
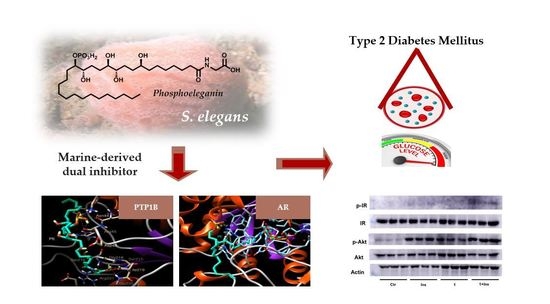
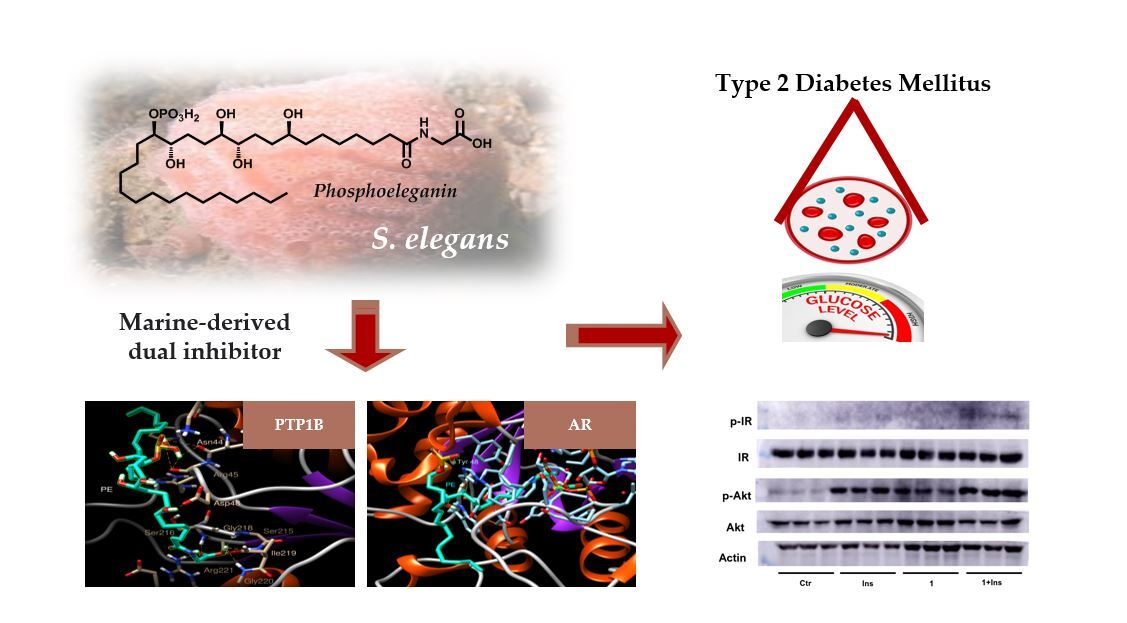
Dual Targeting of PTP1B and Aldose Reductase with Marine Drug Phosphoeleganin: A Promising Strategy for Treatment of Type 2 Diabetes

 ,
,  ,
,  ,
, 

 ,
,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
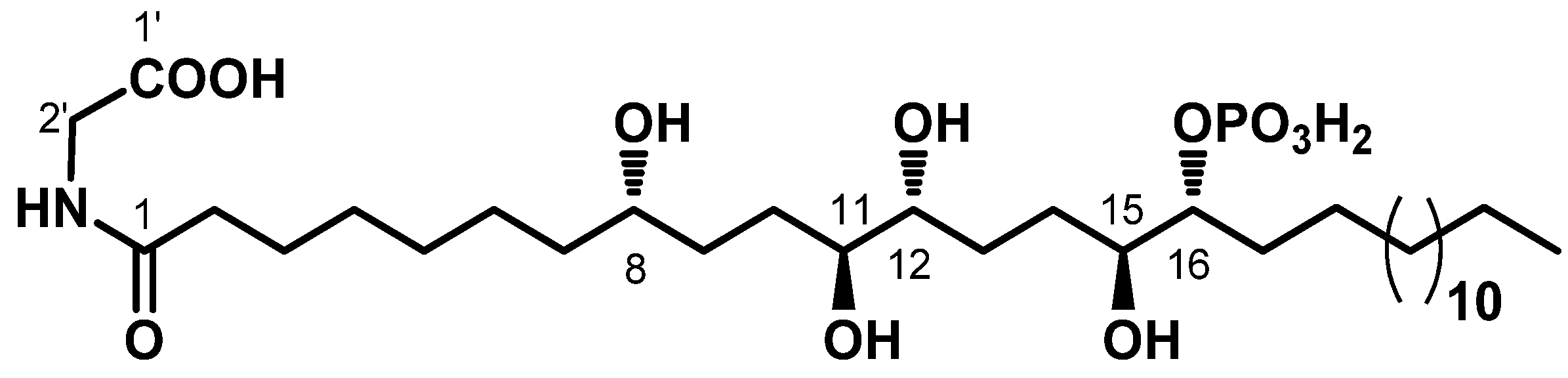
2.1. Chemistry
2.2. Preliminary Screening on PTP1B and AR In Vitro
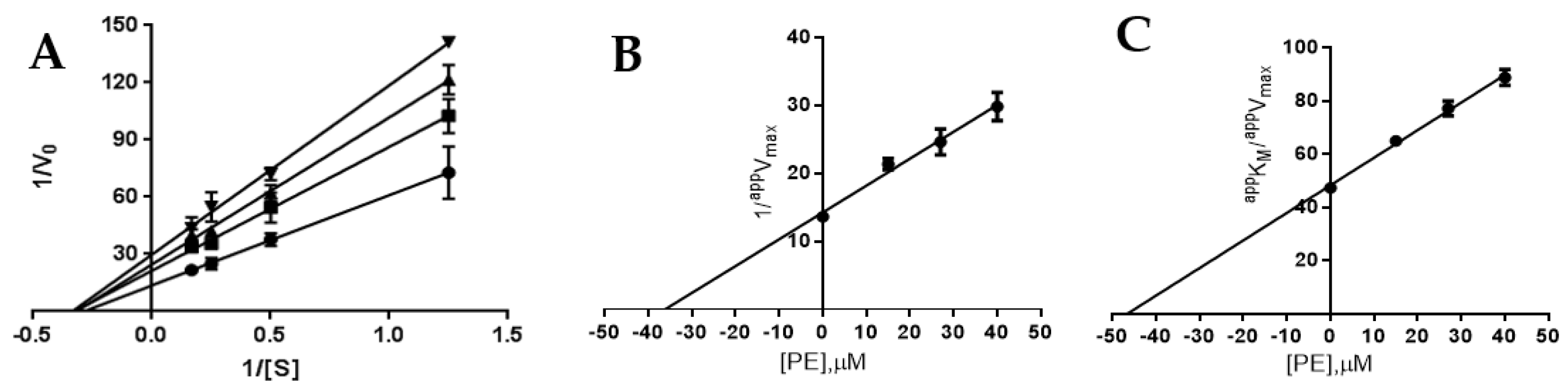
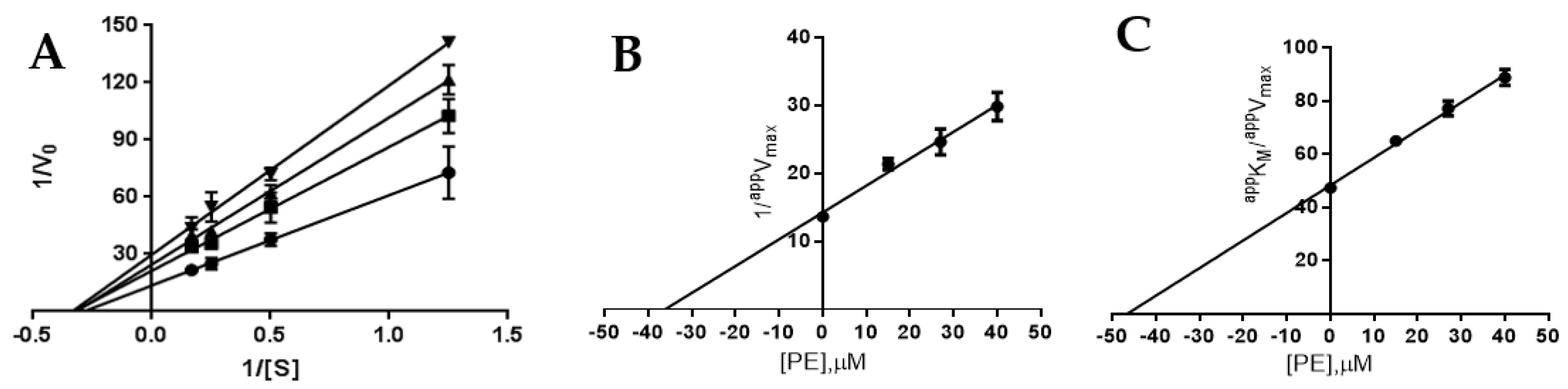
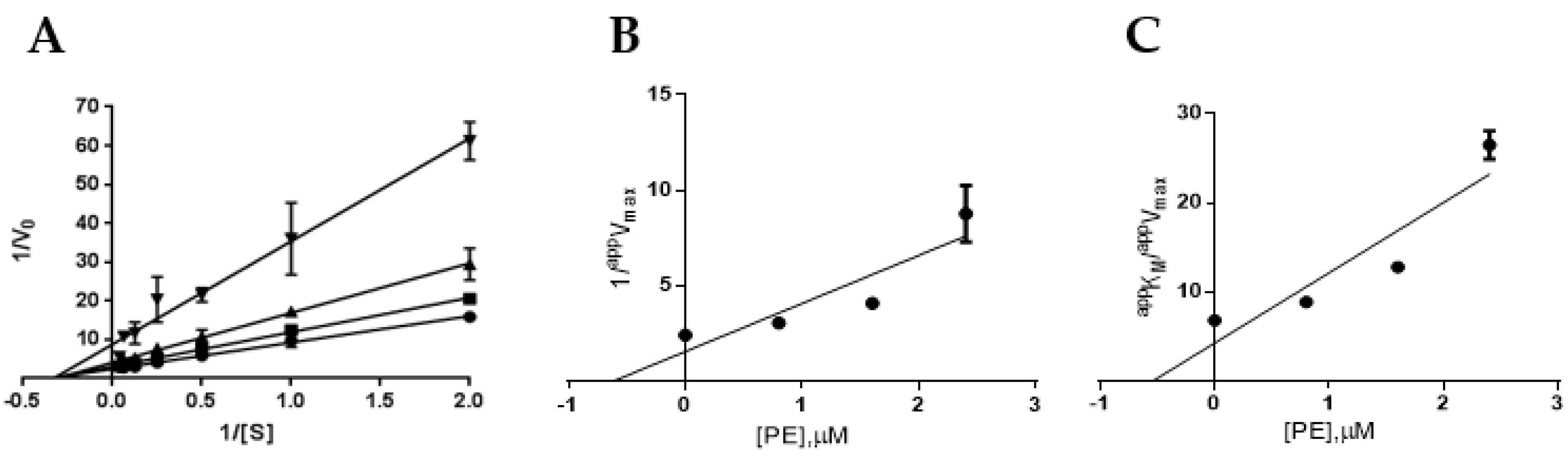
2.3. Definition of PTP1B and AR Inhibition Mechanism by PE
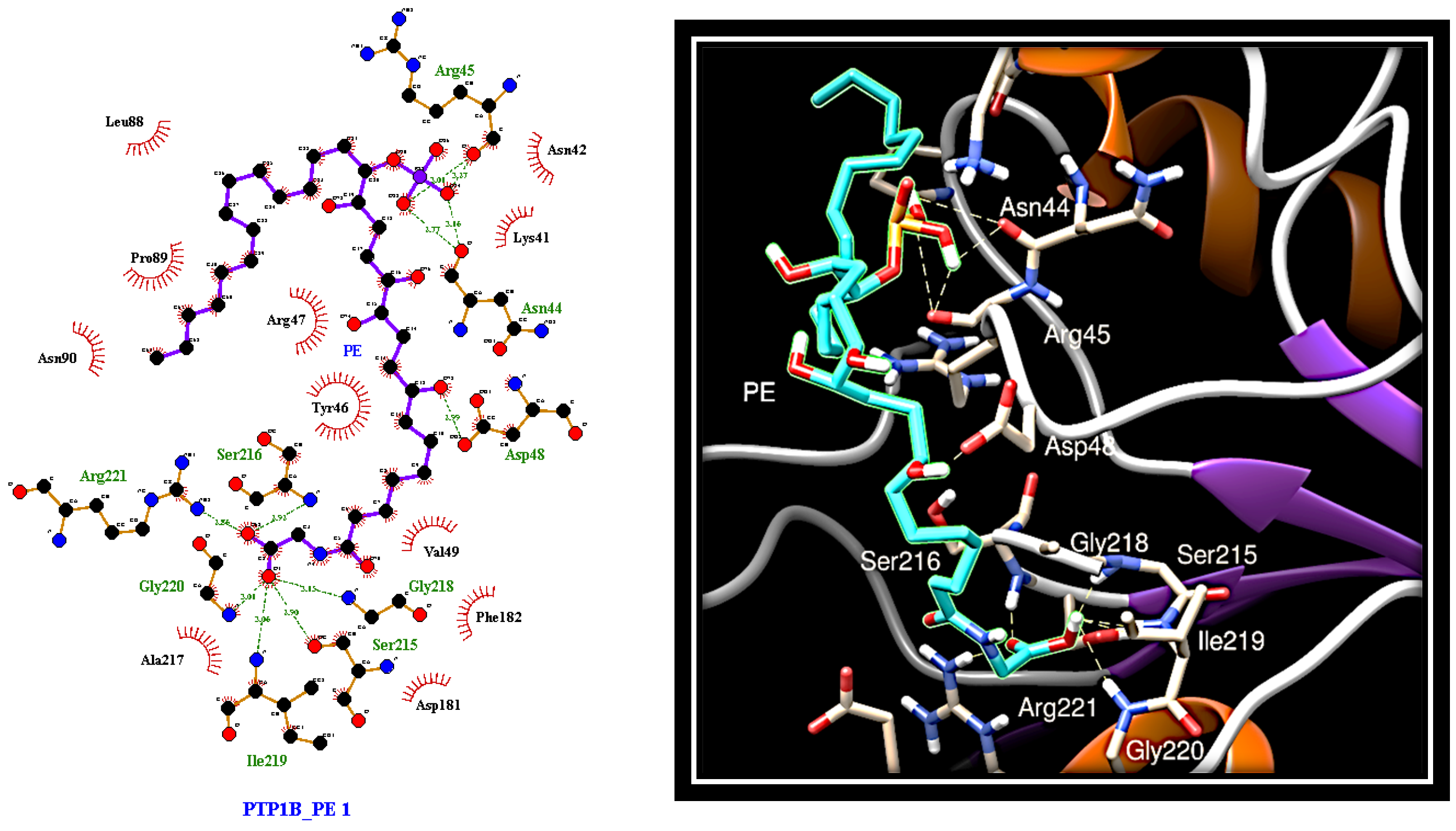
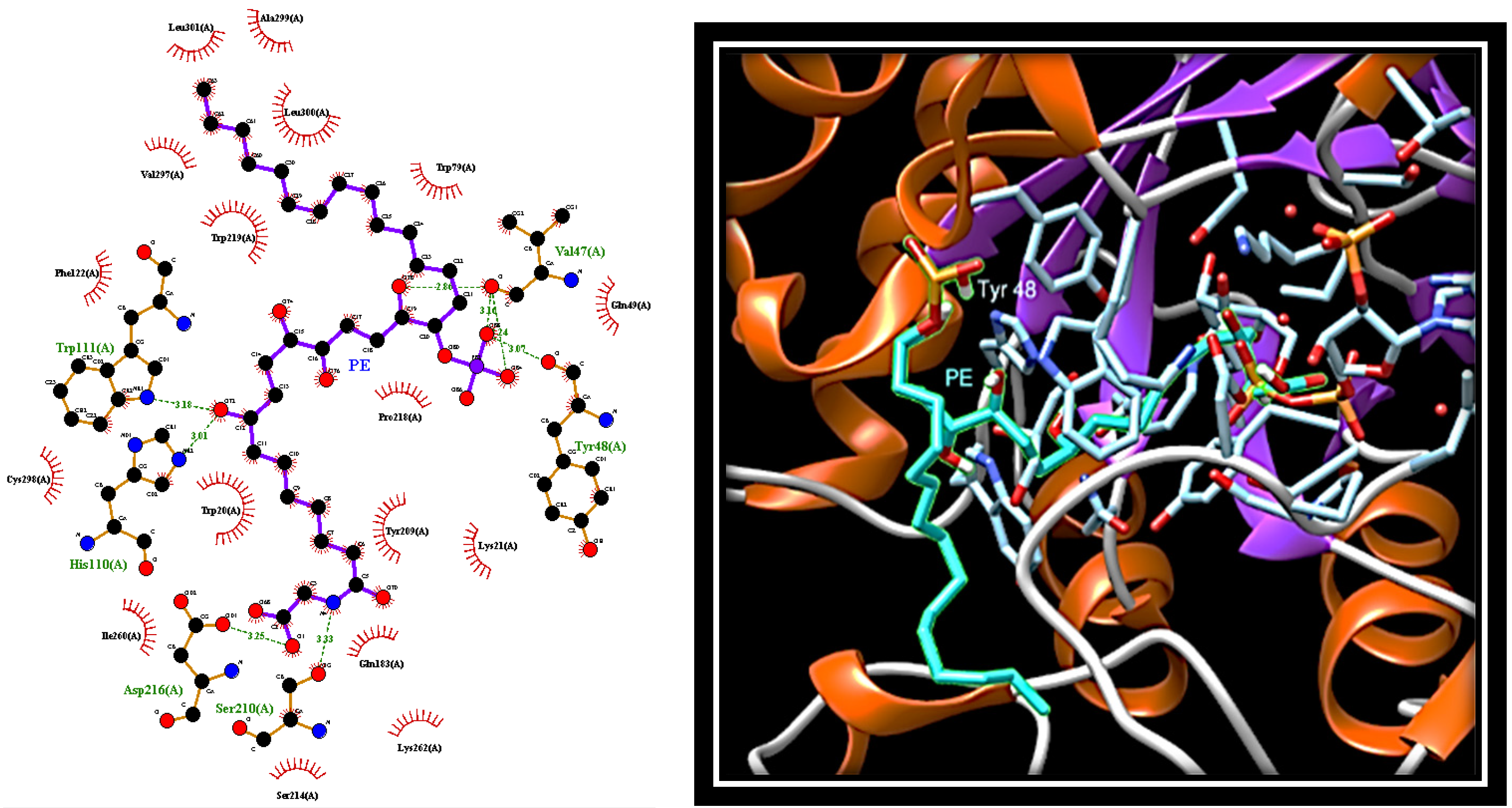
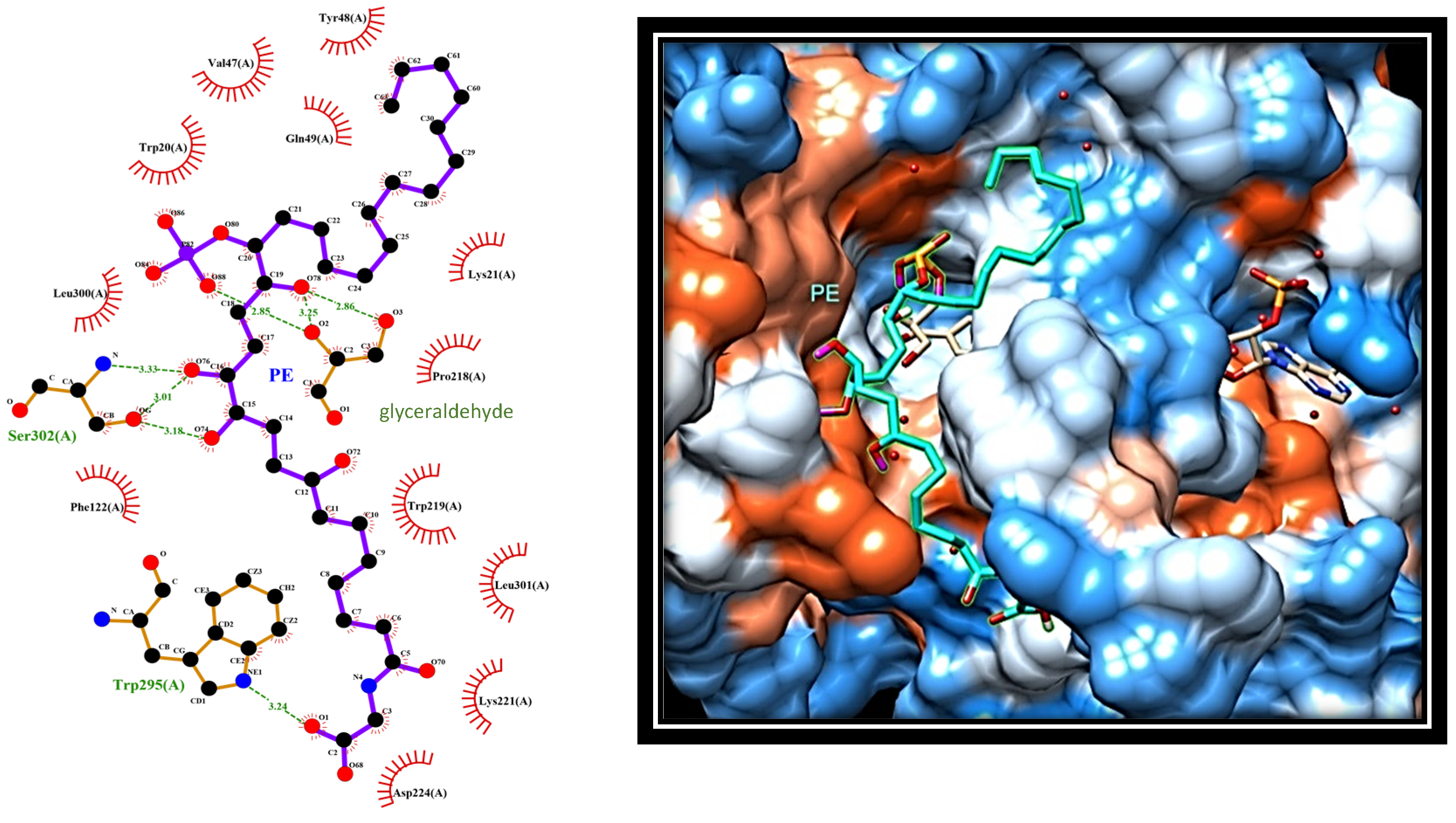
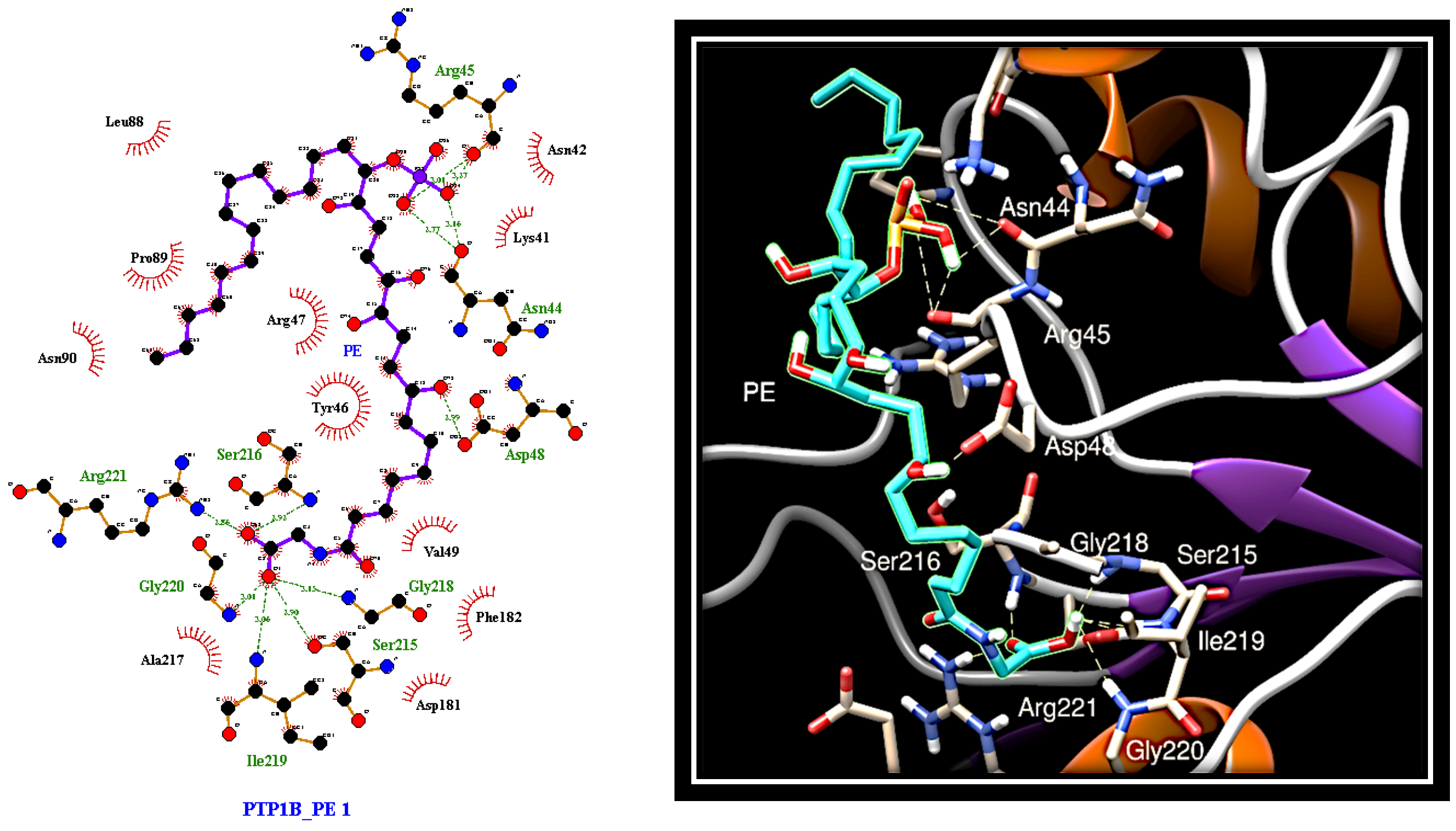
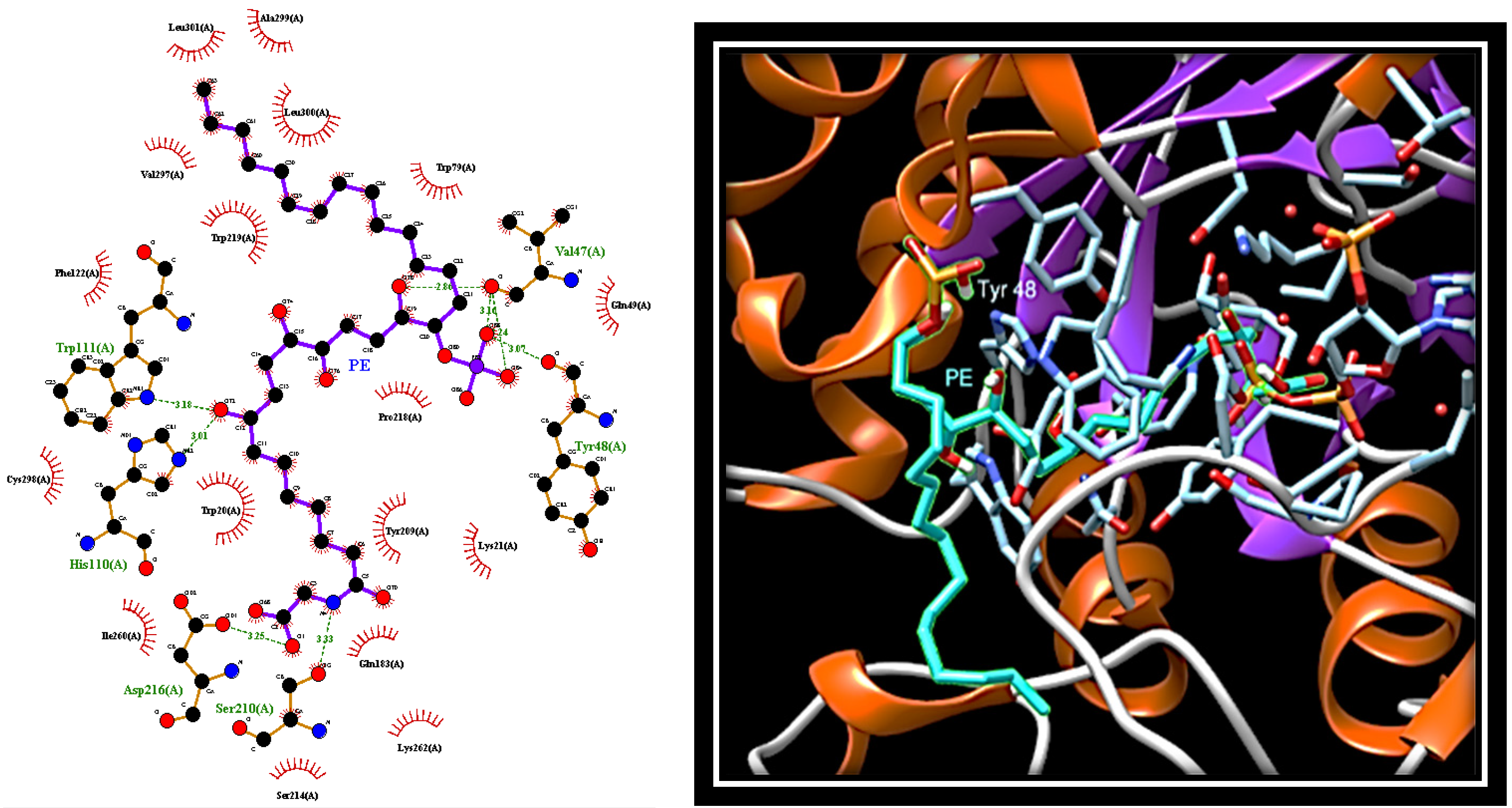
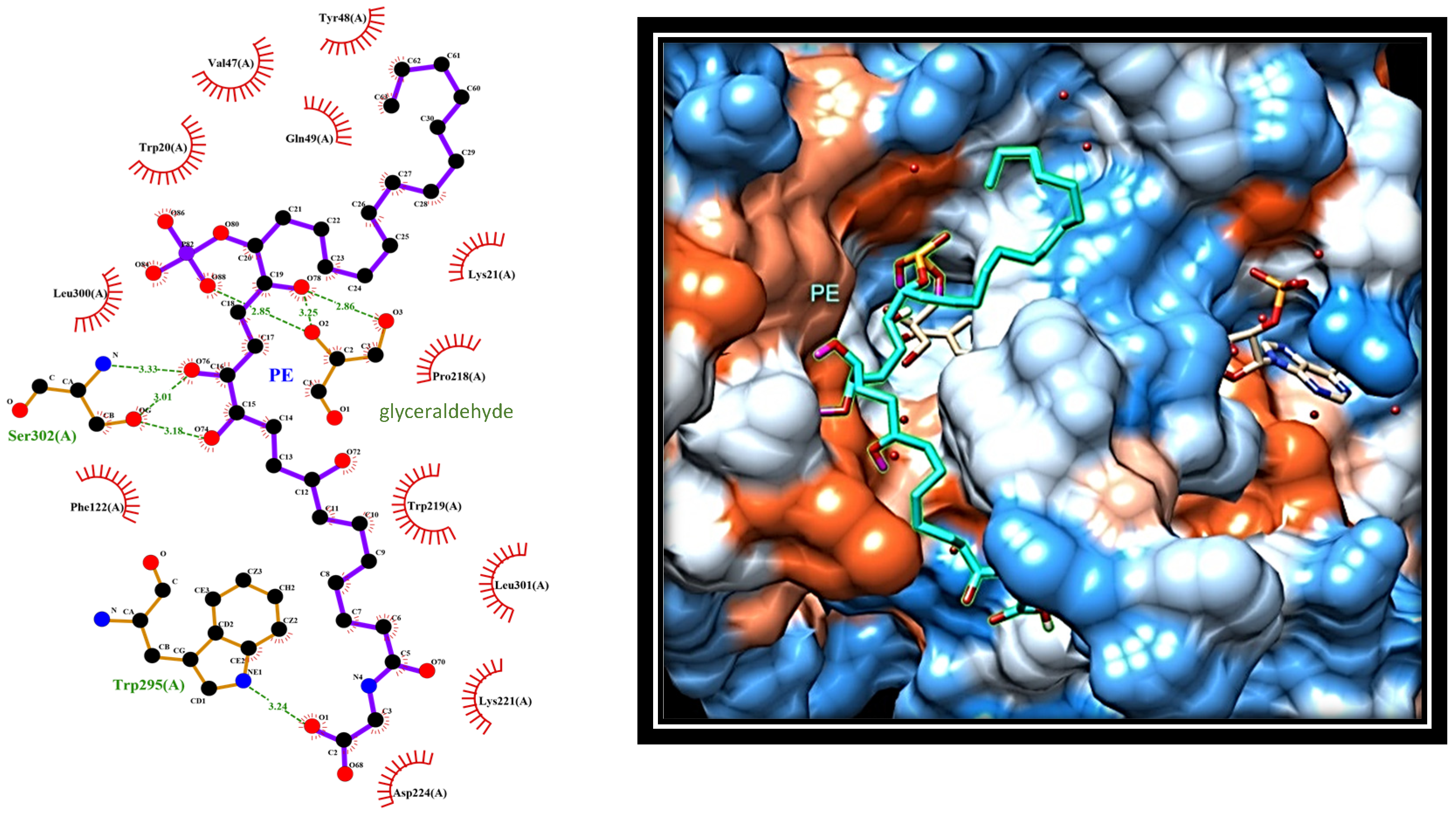
2.4. Dissecting the Interaction of PE with PTP1B and AR by In Silico Docking Analyses
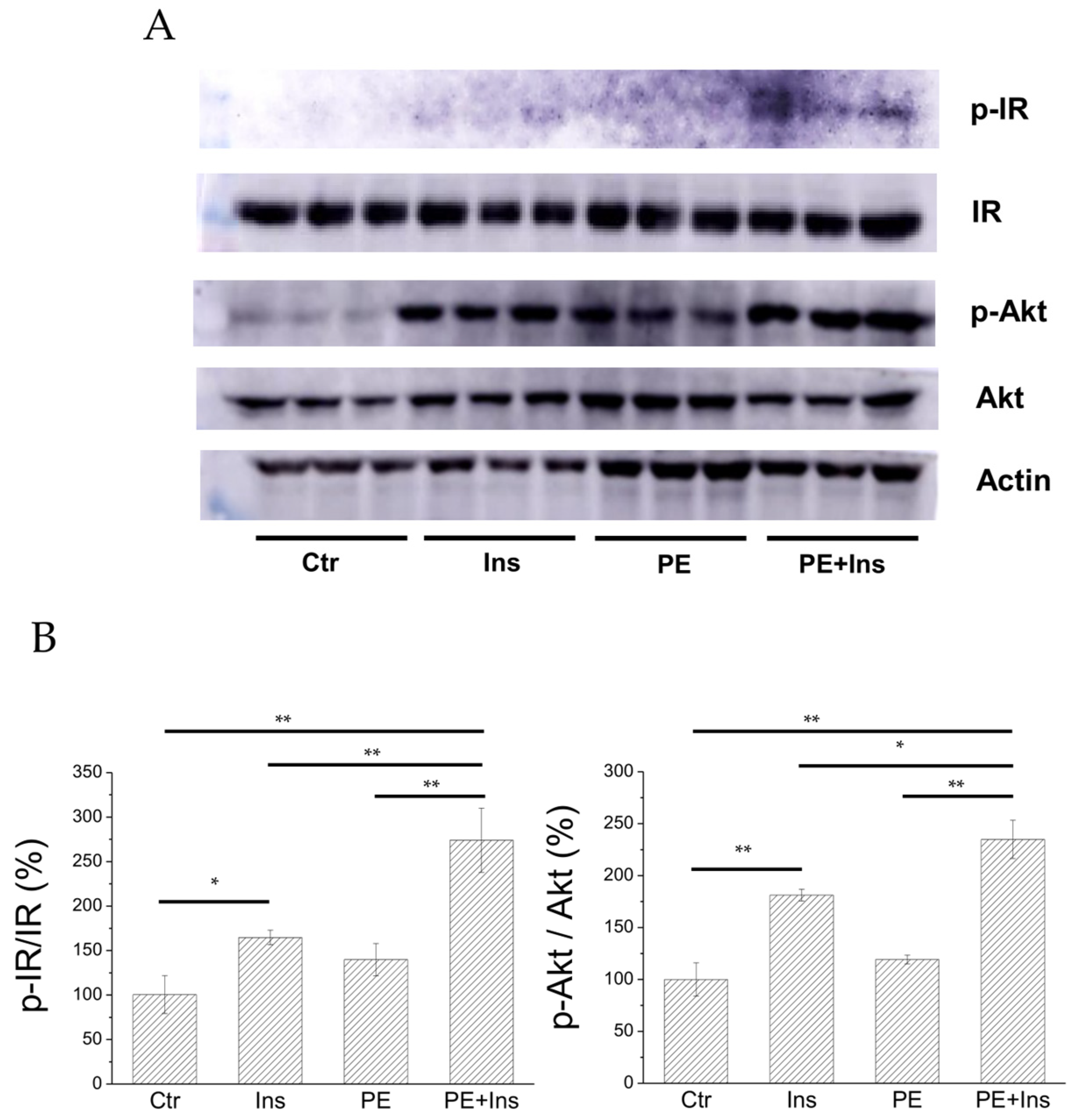
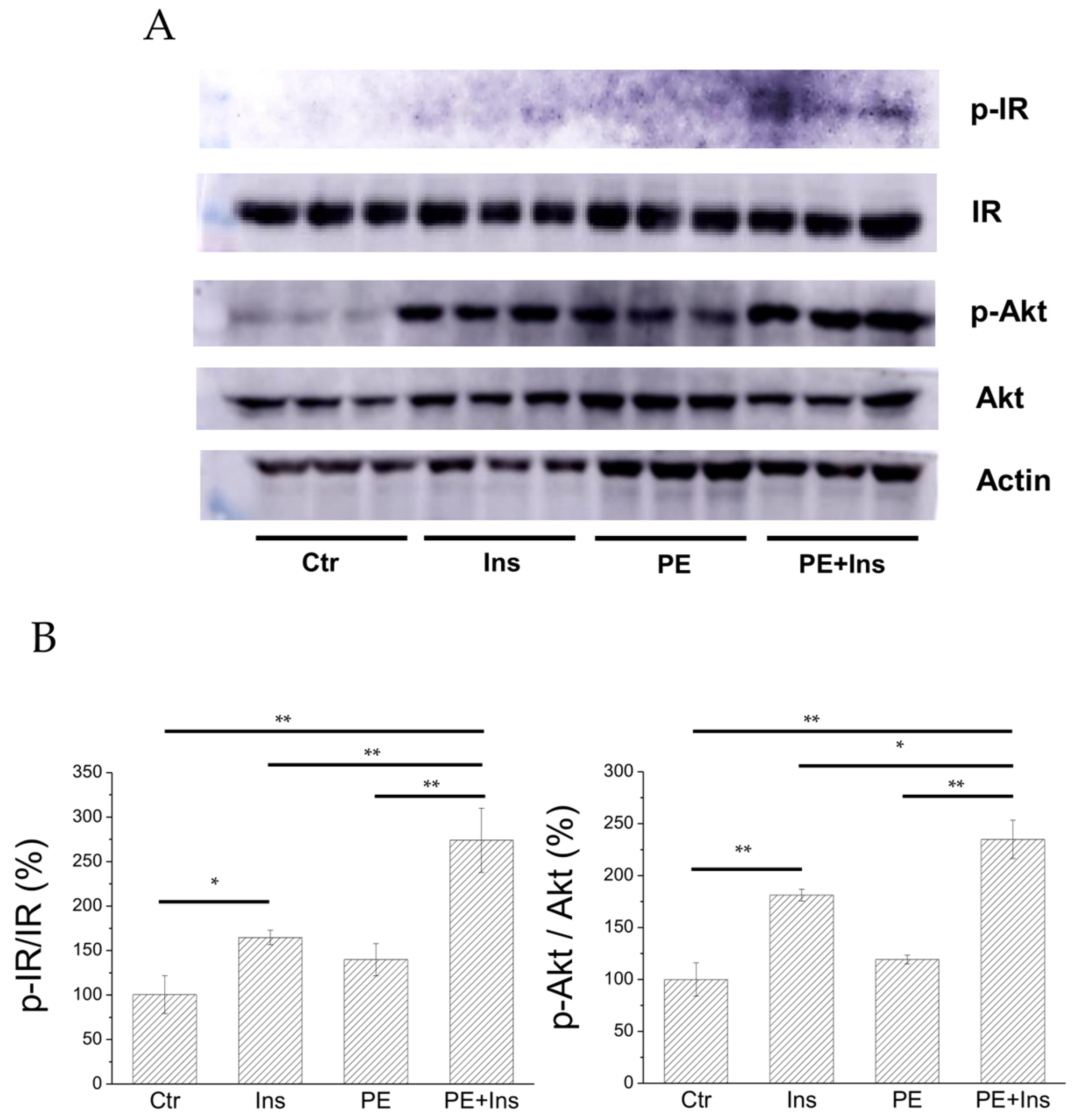
2.5. Analysis of the Insulin Signalling Pathway in Liver Cells Treated with PE
3. Discussion
4. Material and Methods
4.1. Materials
4.2. Collection, Extraction, and Isolation of PE from Specimens of S. elegans
4.3. Expression and Purification of AR and PTP1B
4.4. Determination of Enzymatic Activities
4.5. Inhibition Studies
4.6. Cell Cultures
4.7. Analysis of the Insulin Signalling Pathway
4.8. Molecular Docking
4.9. Statistical Analysis of Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Costantino, L.; Barlocco, D. Challenges in the design of multitarget drugs against multifactorial pathologies: A new life for medicinal chemistry? Future Med. Chem. 2013, 5, 5–7. [Google Scholar] [CrossRef] [Green Version]
- Bolognesi, M.L. Polypharmacology in a single drug: Multitarget drugs. Curr. Med. Chem. 2013, 20, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Cavalli, A. Multitarget drug discovery and polypharmacology. Chem. Med. Chem. 2016, 11, 1190–1192. [Google Scholar] [CrossRef] [Green Version]
- Csermely, P.; Agoston, V.; Pongor, S. The efficiency of multi-target drugs: The network approach might help drug design. Trends Pharmacol. Sci. 2005, 26, 178–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morphy, R.; Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Hopkins, A. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Rankovic, Z. Designing multiple ligands-medicinal chemistry strategies and challenges. Curr. Pharm. Des. 2009, 15, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Bagetta, D.; Maruca, A.; Lupia, A.; Mesiti, F.; Catalano, R.; Romeo, I.; Moraca, F.; Ambrosio, F.A.; Costa, G.; Artese, A.; et al. Mediterranean products as promising source of multi-target agents in the treatment of metabolic syndrome. Eur. J. Med. Chem. 2020, 186, 111903. [Google Scholar] [CrossRef]
- Shaw, J.E.; Sicree, R.A.; Zimmet, P.Z. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 2010, 87, 4–14. [Google Scholar] [CrossRef]
- Nolan, C.J.; Damm, P.; Prentki, M. Type 2 diabetes across generations: From pathophysiology to prevention and management. Lancet 2011, 378, 169–181. [Google Scholar] [CrossRef]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Inv. 2000, 106, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Vela, M.E.; Torres, N.; Tovar, A.R. White adipose tissue as endocrine organ and its role in obesity. Arch. Med. Res. 2008, 39, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Mechanick, J.I.; Zhaoz, S.; Garvey, W.T. Leptin, an Adipokine with Central Importance in the Global Obesity Problem. Glob. Heart 2018, 13, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Elchebly, M.; Payette, P.; Michaliszyn, E.; Cromlish, W.; Collins, S.; Loy, A.L.; Normandin, D.; Cheng, A.; Himms-Hagen, J.; Chan, C.C.; et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Z.Y. PTP1B as a drug target: Recent developments in PTP1B inhibitor discovery. Drug Discov. 2007, 12, 373–381. [Google Scholar] [CrossRef]
- Chung, S.S.; Ho, E.C.; Lam, K.S.; Chung, S.K. Contribution of polyol pathway to diabetes-induced oxidative stress. J. Am. Soc. Nephrol. 2003, 14, 233–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotfy, M.; Adeghate, J.; Kalasz, H.; Singh, J.; Adeghate, E. Chronic Complications of Diabetes Mellitus: A Mini Review. Curr. Diabetes Rev. 2017, 13, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottanà, R.; Paoli, P.; Cappiello, M.; Nguyen, T.N.; Adornato, I.; Del Corso, A.; Genovese, M.; Nesi, I.; Moschini, R.; Naß, A.; et al. In Search for Multi-Target Ligands as Potential Agents for Diabetes Mellitus and Its Complications- A Structure-Activity Relationship Study on Inhibitors of Aldose Reductase and Protein Tyrosine Phosphatase 1B. Molecules 2021, 26, 330. [Google Scholar] [CrossRef]
- Maccari, R.; Del Corso, A.; Paoli, P.; Adornato, I.; Lori, G.; Balestri, F.; Cappiello, M.; Naß, A.; Wolber, G.; Ottanà, R. An investigation on 4-thiazolidinone derivatives as dual inhibitors of aldose reductase and protein tyrosine phosphatase 1B, in the search for potential agents for the treatment of type 2 diabetes mellitus and its complications. Bioorg. Med. Chem. Lett. 2018, 28, 3712–3720. [Google Scholar] [CrossRef]
- Kousaxidis, A.; Petrou, A.; Lavrentaki, V.; Fesatidou, M.; Nicolaou, J.; Geronikaki, A. Aldose reductase and protein tyrosine phosphatase 1B inhibitors as a promising therapeutic approach for diabetes mellitus. Eur. J. Med. Chem. 2020, 207, 112742. [Google Scholar] [CrossRef]
- International Diabetes Federation. Access to Medicines and Supplies for People with Diabetes. Brussels, Belgium: International Diabetes Federation. 2016. Available online: www.idf.org/accesstomedicine (accessed on 22 September 2021).
- Eddouks, M.; Bidi, A.; El Bouhali, B.; Hajji, L.; Zeggwagh, N.A. Antidiabetic plants improving insulin sensitivity. J. Pharm. Pharmacol. 2014, 66, 1197–1214. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, H.; Wang, S.; Wang, W.; Chen, Q.; Ma, Y.; Zhang, Y.; Wang, T. Natural products, an important resource for discovery of multitarget drugs and functional food for regulation of hepatic glucose metabolism. Drug Des. Devel. Ther. 2018, 10, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Genovese, M.; Nesi, I.; Caselli, A.; Paoli, P. Natural α-Glucosidase and Protein Tyrosine Phosphatase 1B Inhibitors: A Source of Scaffold Molecules for Synthesis of New Multitarget Antidiabetic Drugs. Molecules 2021, 26, 4818. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.; Preto, M.; Vasconcelos, V.; Urbatzka, R. Obesity: The Metabolic Disease, Advances on Drug Discovery and Natural Product Research. Curr. Top. Med. Chem. 2016, 16, 2577–2604. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Imperatore, C.; Della Sala, G.; Casertano, M.; Luciano, P.; Aiello, A.; Laurenzana, I.; Piccoli, P.; Menna, M. In Vitro Antiproliferative Evaluation of Synthetic Meroterpenes Inspired by Marine Natural Products. Mar. Drugs 2019, 17, 684. [Google Scholar] [CrossRef] [Green Version]
- Imperatore, C.; Gimmelli, R.; Persico, M.; Casertano, M.; Guidi, A.; Saccoccia, F.; Ruberti, G.; Luciano, P.; Aiello, A.; Parapini, S.; et al. Investigating the Antiparasitic Potential of the Marine Sesquiterpene Avarone, Its Reduced Form Avarol, and the Novel Semisynthetic Thiazinoquinone Analogue Thiazoavarone. Mar. Drugs 2020, 18, 112. [Google Scholar] [CrossRef] [Green Version]
- Gunathilaka, T.L.; Samarakoon, K.; Ranasinghe, P.; Peiris, L.D.C. Antidiabetic Potential of Marine Brown Algae-a Mini Review. J. Diabetes Res. 2020, 1230218. [Google Scholar] [CrossRef]
- Ezzat, S.M.; Bishbishy, M.H.E.; Habtemariam, S.; Salehi, B.; Sharifi-Rad, M.; Martins, N.; Sharifi-Rad, J. Looking at Marine-Derived Bioactive Molecules as Upcoming Anti-Diabetic Agents: A Special Emphasis on PTP1B Inhibitors. Molecules 2018, 23, 3334. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.T.; Nguyen, D.H.; Le, D.D.; Choi, J.S.; Min, B.S.; Woo, M.H. Protein tyrosine phosphatase 1B inhibitors from natural sources. Arch. Pharm. Res. 2018, 41, 130–161. [Google Scholar] [CrossRef]
- Manzanaro, S.; Salva, J.; de la Fuente, J.A. Phenolic marine natural products as aldose reductase inhibitors. J. Nat. Prod. 2006, 69, 1485–1487. [Google Scholar] [CrossRef]
- Imperatore, C.; Luciano, P.; Aiello, A.; Vitalone, R.; Irace, C.; Santamaria, R.; Li, J.; Guo, Y.W.; Menna, M. Structure and Configuration of Phosphoeleganin, a Protein Tyrosine Phosphatase 1B Inhibitor from the Mediterranean Ascidian Sidnyum elegans. J. Nat. Prod. 2016, 79, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Luciano, P.; Imperatore, C.; Senese, M.; Aiello, A.; Casertano, M.; Guo, Y.W.; Menna, M. Assignment of the Absolute Configuration of Phosphoeleganin via Synthesis of Model Compounds. J. Nat. Prod. 2017, 80, 2118–2123. [Google Scholar] [CrossRef] [Green Version]
- Asante-Appiah, E.; Patel, S.; Dufresne, C.; Roy, P.; Wang, Q.; Patel, V.; Friesen, R.W.; Ramachandran, C.; Becker, J.W.; Leblanc, Y.; et al. The structure of PTP-1B in complex with a peptide inhibitor reveals an alternative binding mode for bisphosphonates. Biochemistry 2002, 41, 9043–9051. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.P.; Fedorov, A.A.; Lee, S.Y.; Guo, X.L.; Shen, K.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.Y. Crystal structure of PTP1B complexed with a potent and selective bidentate inhibitor. J. Biol. Chem. 2003, 278, 12406–12414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinajero, M.G.; Malik, V.S. An Update on the Epidemiology of Type 2 Diabetes: A Global Perspective. Endocrinol. Metab. Clin. North. Am. 2021, 50, 337–355. [Google Scholar] [CrossRef]
- Thakur, S.; Gupta, S.K.; Ali, V.; Singh, P.; Verma, M. Aldose Reductase: A cause and a potential target for the treatment of diabetic complications. Arch. Pharm. Res. 2021. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Lee, S.Y.; Andersen, J.N.; Waters, S.; Shen, K.; Guo, X.L.; Moller, N.P.; Olefsky, J.M.; Lawrence, D.S.; Zhang, Z.Y. Cellular effects of small molecule PTP1B inhibitors on insulin signaling. Biochemistry 2003, 42, 12792–12804. [Google Scholar] [CrossRef]
- Ottanà, R.; Maccari, R.; Mortier, J.; Caselli, A.; Amuso, S.; Camici, G.; Rotondo, A.; Wolber, G.; Paoli, P. Synthesis, biological activity and structure-activity relationships of new benzoic acid-based protein tyrosine phosphatase inhibitors endowed with insulinomimetic effects in mouse C2C12 skeletal muscle cells. Eur. J. Med. Chem. 2014, 71, 112–127. [Google Scholar] [CrossRef]
- Qin, Z.; Pandey, N.R.; Zhou, X.; Stewart, C.A.; Hari, A.; Huang, H.; Stewart, A.F.; Brunel, J.M.; Chen, H.H. Functional properties of Claramine: A novel PTP1B inhibitor and insulin-mimetic compound. Biochem. Biophys. Res. Commun. 2015, 458, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randal, M.; et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730–737. [Google Scholar] [CrossRef]
- Hooft van Huijsduijnen, R.; Sauer, W.H.; Bombrun, A.; Swinnen, D. Prospects for inhibitors of protein tyrosine phosphatase 1B as antidiabetic drugs. J. Med. Chem. 2004, 47, 4142–4146. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Cappiello, M.; Moschini, R.; Rotondo, R.; Buggiani, I.; Pelosi, P.; Mura, U.; Del Corso, A. L-Idose: An attractive substrate alternative to D-glucose for measuring aldose reductase activity. Biochem. Biophys. Res. Commun. 2015, 456, 891–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestri, F.; Rotondo, R.; Moschini, R.; Pellegrino, M.; Cappiello, M.; Barracco, V.; Misuri, L.; Sorce, C.; Andreucci, A.; Del Corso, A.; et al. Zolfino landrace (Phaseolus vulgaris L.) from Pratomagno: General and specific features of a functional food. Food Nutr. Res. 2016, 60, 31792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottanà, R.; Paoli, P.; Lori, G.; Adornato, I.; Previti, S.; Naß, A.; Wolber, G.; Maccari, R. Design and evaluation of non-carboxylate 5-arylidene-2-thioxo-4-imidazolidinones as novel non-competitive inhibitors of protein tyrosine phosphatase 1B. Bioorg. Chem. 2019, 92, 103211. [Google Scholar] [CrossRef]
- Misuri, L.; Cappiello, M.; Balestri, F.; Moschini, R.; Barracco, V.; Mura, U.; Del Corso, A. The use of dimethylsulfoxide as a solvent in enzyme inhibition studies: The case of aldose reductase. J. Enz. Inhib. Med. Chem. 2017, 32, 1152–1158. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scapin, G.; Patel, S.B.; Becker, J.W.; Wang, Q.; Desponts, C.; Waddleton, D.; Skorey, K.; Cromlish, W.; Bayly, C.; Therien, M.; et al. The Structural Basis for the Selectivity of Benzotriazole Inhibitors of PTP1B. Biochemistry 2003, 42, 11451–11459. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, L.; Chen, W.; Chen, Y.; Xie, W.; Hu, X. Partial Inhibition of Aldose Reductase by Nitazoxanide and Its Molecular Basis. Chem. Med. Chem. 2012, 7, 1921–1923. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.S.; Liang, L.F.; Guo, Y.W. Natural products possessing protein tyrosine phosphatase 1B (PTP1B) inhibitory activity found in the last decades. Acta Pharmacol. Sin. 2012, 33, 1217–1245. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Martin, K.A.; Hwa, J. Aldose reductase, oxidative stress, and diabetic mellitus. Front. Pharmacol. 2012, 3, 87. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genovese, M.; Imperatore, C.; Casertano, M.; Aiello, A.; Balestri, F.; Piazza, L.; Menna, M.; Del Corso, A.; Paoli, P. Dual Targeting of PTP1B and Aldose Reductase with Marine Drug Phosphoeleganin: A Promising Strategy for Treatment of Type 2 Diabetes. Mar. Drugs 2021, 19, 535. https://doi.org/10.3390/md19100535
Genovese M, Imperatore C, Casertano M, Aiello A, Balestri F, Piazza L, Menna M, Del Corso A, Paoli P. Dual Targeting of PTP1B and Aldose Reductase with Marine Drug Phosphoeleganin: A Promising Strategy for Treatment of Type 2 Diabetes. Marine Drugs. 2021; 19(10):535. https://doi.org/10.3390/md19100535
Chicago/Turabian StyleGenovese, Massimo, Concetta Imperatore, Marcello Casertano, Anna Aiello, Francesco Balestri, Lucia Piazza, Marialuisa Menna, Antonella Del Corso, and Paolo Paoli. 2021. "Dual Targeting of PTP1B and Aldose Reductase with Marine Drug Phosphoeleganin: A Promising Strategy for Treatment of Type 2 Diabetes" Marine Drugs 19, no. 10: 535. https://doi.org/10.3390/md19100535
APA StyleGenovese, M., Imperatore, C., Casertano, M., Aiello, A., Balestri, F., Piazza, L., Menna, M., Del Corso, A., & Paoli, P. (2021). Dual Targeting of PTP1B and Aldose Reductase with Marine Drug Phosphoeleganin: A Promising Strategy for Treatment of Type 2 Diabetes. Marine Drugs, 19(10), 535. https://doi.org/10.3390/md19100535