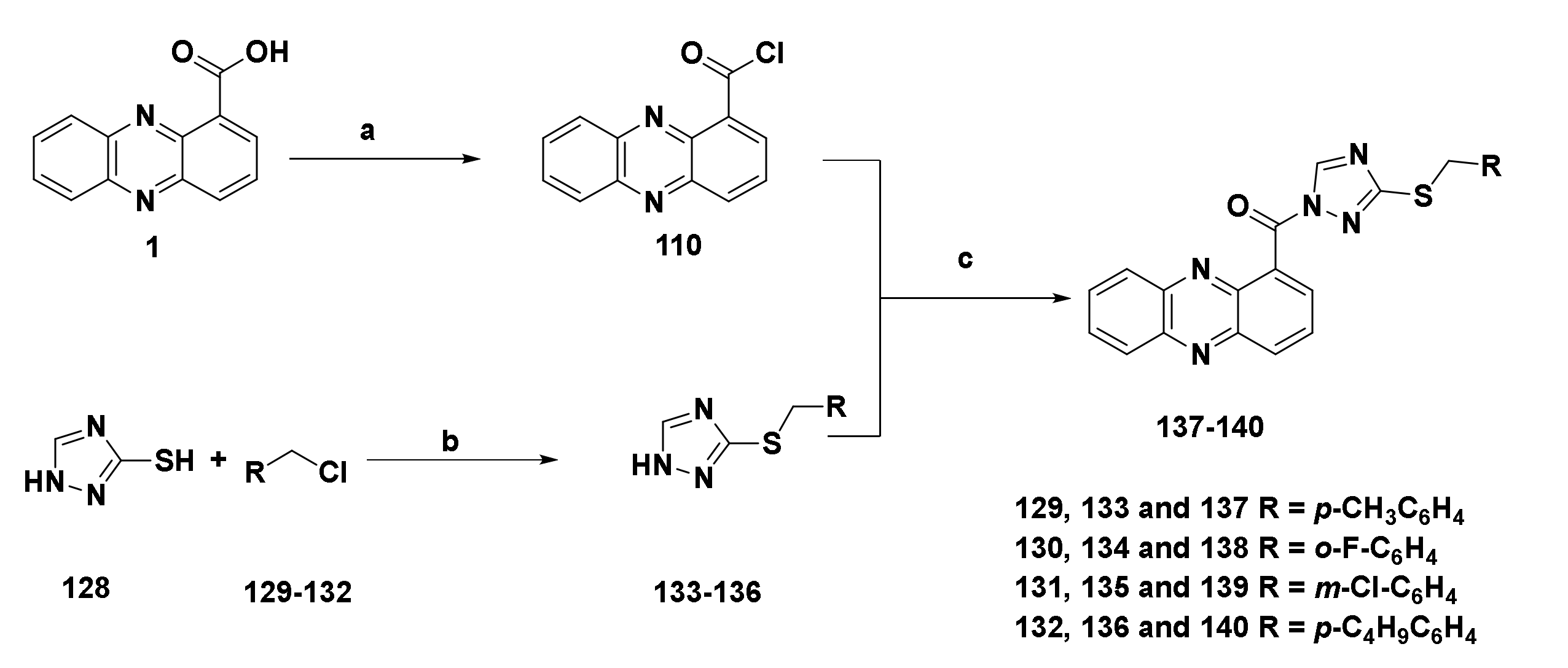
4.1.4. Water-Soluble Triazole Phenazine Derivatives
4.2. Insecticidal Activity
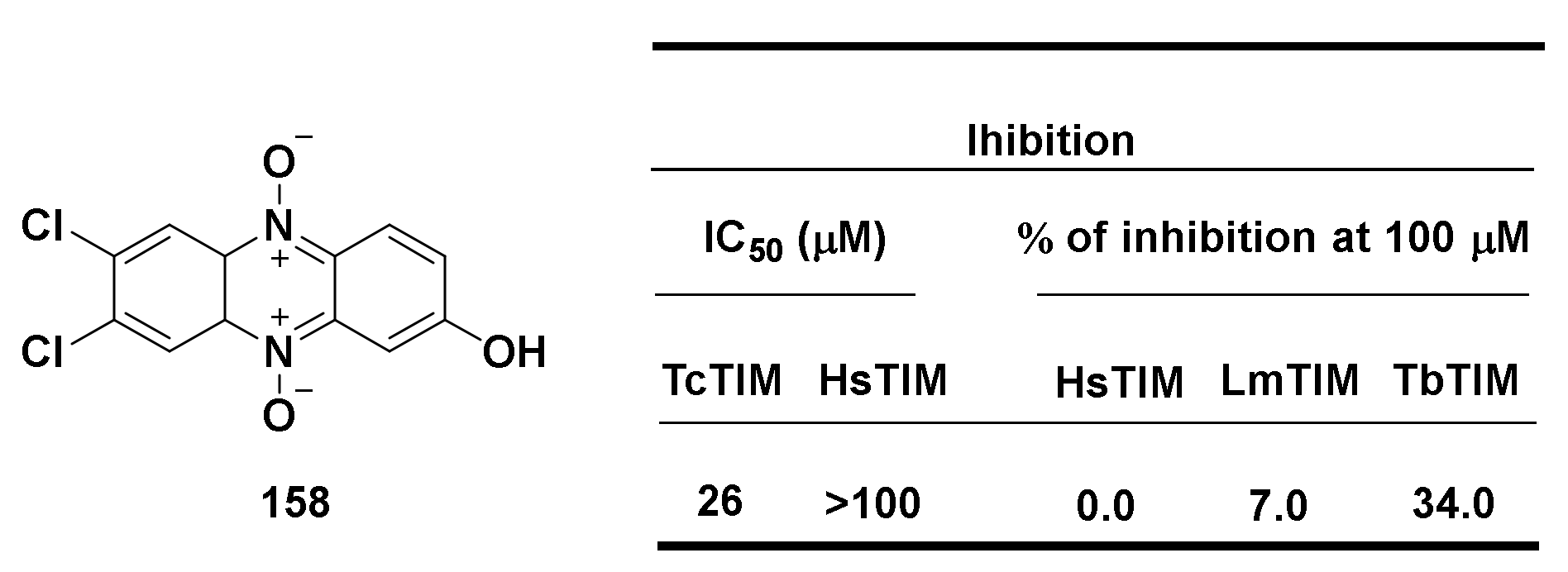
4.3. Antiparasitic Activity
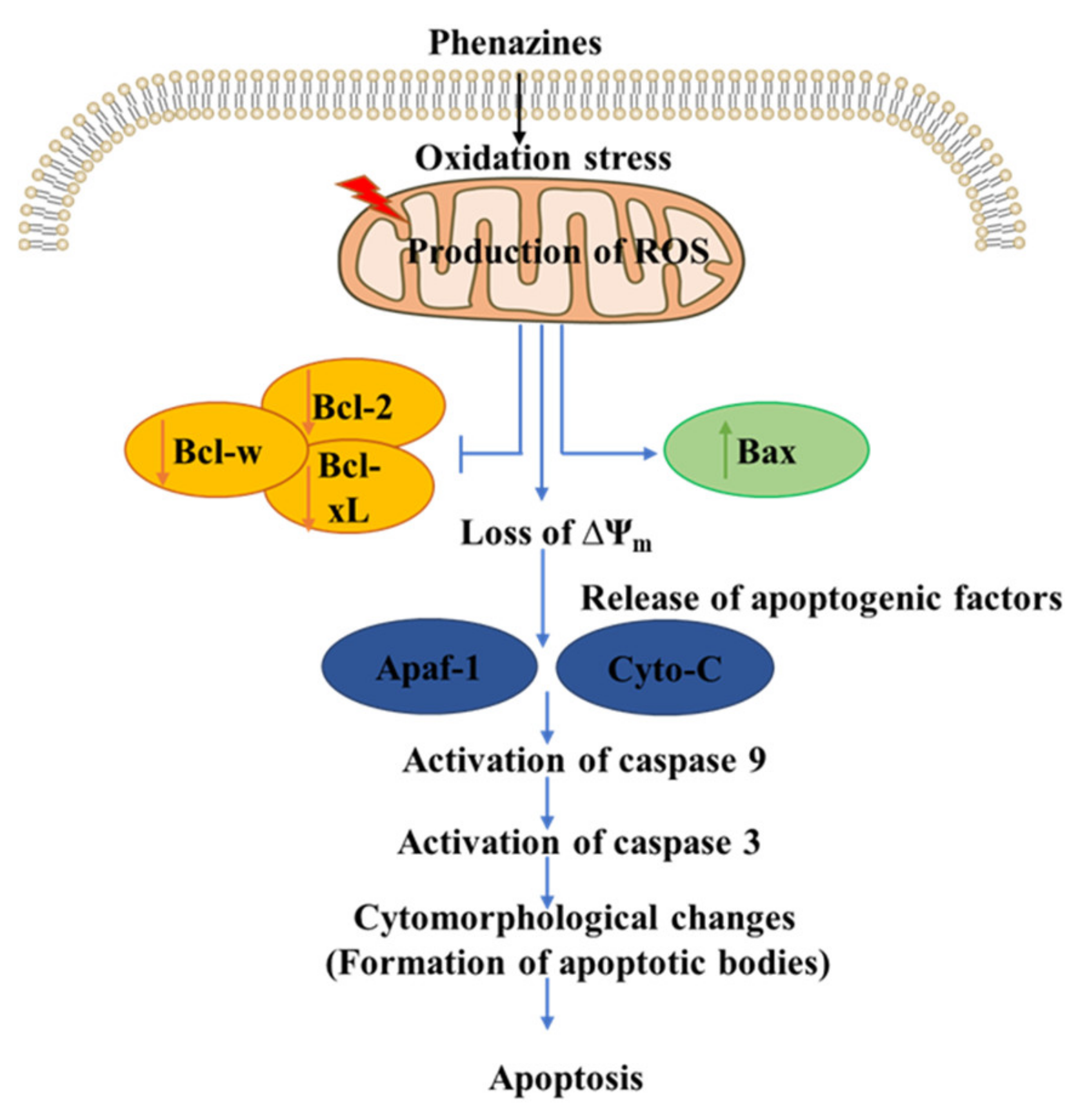
4.4. Anticancer Activity
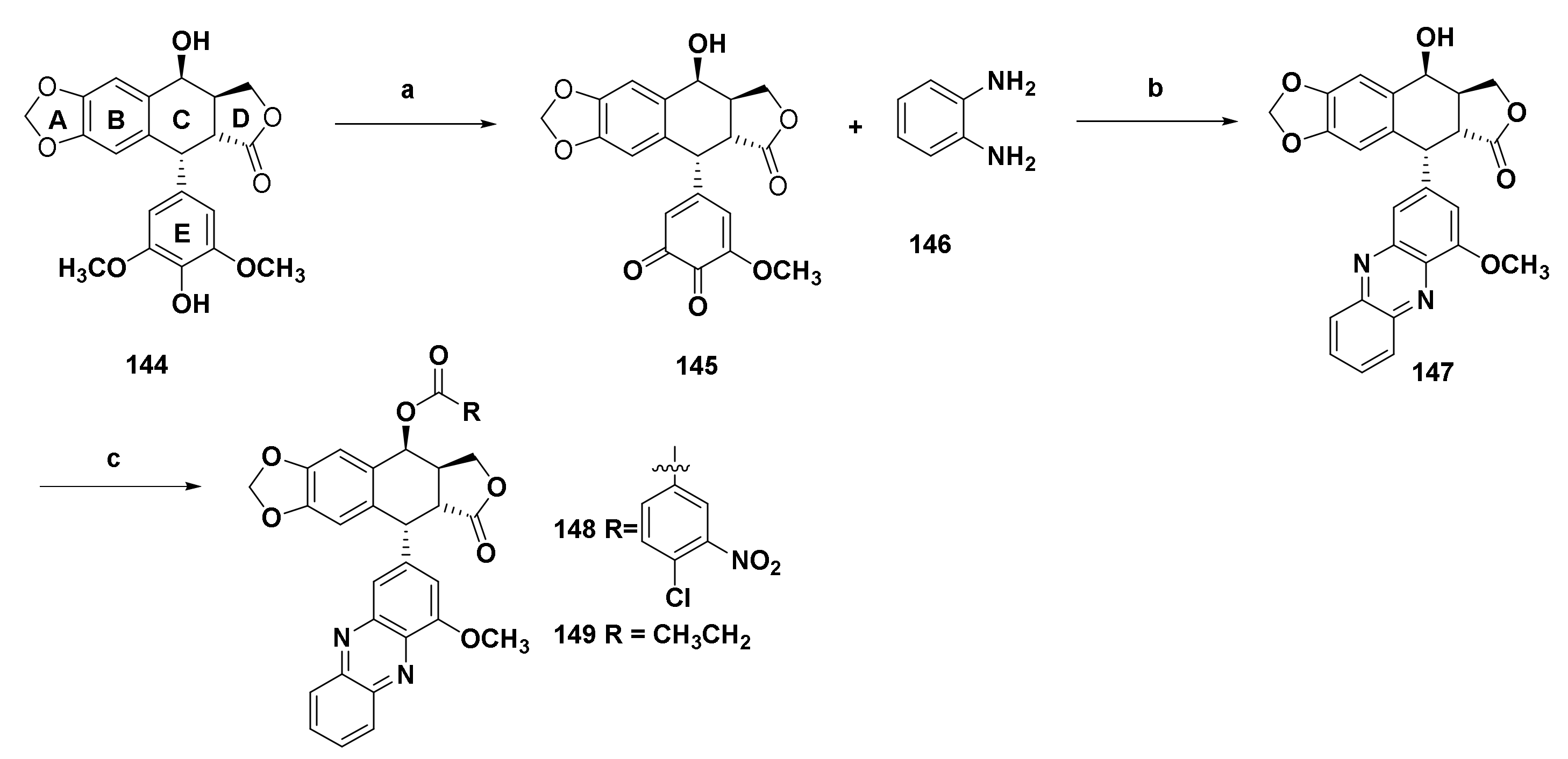
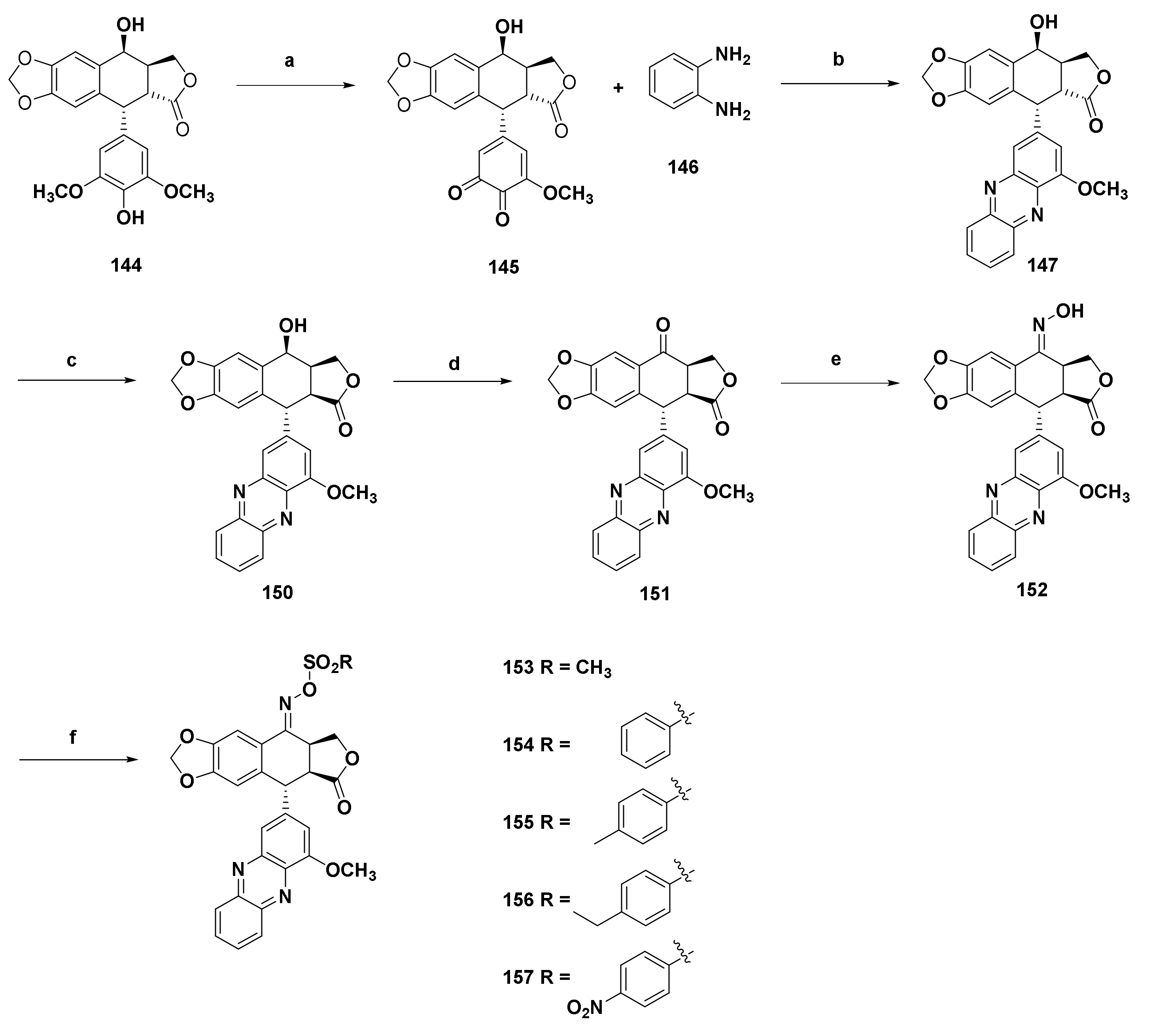
4.4.1. Phenazine 5,10-dioxide Derivatives
4.4.2. Benzo[a]phenazine Derivatives
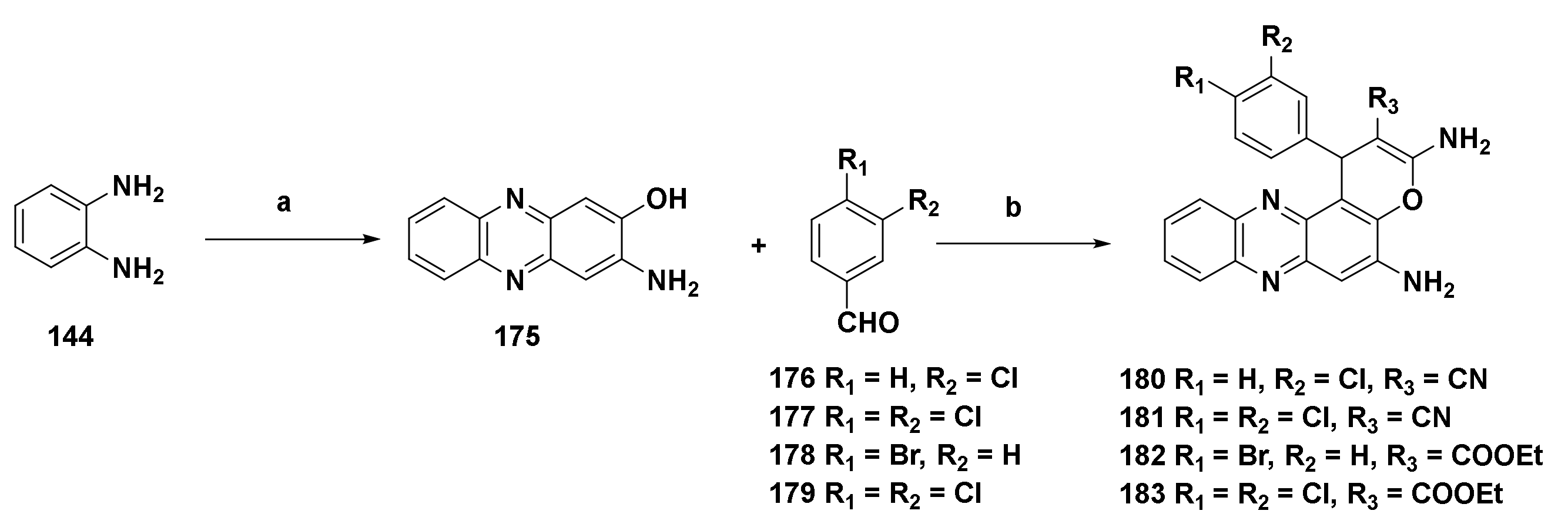
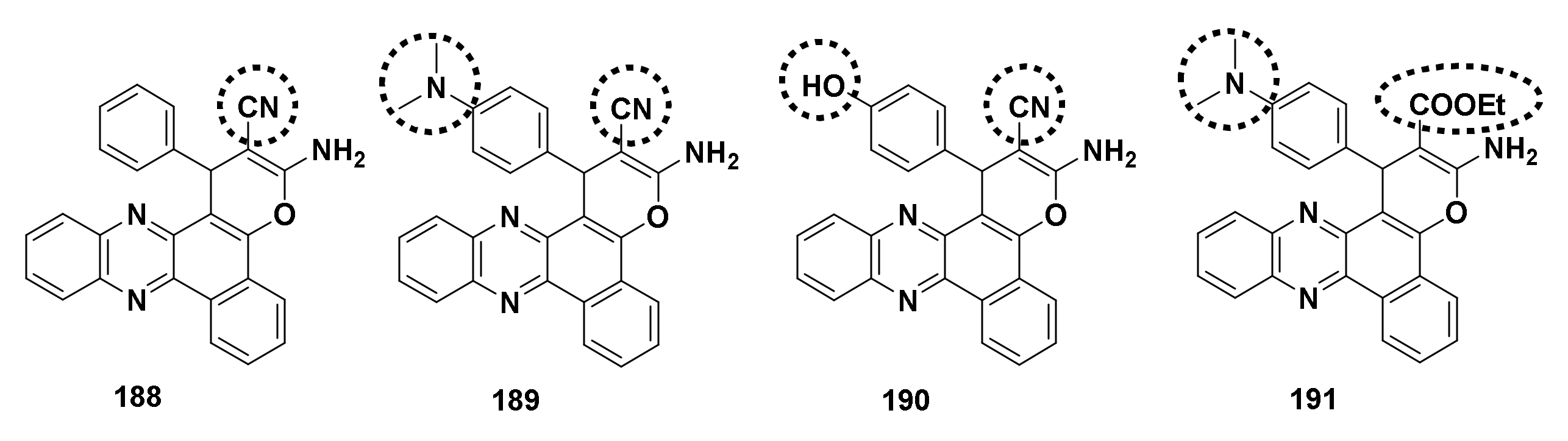
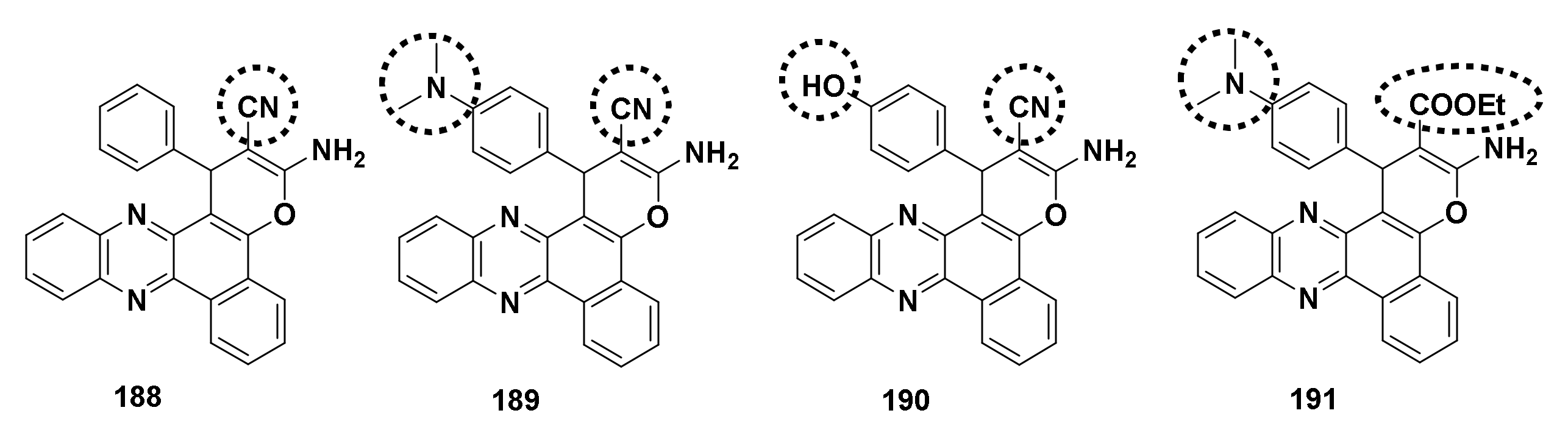
4.4.3. Pyran[2,3-c]phenazine Derivatives
4.4.4. Benzo[a]pyran[2,3-c]phenazine Derivatives
4.4.5. Benzo[a]chromeno[2,3-c] phenazine Derivatives
4.4.6. Derivatives Derived from 2-Phenazinamine
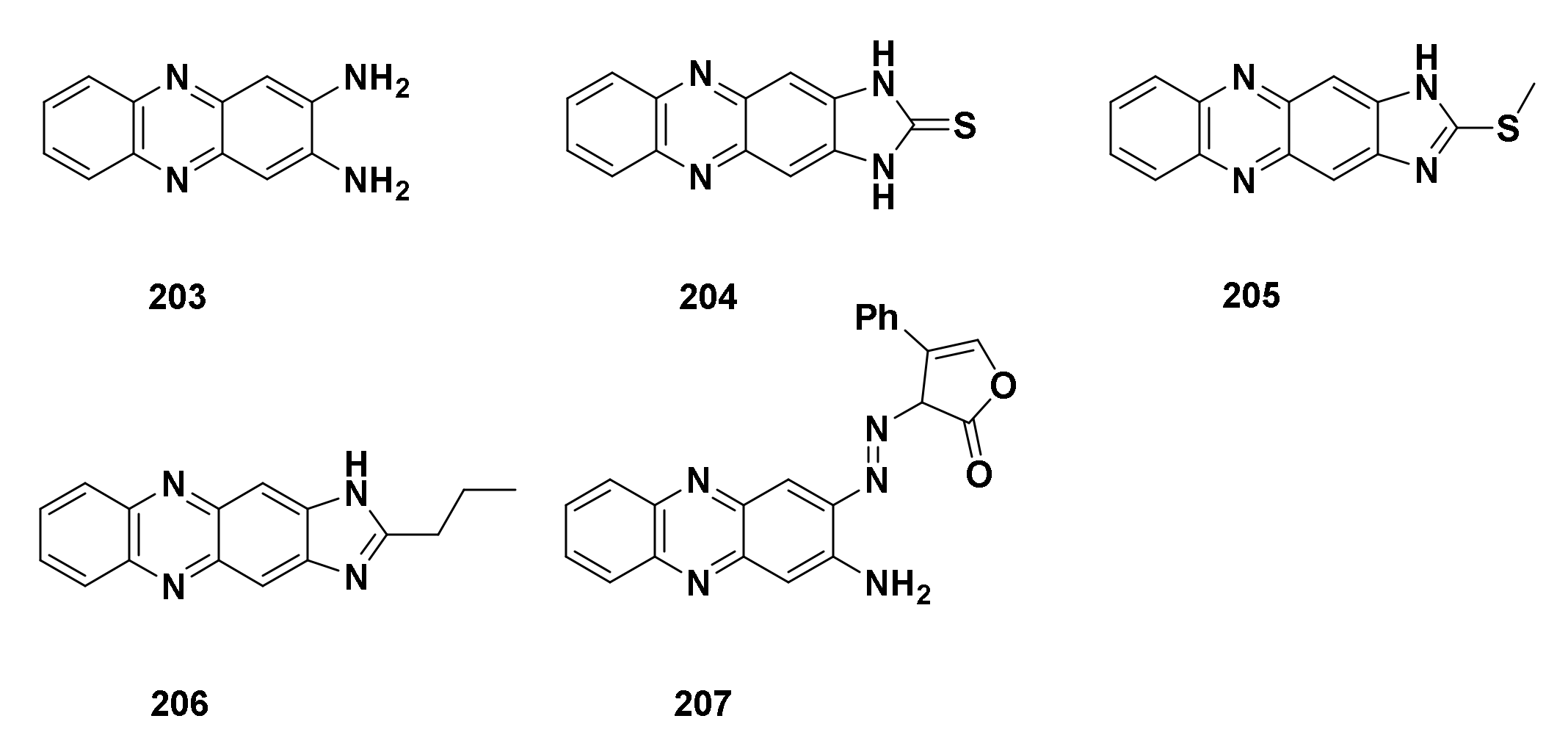
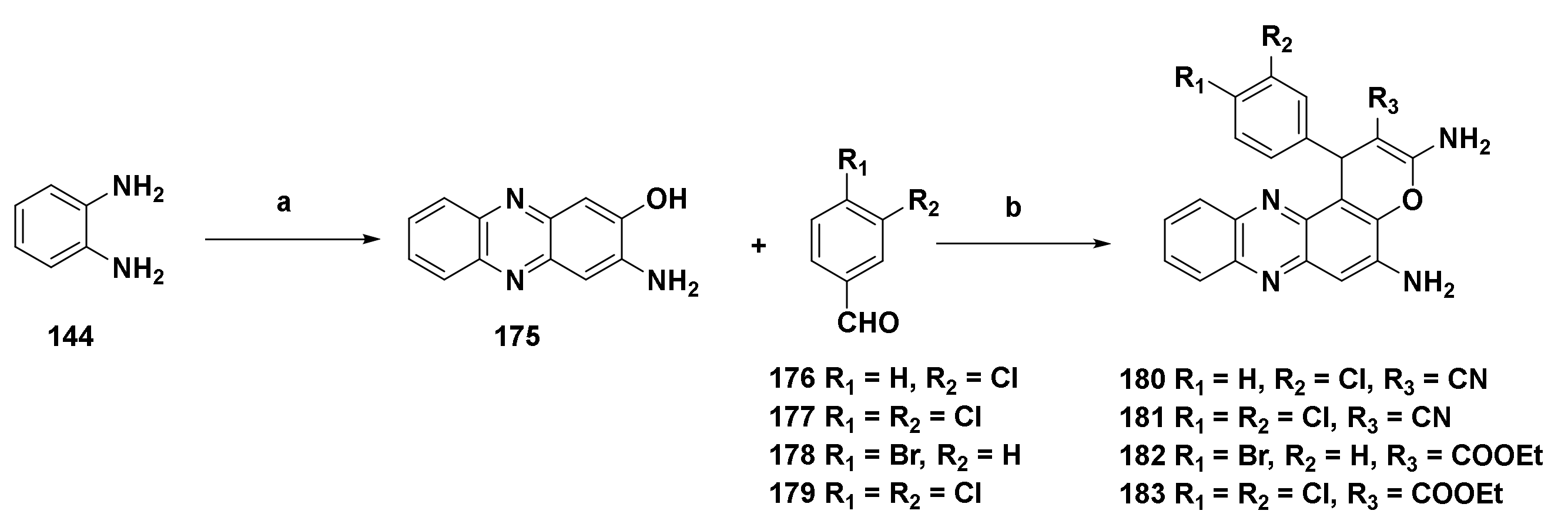
4.4.7. Derivatives Derived from 2,3-Diaminophenazine
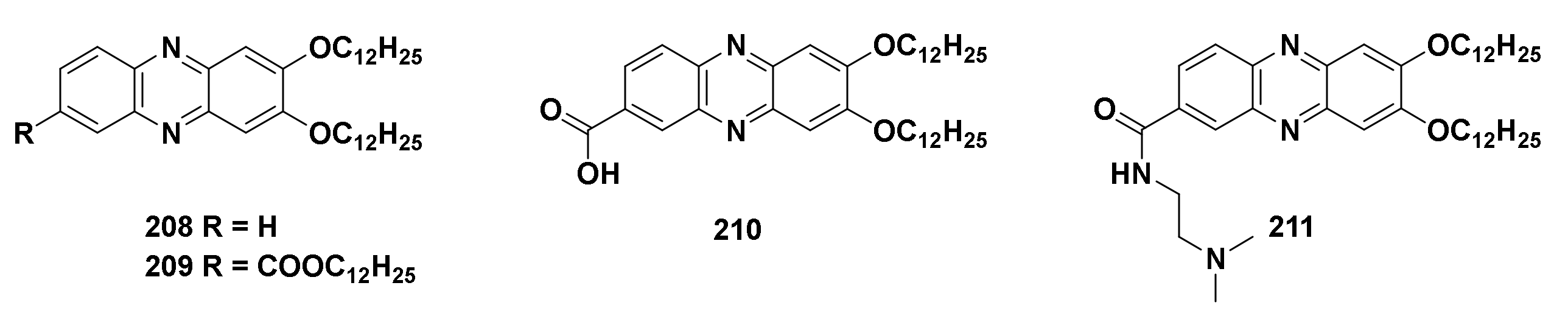
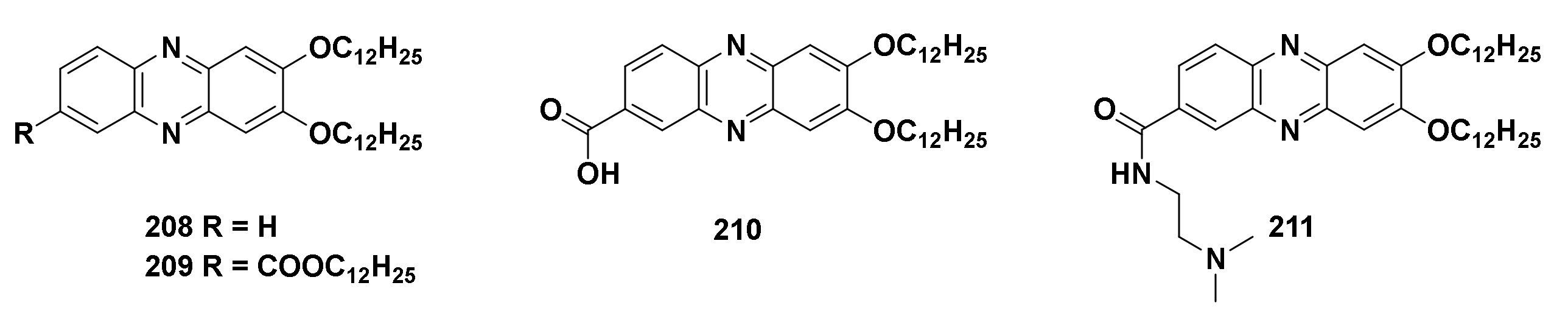
4.4.8. 2,3-Dialkoxyphenazine Derivatives
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Guttenberger, N.; Blankenfeldt, W.; Breinbauer, R. Recent developments in the isolation, biological function, biosynthesis, and synthesis of phenazine natural products. Bioorg. Med. Chem. 2017, 25, 6149–6166. [Google Scholar] [CrossRef] [PubMed]
- Valliappan, K.; Sun, W.; Li, Z. Marine actinobacteria associated with marine organisms and their potentials in producing pharmaceutical natural products. Appl. Microbiol. Biotechnol. 2014, 98, 7365–7377. [Google Scholar] [CrossRef]
- Yang, P.; Yang, Q.; Qian, X.; Cui, J. Novel synthetic Aisoquinolino[5,4-ab] phenazines: Inhibition toward topoisomerase I, antitumor and DNA photo-cleaving activities. Bioorg. Med. Chem. 2005, 13, 5909–5914. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Zhang, P.; Bilal, M.; Wang, W.; Hu, H.; Zhang, X. Enhanced biosynthesis of phenazine-1-carboxamide by engineered Pseudomonas chlororaphis HT66. Microb. Cell Fact. 2018, 17, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Blankenfeldt, W.; Parsons, J.F. The structural biology of phenazine biosynthesis. Curr. Opin. Struct. Biol. 2014, 29, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Laursen, J.B.; Nielsen, J. Phenazine natural products: Biosynthesis, synthetic analogues, and biological activity. Chem. Rev. 2004, 104, 1663–1685. [Google Scholar] [CrossRef] [PubMed]
- Hari Narayana Moorthy, N.S.; Pratheepa, V.; Maria, J.R.; Vitor, V.; Pedro, A.F. Fused aryl-phenazines: Scaffold for the development of bioactive molecules. Curr. Drug Targets. 2014, 15, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Zhao, Y.; Zhang, J.; Wang, C.; Sun, X.; He, J.; Xu, Q.; Lu, J.; Zhang, Q. Synthesis, physical properties, and light-emitting diode performance of phenazine-based derivatives with three, five, and nine fused six-membered rings. J. Org. Chem. 2015, 806, 3030–3035. [Google Scholar] [CrossRef]
- Raymond, Z.; Laure, B.; Pascal, R.; Gilles, U. Isocyanate-, isothiocyanate-, urea-, and thiourea-substituted boron dipyrromethene dyes as fluorescent probes. J. Org. Chem. 2006, 718, 3093–3102. [Google Scholar]
- Tagele, S.B.; Lee, H.G.; Kim, S.W.; Lee, Y.S. Phenazine and 1-undecene producing Pseudomonas chlororaphis subsp. aurantiaca strain KNU17Pc1 for growth promotion and disease suppression in Korean maize cultivars. J. Microbiol. Biotechnol. 2018, 29, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Mageed, W.M.; Milne, B.F.; Wagner, M.; Schumacher, M.; Sandor, P.; Pathom-aree, W.; Goodfellow, M.; Bull, A.T.; Horikoshi, K.; Ebel, R. Dermacozines, a new phenazine family from deep-sea dermacocci isolated from a Mariana Trench sediment. Org. Biomol. Chem. 2010, 8, 2352–2362. [Google Scholar] [CrossRef]
- Lavaggi, M.L.; Aguirre, G.A.; Boiani, L.; Orelli, L.; García, B.; Cerecetto, H.; González, M. Pyrimido[1,2-a] quinoxaline 6-oxide and phenazine 5,10-dioxide derivatives and related compounds as growth inhibitors of Trypanosoma cruzi. Eur. J. Med. Chem. 2008, 43, 1737–1741. [Google Scholar] [CrossRef]
- Krishnaiah, M.; Almeida, N.R.; Udumula, V.; Song, Z.; Chhonker, Y.S.; Abdelmoaty, M.M.; Nascimento, V.A.; Murry, D.J.; Conda-Sheridan, M. Synthesis, biological evaluation, and metabolic stability of phenazine derivatives as antibacterial agents. Eur. J. Med. Chem. 2018, 143, 936–947. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.W.; Lee, S.I.; Kim, M.C.; Thida, M.; Lee, J.W.; Park, J.; Kwon, H.C. Pontemazines A and B, phenazine derivatives containing a methylamine linkage from Streptomyces sp. UT1123 and their protective effect to HT-22 neuronal cells. Bioorg. Med. Chem. Lett. 2015, 25, 5083–5086. [Google Scholar] [CrossRef]
- Pachón, O.G.; Azqueta, A.; Lavaggi, M.L.; Cerain, A.L.; Creppy, E.; Collins, A.; Cerecetto, H.; González, M.; Centelles, J.J.; Cascante, M. Antitumoral effect of phenazine N5, N10-dioxide derivatives on Caco-2 Cells. Chem. Res. Toxicol. 2008, 21, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Makgatho, E.M.; Mbajiorgu, E.F. In vitro investigation of clofazimine analogues for antiplasmodial, cytotoxic and pro-oxidative activities. Afr. Health Sci. 2017, 17, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierson, S.L.; Pierson, E.A. Metabolism and function of phenazines in bacteria: Impacts on the behavior of bacteria in the environment and biotechnological processes. Appl. Microbiol. Biotechnol. 2010, 86, 1659. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Wang, L.; Wang, X.; Xi, T.; Liao, J.; Wang, Z.; Jiang, F. Design, combinatorial synthesis and biological evaluations of novel 3-amino-1’-((1-aryl-1H-1,2,3-triazol-5-yl) methyl)-2’-oxospiro[benzo[a] pyrano[2,3-c]phenazine-1,3’-indoline]-2-carbonitrile antitumor hybrid molecules. Eur. J. Med. Chem. 2017, 135, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.V.; Valasani, K.R.; Lim, K.T.; Jeong, Y.T. Tetramethylguanidiniumchlorosulfonate ionic liquid (TMG IL): An efficient reusable catalyst for the synthesis of tetrahydro-1H-benzo[a]chromeno[2,3-c] phenazin-1-ones under solvent-free conditions and evaluation for their in vitro bioassay activity. New J. Chem. 2015, 39, 9931–9941. [Google Scholar] [CrossRef]
- Mahran, A.M.; Ragab, S.S.; Hashem, A.I.; Ali, M.M.; Nada, A.A. Synthesis and antiproliferative activity of novel polynuclear heterocyclic compounds derived from 2,3-diaminophenazine. Eur. J. Med. Chem. 2015, 90, 568–576. [Google Scholar] [CrossRef]
- Moris, M.A.; Andrieu, C.; Rocchi, P.; Seillan, C.; Acunzo, J.; Brunel, F.; Garzino, F.; Siri, O.; Camplo, M. 2,3-Dialkoxyphenazines as anticancer agents. Tetrahedron Lett. 2015, 56, 2695–2698. [Google Scholar] [CrossRef]
- Yao, B.; Mai, Y.; Chen, S.; Xie, H.; Yao, P.; Ou, T.; Tan, J.; Wang, H.; Li, D.; Huang, S. Design, synthesis and biological evaluation of novel 7-alkylamino substituted benzo[a]phenazin derivatives as dual topoisomerase I/II inhibitors. Eur. J. Med. Chem. 2015, 92, 540–553. [Google Scholar] [CrossRef]
- Lu, Y.; Yan, Y.; Wang, L.; Wang, X.; Gao, J.; Xi, T.; Wang, Z.; Jiang, F. Design, facile synthesis and biological evaluations of novel pyrano[3,2-a]phenazine hybrid molecules as antitumor agents. Eur. J. Med. Chem. 2016, 127, 928–943. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Wang, L.; Wu, Z.; Wang, Z.; Chen, J.; Zhong, Y.; Jiang, F.; Lu, Y. Identification of phenazine analogue as a novel scaffold for thioredoxin reductase I inhibitors against Hep G2 cancer cell lines. J. Enzym. Inhib. Med. Chem. 2019, 34, 1158–1163. [Google Scholar] [CrossRef]
- Kaleza, M.; Sonwane, G.; Choudhari, Y. Searching for potential novel BCR-ABL tyrosine kinase inhibitors through G-QSAR and docking studies of some novel 2-phenazinamine derivatives. Curr. Comput.-Aid. Drug. 2020, 16, 501–510. [Google Scholar]
- Cimmino, A.; Evidente, A.; Mathieu, V.; Andolfi, A.; Lefranc, F.; Kornienko, A.; Kiss, R. Phenazines and cancer. Nat. Prod. Rep. 2012, 29, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Gorantla, J.N.; Nishanth Kumar, S.; Nisha, G.V.; Sumandu, A.S.; Dileep, C.; Sudaresan, A.; Sree Kumar, M.M.; Lankalapalli, R.S.; Dileep Kumar, B.S. Purification and characterization of antifungal phenazines from a fluorescent Pseudomonas strain FPO4 against medically important fungi. J. Mycol. Med. 2014, 24, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Varsha, K.K.; Nishant, G.; Sneha, S.; Shilpa, G.; Devendra, L.; Priya, S.; Nampoothiri, K. Antifungal, anticancer and aminopeptidase inhibitory potential of a phenazine compound produced by Lactococcus BSN307. Indian J. Microbiol. 2016, 56, 411–416. [Google Scholar] [CrossRef] [Green Version]
- Cardozo, V.F.; Oliveira, A.G.; Nishio, E.K.; Perugini, M.R.; Andrade, C.G.; Silveira, W.; Durán, N.; Andrade, G.; Kobayashi, R.K.; Nakazato, G. Antibacterial activity of extracellular compounds produced by a Pseudomonas strain against methicillin-resistant Staphylococcus aureus (MRSA) strains. Ann. Clin. Microbiol. Antimicrob. 2013, 12, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thanabalasingama, D.; Kumara, N.S.; Jayasinghea, L.; Fujimotoa, Y. Endophytic fungus Nigrospora oryzae from a medicinal plant Coccinia grandis, a high yielding new source of phenazine-1-carboxamide. Nat. Prod. Commun. 2015, 10, 1659–1660. [Google Scholar] [CrossRef] [Green Version]
- Tupe, S.G.; Kulkarni, R.R.; Shirazi, F.; Sant, D.G.; Joshi, S.P.; Deshpande, M.V. Possible mechanism of antifungal phenazine-1-carboxamide from Pseudomonas sp. against dimorphic fungi Benjaminiella poitrasii and human pathogen Candida albicans. J. Appl. Microbiol. 2015, 118, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R.K.; Veena, V.; Naik, P.R.; Lakshmi, P.; Krishna, R.; Sudharani, S.; Sakthivel, N. Phenazine-1-carboxamide (PCN) from Pseudomonas sp. strain PUP6 selectively induced apoptosis in lung (A549) and breast (MDA MB-231) cancer cells by inhibition of antiapoptotic Bcl-2 family proteins. Apoptosis 2015, 20, 858–868. [Google Scholar] [CrossRef]
- Ali, H.M.; El-Shikhl, H.; Salem, M.Z.M.; Muzaheed, M. Isolation of bioactive phenazine-1-carboxamide from the soil bacterium Pantoea agglomerans and study of its anticancer potency on different cancer cell lines. J. AOAC Int. 2016, 99, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Chai, W.; Zhang, J.; Zhu, Z.; Liu, W.; Pan, D.; Li, Y.; Chen, B. Pyocyanin from Pseudomonas induces IL-8 production through the PKC and NF-κB pathways in U937 cells. Mol. Med. Rep. 2013, 8, 1404–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, A.; Davey, A.K.; Perkins, A.V.; Grant, G.D.; McFarland, A.J.; McDermott, C.M.; Anoopkumar-Dukie, S. ERK1/2 activation modulates pyocyanin-induced toxicity in A549 respiratory epithelial cells. Chem. Biol. Interact. 2014, 208, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Sletta, H.; Degnes, K.F.; Herfindal, L.; Klinkenberg, G.; Fjærvik, E.; Zahlsen, K.; Brunsvik, A.; Nygaard, G.; Aachmann, F.L.; Ellingsen, T.E.; et al. Anti-microbial and cytotoxic 1,6-dihydroxyphenazine-5,10-dioxide (iodinin) produced by Streptosporangium sp. DSM 45942 isolated from the fjord sediment. Appl. Microbiol. Biotechnol. 2014, 98, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Myhren, L.E.; Nygaard, G.; Gausdal, G.; Sletta, H.; Teigen, K.; Degnes, K.F.; Zahlsen, K.; Brunsvik, A.; Bruserud, Ø.; Ove Døskeland, S.; et al. Iodinin (1,6-dihydroxyphenazine 5,10-dioxide) from Streptosporangium sp. induces apoptosis selectively in myeloid leukemia cell lines and patient cells. Mar. Drugs 2013, 11, 332–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.S.; Kang, J.; Choi, B.K.; Lee, H.S.; Lee, Y.J.; Lee, J.; Shin, H.J. Phenazine derivatives with anti-inflammatory activity from the deep-sea sediment-derived yeast-like fungus Cystobasidium laryngis IV17-028. Mar. Drugs 2019, 17, 482. [Google Scholar] [CrossRef] [Green Version]
- Hifnawy, M.S.; Hassan, H.M.; Mohammed, R.; Fouda, M.M.; Sayed, A.M.; Hamed, A.A.; Abouzid, S.F.; Rateb, M.E.; Alhadrami, H.A.; Abdelmohsen, U.R. Induction of antibacterial metabolites by co-cultivation of two red-sea-sponge-associated Actinomycetes Micromonospora sp. UR56 and Actinokinespora sp. EG49. Mar. Drugs 2020, 18, 243. [Google Scholar] [CrossRef]
- Kondratyuk, T.P.; Park, E.J.; Yu, R.; van Breemen, R.B.; Asolkar, R.N.; Murphy, B.T.; Fenical, W.; Pezzuto, J.M. Novel marine phenazines as potential cancer chemopreventive and anti-inflammatory agents. Mar. Drugs 2012, 10, 451–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlendorf, B.; Schulz, D.; Erhard, A.; Nagel, K.; Imhoff, J.F. Geranylphenazinediol, an acetylcholinesterase inhibitor produced by a Streptomyces Species. J. Nat. Prod. 2012, 75, 1400–1404. [Google Scholar] [CrossRef]
- Song, Y.; Huang, H.; Chen, Y.; Ding, J.; Zhang, Y.; Sun, A.; Zhang, W.; Ju, J. Cytotoxic and antibacterial marfuraquinocins from the deep south China sea-derived Streptomyces niveus SCSIO 3406. J. Nat. Prod. 2013, 76, 2263–2268. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; van Wezel, G.P.; Choi, Y.H. Identification of novel endophenaside antibiotics produced by Kitasatospora sp. MBT66. J. Antibiot. 2015, 68, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Guo, Z.; Zhang, B.; Zhang, M.; Shi, J.; Li, W.; Jiao, R.; Tan, R.; Ge, H. Bioactive phenazines from an earwig-associated Streptomyces sp. Chin. J. Nat. Med. 2019, 17, 475–480. [Google Scholar] [CrossRef]
- Rusman, Y.; Oppegard, L.M.; Hiasa, H.; Gelbmann, C.; Salomon, C.E. Solphenazines A−F, glycosylated phenazines from Streptomyces sp. Strain DL-93. J. Nat. Prod. 2013, 76, 91–96. [Google Scholar] [CrossRef]
- Li, Y.; Han, L.; Rong, H.; Li, L.; Zhao, L.; Wu, L.; Xu, L.; Jiang, Y.; Huang, X. Diastaphenazine, a new dimeric phenazine from an endophytic Streptomyces diastaticus subsp. Ardesiacus. J. Antibiot. 2015, 68, 210–212. [Google Scholar] [CrossRef]
- Wang, X.; Abbas, M.; Zhang, Y.; Elshahawi, S.I.; Ponomareva, L.V.; Cui, Z.; Van Lanen, S.G.; Sajid, I.; Voss, S.R.; Shaaban, K.A.; et al. Baraphenazines A−G, divergent fused phenazine-based metabolites from a himalayan Streptomyces. J. Nat. Prod. 2019, 82, 1686–1693. [Google Scholar] [CrossRef]
- Deng, R.; Zhang, Z.; Li, H.; Wang, W.; Hu, H.; Zhang, X. Identification of a novel bioactive phenazine derivative and regulation of phoP on its production in Streptomyces lomondensis S015. J. Agric. Food Chem. 2021, 69, 974–981. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.; Mavrodi, D.V.; Thomashow, L.S.; Floss, H.G. Phenazine biosynthesis in Pseudomonas fluorescens: Branchpoint from the primary shikimate biosynthetic pathway and role of phenazine-1,6-dicarboxylic acid. J. Am. Chem. Soc. 2001, 123, 9459–9460. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Ahuja, E.G.; Janning, P.; Mavrodi, D.V.; Thomashow, L.S.; Blankenfeldt, W. Trapped intermediates in crystals of the FMN-dependent oxidase phzG provide insight into the final steps of phenazine biosynthesis. Acta Crystallogr. Sect. D 2013, 69, 1403–1413. [Google Scholar] [CrossRef]
- Shi, Y.; Brachmann, A.O.; Westphalen, M.A.; Neubacher, N.; Tobias, N.J.; Bode, H.B. Dual phenazine gene clusters enable diversification during biosynthesis. Nat. Chem. Biol. 2019, 15, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Liu, R.; Wang, W.; Hu, H.; Li, Z.; Zhang, X. Designing an artificial pathway for the biosynthesis of a novel phenazine N-Oxide in Pseudomonas chlororaphis HT66. ACS Synth. Biol. 2020, 9, 883–892. [Google Scholar] [CrossRef] [PubMed]
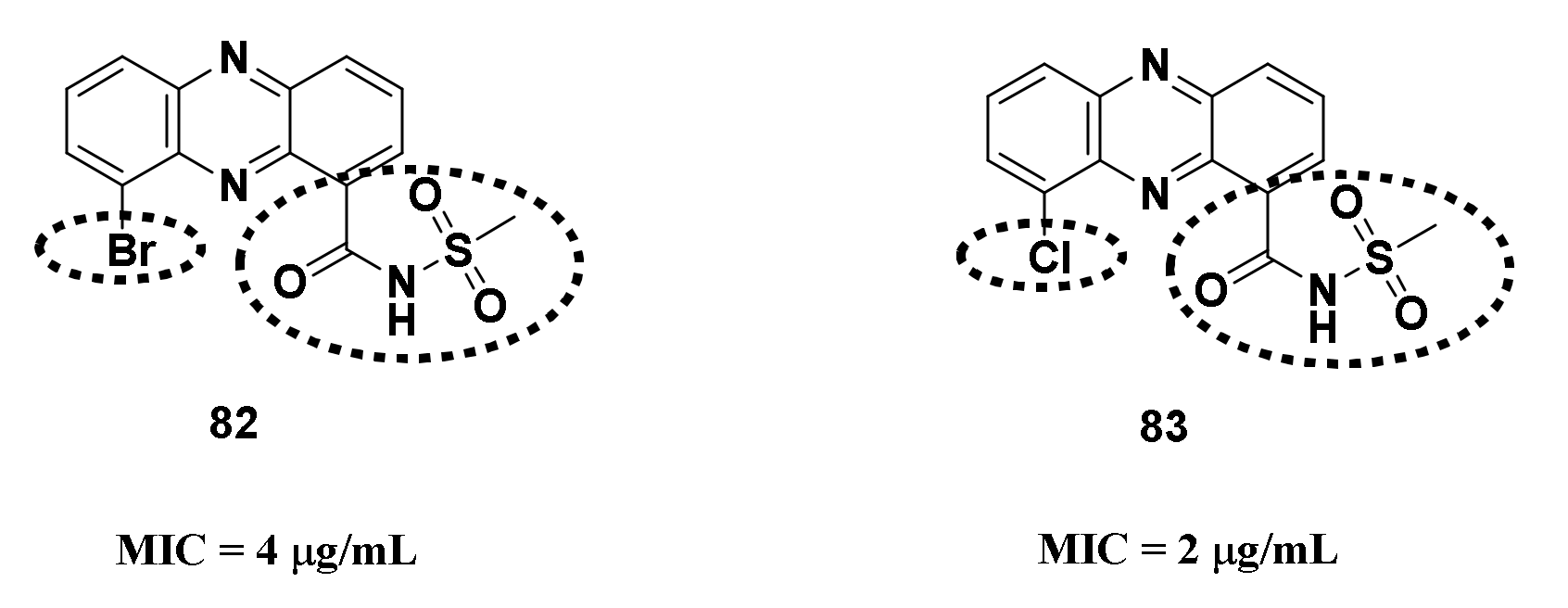
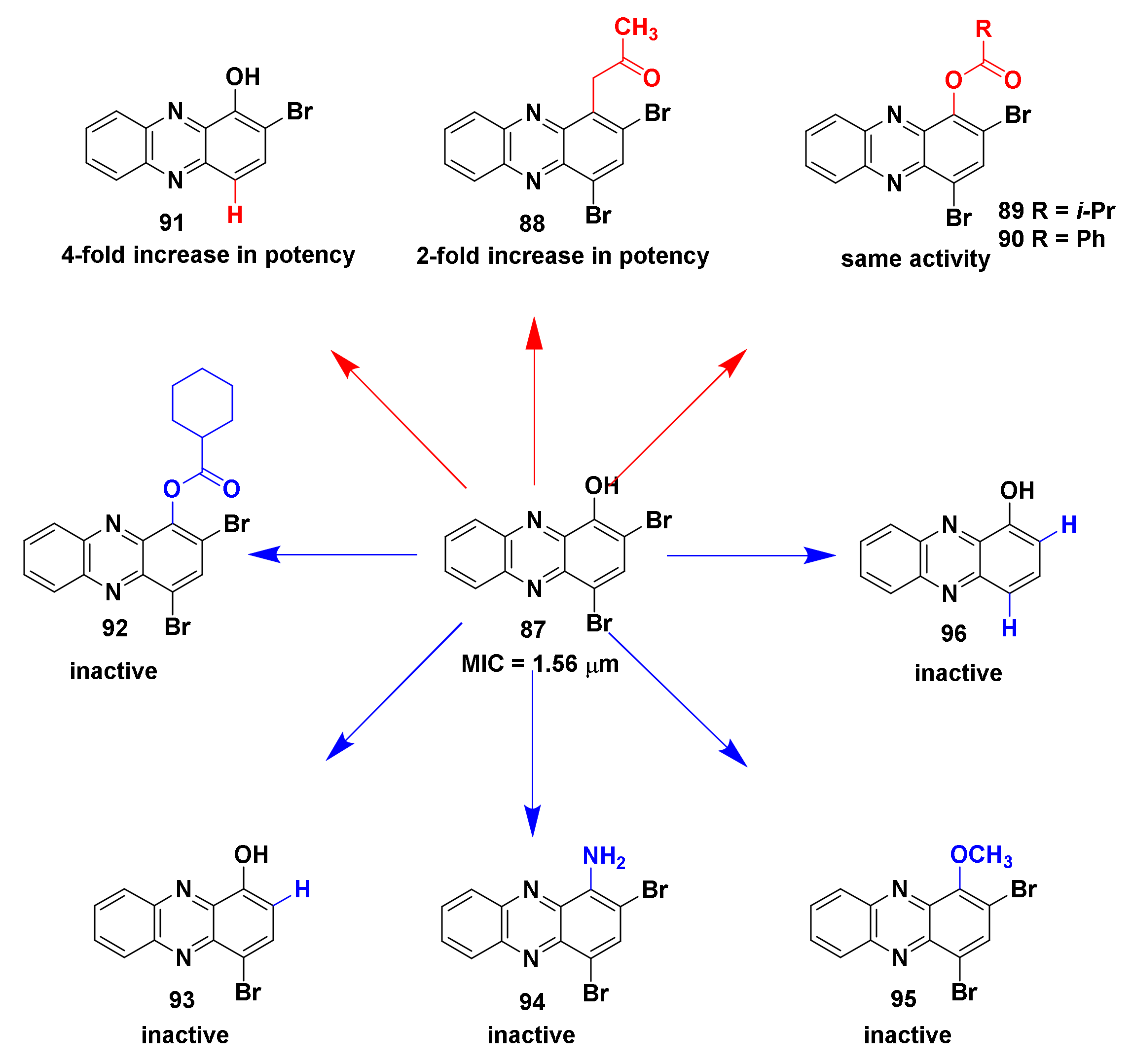
- Yang, H.; Abouelhassan, Y.; Burch, G.M.; Kallifidas, D.; Huang, G.; Yousaf, H.; Jin, S.; Luesch, H.; Huigens, R.W. A highly potent class of halogenated phenazine antibacterial and biofilm-eradicating agents accessed through a modular Wohl-Aue synthesis. Sci. Rep. 2017, 7, 2003–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conda-Sheridan, M.; Udumula, V.; Endres, J.L.; Harper, C.N.; Jaramillo, L.; Zhong, H.A.; Bayles, K.W. Simple synthesis of endophenazine G and other phenazines and their evaluation as anti-methicillin-resistant Staphylococcus aureus agent. Eur. J. Med. Chem. 2017, 125, 710–721. [Google Scholar]
- Garrison, A.T.; Abouelhassan, Y.; Kallifidas, D.; Tan, H.; Kim, Y.S.; Jin, S.; Luesch, H. An efficient buchwald-hartwig/reductive cyclization for the scaffold diversification of halogenated phenazines: Potent antibacterial targeting, biofilm eradication, and prodrug exploration. J. Med. Chem. 2018, 61, 3962–3983. [Google Scholar] [CrossRef] [PubMed]
- Borrero, N.V.; Bai, F.; Perez, C.; Duong, B.Q.; Rocca, J.R.; Jin, S.; Huigens, R.W. Phenazine antibiotic inspired discovery of potent bromophenazine antibacterial agents against Staphylococcus aureus and Staphylococcus epidermidis. Org. Biomol. Chem. 2014, 12, 881–886. [Google Scholar] [CrossRef]
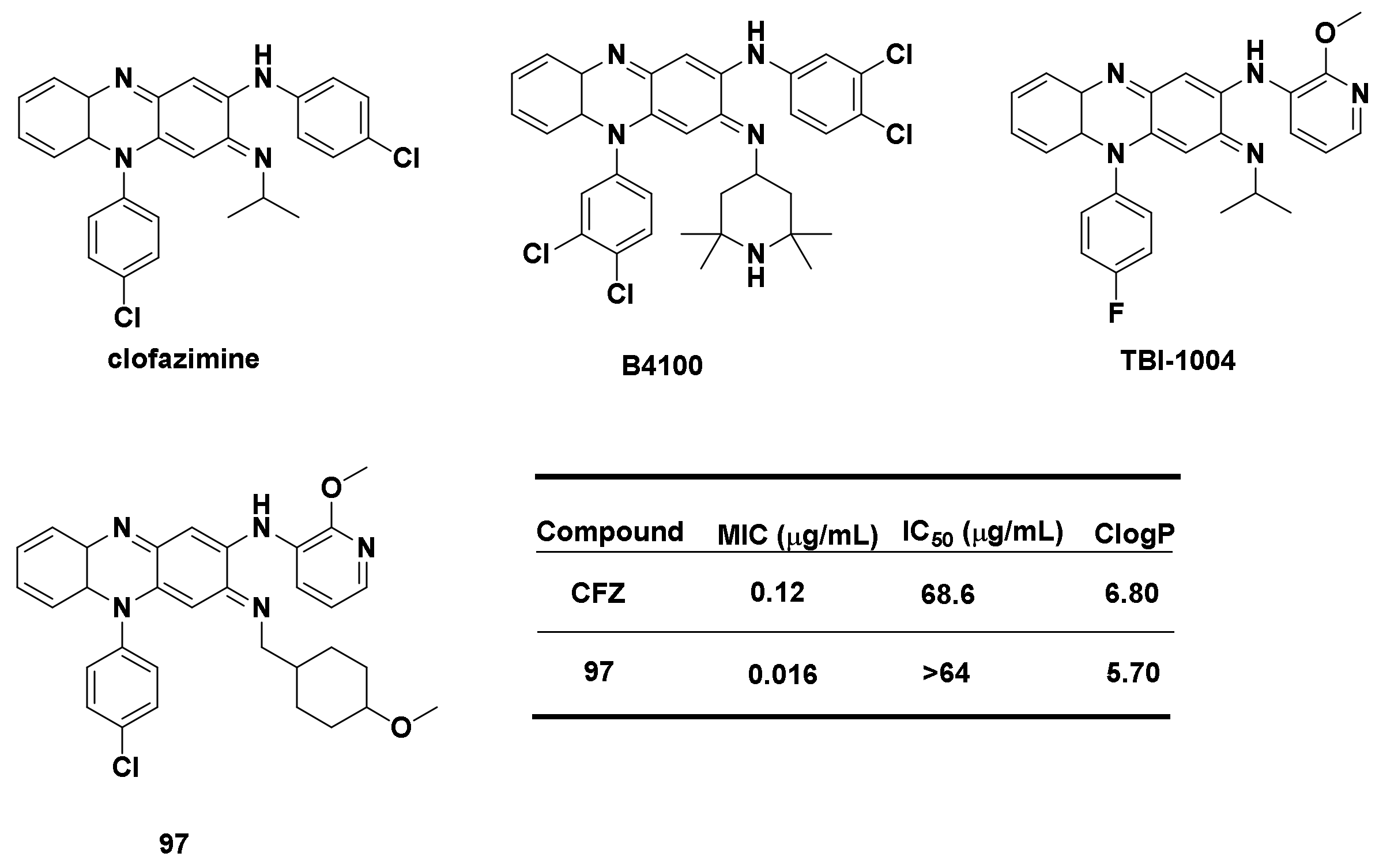
- Zhang, D.; Lu, Y.; Liu, K.; Liu, B.; Wang, J.; Zhang, G.; Zhang, H.; Liu, Y.; Wang, B.; Zheng, M.; et al. Identification of less lipophilic riminophenazine derivatives for the treatment of drug-resistant tuberculosis. J. Med. Chem. 2012, 55, 8409–8417. [Google Scholar] [CrossRef]
- Tonelli, M.; Novelli, F.; Tasso, B.; Sparatore, A.; Boido, V.; Sparatore, F.; Cannas, S.; Molicotti, P.; Zanetti, S.; Parapini, S.; et al. Antitubercular activity of quinolizidinyl/pyrrolizidinylalkyliminophenazines. Bioorgan. Med. Chem. 2014, 22, 6837–6845. [Google Scholar] [CrossRef]
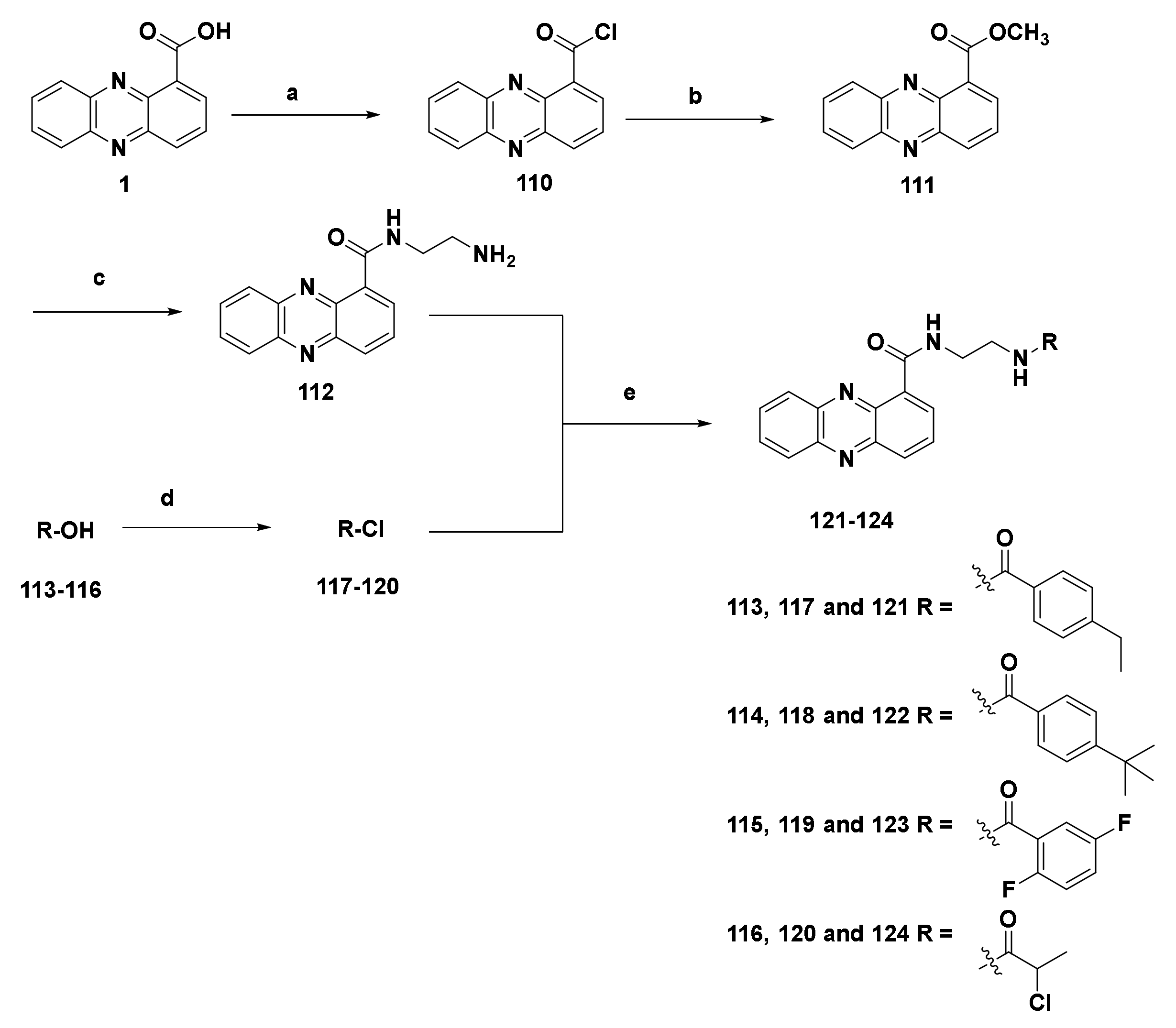
- Niu, J.; Chen, J.; Xu, Z.; Zhu, X.; Wu, Q.; Li, J. Synthesis and bioactivities of amino acid ester conjugates of phenazine-1-carboxylic acid. Bioorg. Med. Lett. 2016, 26, 5384–5386. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, M.; Yu, L.; Xu, Z.; Yang, D.; Du, X.; Wu, Q.; Li, J. Synthesis and bioactivities of diamide derivatives containing a phenazine-1-carboxamide scaffold. Nat. Prod. Res. 2019, 33, 2453–2460. [Google Scholar] [CrossRef]
- Han, F.; Yan, R.; Zhang, M.; Xiang, Z.; Wu, Q.; Li, J. Synthesis and bioactivities of phenazine-1-carboxylic piperazine derivatives. Nat. Prod. Res. 2020, 34, 1282–1287. [Google Scholar] [CrossRef]
- Lu, X.; Zhu, X.; Zhang, M.; Wu, Q.; Zhou, X.; Li, J. Synthesis and fungicidal activity of 1,3,4-oxadiazol-2-ylthioether derivatives containing a phenazine-1-carboxylic acid scaffold. Nat. Prod. Res. 2019, 33, 2145–2150. [Google Scholar]
- Li, X.; Zhang, W.; Zhao, C.; Wu, Q.; Li, J.; Xu, Z. Synthesis and fungicidal activity of phenazine-1-carboxylic triazole derivatives. J. Asian Nat. Prod. Res. 2021, 23, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Hayden, S.C.; Bryant, J.J.; Mackey, M.A.; Höfer, K.; Lindner, B.D.; Nguyen, V.P.; Jäschke, A.; Bunz, U.H.F. Antimicrobial activity of water-soluble triazole phenazine clickamers against E. coli. Chem. Eur. J. 2014, 20, 719–723. [Google Scholar] [CrossRef]
- Zhi, X.; Yang, C.; Zhang, R.; Hu, Y.; Ke, Y.; Xu, H. Natural products-based insecticidal agents 13. Semisynthesis and insecticidal activity of novel phenazine derivatives of 4 beta-acyloxypodophyllotoxin modified in the E-ring against Mythimna separata Walker in vivo. Ind. Crops Prod. 2013, 42, 520–526. [Google Scholar] [CrossRef]
- Zhi, X.; Yang, C.; Yu, X.; Xu, H. Synthesis and insecticidal activity of new oxime derivatives of podophyllotoxin-based phenazines against Mythimna separata Walker. Bioorg. Med. Chem. Lett. 2014, 24, 5679–5682. [Google Scholar] [CrossRef]
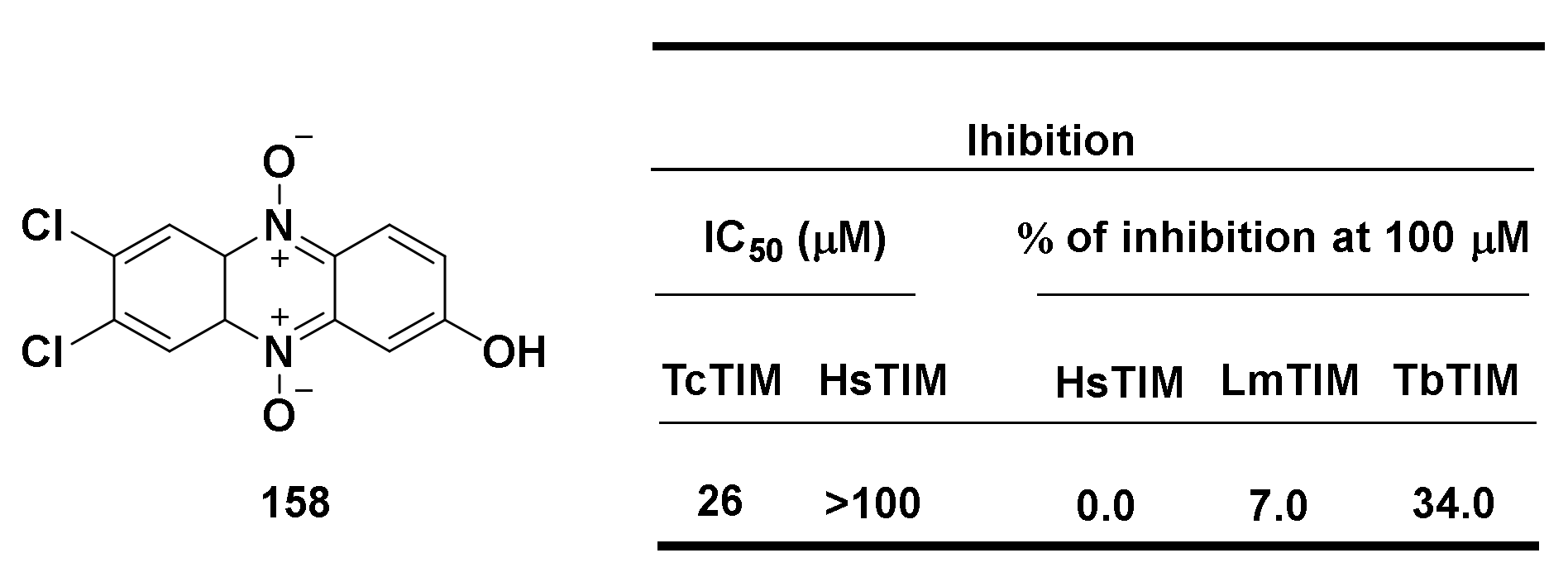
- Alvarez, G.; Martinez, J.; Aguirre-Lopez, B.; Cabrera, N.; Perez-Diaz, L.; Tuena de Gomez-Puyou, M.; Gomez-Puyou, A.; Perez-Montfort, R.; Garat, B.; Merlino, A.; et al. New chemotypes as Trypanosoma cruzi triosephosphate isomerase inhibitors: A deeper insight into the mechanism of inhibition. J. Enzyme Inhib. Med. Chem. 2014, 29, 1–7. [Google Scholar] [CrossRef]
- Minini, L.; Álvarez, G.; González, M.; Cerecetto, H.; Merlino, A. Molecular docking and molecular dynamics simulation studies of Trypanosoma cruzi triosephosphate isomerase inhibitors. Insights into the inhibition mechanism and selectivity. J. Mol. Graph. Model. 2015, 58, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Hernández, P.; Alem, D.; Nieves, M.; Cerecetto, H.; González, M.; Martínez-López, W.; Lavaggi, M.L. Chemosensitizer effect of cisplatin-treated bladder cancer cells by phenazine-5,10-dioxides. Environ. Toxicol. Pharmacol. 2019, 69, 9–15. [Google Scholar] [CrossRef]
- Viktorsson, E.Ö.; Grøthe, B.M.; Aesoy, R.; Sabir, M.; Snellingen, S.; Prandina, A.; Høgmoen Åstrand, O.A.; Bonge-Hansen, T.; Døskeland, S.O.; Herfindal, L.; et al. Total synthesis and antileukemic evaluations of the phenazine 5,10-dioxide natural products iodinin, myxin and their derivatives. Bioorg. Med. Chem. 2017, 25, 2285–2293. [Google Scholar] [CrossRef]
- Chowdhury, G.; Sarkar, U.; Pullen, S. DNA strand cleavage by the phenazine di-N-oxide natural product myxin under both aerobic and anaerobic conditions. Chem. Res. Toxicol. 2012, 25, 197–206. [Google Scholar] [CrossRef]
- Lavaggi, M.L.; Cabrera, M.; Celano, L.; Thomson, L.; Cerecetto, H.; González, M. Biotransformation of phenazine 5,10-dioxides under hypoxic conditions as an example of activation of anticancer prodrug: An interdisciplinary experiment for biochemistry or organic chemistry. J. Chem. Educ. 2013, 90, 1388–1391. [Google Scholar] [CrossRef]
- Gonda, M.; Nieves, M.; Nunes, E.; López De Ceráin, A.; Monge, A.; Lavaggi, M.L.; González, M.; Cerecetto, H.; Phenazine, N. N’-dioxide scaffold as selective hypoxic cytotoxin pharmacophore. Structural modifications looking for further DNA topoisomerase II-inhibition activity. Med. Chem. Commun. 2013, 4, 595–607. [Google Scholar] [CrossRef] [Green Version]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuo, S.; Li, C.; Hu, M.; Chen, S.; Yao, P.; Huang, S.; Ou, T.; Tan, J.; An, L.; Li, D. Synthesis and biological evaluation of benzo[a]phenazine derivatives as a dual inhibitor of topoisomerase I and II. Org. Biomol. Chem. 2013, 11, 3989–4005. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef]
- Gamage, S.A.; Spicer, J.A.; Rewcastle, G.W.; Milton, J.; Sohal, S.; Dangerfield, W.; Mistry, P.; Vicker, N.; Charlton, P.A.; Denny, W.A. Structure-activity relationships for pyrido-, imidazo-, pyrazolo-, pyrazino-, and pyrrolophenazinecarboxamides as topoisomerase-targeted anticancer agents. J. Med. Chem. 2002, 45, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, M.; Tong, X.; Zhu, H.; Yan, H.; Liu, D.; Li, W.; Qi, S.; Xiao, D.; Wang, Y.; et al. Synthesis, antitumor activity, and structure-activity relationship of some benzo[a]pyrano[2,3-c] phenazine derivatives. Comb. Chem. High Throughput Screen. 2015, 18, 960–974. [Google Scholar] [CrossRef]
- Whelan, P.; Dietrich, L.E.P.; Newman, D.K. Rethinking ‘secondary’ metabolism: Physiological roles for phenazine antibiotics. Nat. Chem. Biol. 2006, 2, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lu, Y.; Fang, L.; Fang, X.; Xing, Y.; Gou, S.; Xi, T. Synthesis and anticancer activity of some novel 2-phenazinamine Derivatives. Eur. J. Med. Chem. 2013, 69, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Khafagy, M.M.; El-Wahab, A.; Eid, F.A.; El-Agrody, A.M. Synthesis of halogen derivatives of benzo[h]chromene and benzo[a]anthracene with promising antimicrobial activities. Farmaco 2002, 57, 715–772. [Google Scholar] [CrossRef]
- Shchemelinin, I.; Sefc, L.; Nečas, E. Protein kinases, their function and implication in cancer and other diseases. Folia Biol. 2006, 52, 81–100. [Google Scholar]
- Endo, H.; Tada, M.; Katagiri, K. Biological characteristics of phenazine derivatives. IX. Effect on the fungal plant pathogen, Piricularia oryzae. Sci. Rept. Res. Inst. Tohoku Univ. Ser. C 1965, 12, 53. [Google Scholar]
- Dai, J.; Punchihewa, C.; Mistry, P.; Ooi, A.T.; Yang, D. Novel DNA bis-intercalation by MLN944, a potent clinical bisphenazine anticancer drug. J. Biol. Chem. 2004, 279, 46096–46103. [Google Scholar] [CrossRef] [PubMed] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure/Name | SF a,b,c | |
|---|---|---|
| Norm | Hypox | |
 159 R = NH2, Z 160 R = OH, Z | (159): 100 ± 10 (160): 100 ± 7 | (159): 45 ± 4 (160): 75 ± 2 |
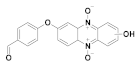 161 | Nd. | Nd. |
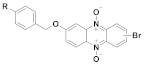 162 R = NO2, 163 R = Cl, 164 R = SCH3 | (162): 89 ± 10 (163): 100 ± 6 (164): 100 ± 6 | (162): 58 ± 4 (163): 44 ± 5 (164): 55 ± 3 |
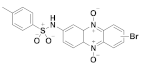 165 | (165): 57 ± 7 | (165): 66 ± 6 |
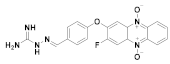 166 | (166): 100 ± 8 | (166): 73 ± 4 |
| Structure/Name | In Vitro Growth Inhibitory a IC50 Values | Relative Activity | |
|---|---|---|---|
| Topo I Cleavage b | Topo II ATPase Inhibition c | ||
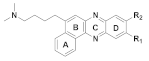 167 R1 = R2= H 168 R1 = H, R2 = OCH3 169 R1 = OCH3, R2= H | (167): IC50 = 3.99 μM (HeLa), 3.65 μM (A549), 5.01 μM (MCF-7), 1.40 μM (HL-60). | (167): +++ | (167): ++ |
| (168): IC50 = 4.36 μM (HeLa), 5.45 μM (A549), 2.83 μM (MCF-7), 2.36 μM (HL-60). | (168): − | (168): +++ | |
| (169): IC50 = 2.26 μM (HeLa), 2.24 μM (A549), 2.27 μM (MCF-7), 1.04 μM (HL-60). | (169): − | (169): ++ | |
 170 n = 1 171 n = 2 172 n = 3 | (170): IC50 = 0.31 μM (HL-60), 24.56 μM (K562), 9.91 μM (Hela), 22.31 μM (A549). | (170): Nd. | (170): Nd. |
| (171): IC50 = 0.21 μM (HL-60), 5.93 μM (K562), 8.41 μM (Hela), 13.51 μM (A549). | (171): Nd. | (171): Nd. | |
| (172): IC50 = 0.38 μM (HL-60), 4.73 μM (K562), 21.44 μM (Hela), 16.28 μM (A549). | (172): ++ | (172): ++++ | |
 173 R1 = R2 = H, R3= OCH3 174 R1= H, R2 = R3 = OCH3 | (173): IC50 = 0.22 μM (HL-60), 7.13 μM (K562), 29.95 μM (Hela), 32.96 μM (A549). | (173): Nd. | (173): ++++ |
| (174): IC50 = 0.09 μM (HL-60), 8.73 μM (K562), 18.23 μM (Hela), 26.45 μM (A549). | (174): Nd. | (174): ++++ | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, J.; Liu, W.; Cai, J.; Wang, Y.; Li, D.; Hua, H.; Cao, H. Advances in Phenazines over the Past Decade: Review of Their Pharmacological Activities, Mechanisms of Action, Biosynthetic Pathways and Synthetic Strategies. Mar. Drugs 2021, 19, 610. https://doi.org/10.3390/md19110610
Yan J, Liu W, Cai J, Wang Y, Li D, Hua H, Cao H. Advances in Phenazines over the Past Decade: Review of Their Pharmacological Activities, Mechanisms of Action, Biosynthetic Pathways and Synthetic Strategies. Marine Drugs. 2021; 19(11):610. https://doi.org/10.3390/md19110610
Chicago/Turabian StyleYan, Junjie, Weiwei Liu, Jiatong Cai, Yiming Wang, Dahong Li, Huiming Hua, and Hao Cao. 2021. "Advances in Phenazines over the Past Decade: Review of Their Pharmacological Activities, Mechanisms of Action, Biosynthetic Pathways and Synthetic Strategies" Marine Drugs 19, no. 11: 610. https://doi.org/10.3390/md19110610
APA StyleYan, J., Liu, W., Cai, J., Wang, Y., Li, D., Hua, H., & Cao, H. (2021). Advances in Phenazines over the Past Decade: Review of Their Pharmacological Activities, Mechanisms of Action, Biosynthetic Pathways and Synthetic Strategies. Marine Drugs, 19(11), 610. https://doi.org/10.3390/md19110610