Abstract
Four polyene macrolactams including the previously reported niizalactam C (4), and three new ones, streptolactams A–C (1–3) with a 26-membered monocyclic, [4,6,20]-fused tricyclic and 11,23-oxygen bridged [14,16]-bicyclic skeletons, respectively, were isolated from the fermentation broth of the deep-sea sediment-derived Streptomyces sp. OUCMDZ-3159. Their structures were determined based on spectroscopic analysis, X-ray diffraction analysis, and chemical methods. The abiotic formation of compounds 2 and 4 from compound 1 were confirmed by a series of chemical reactions under heat and light conditions. Compounds 1 and 3 showed a selective antifungal activity against Candida albicans ATCC 10231.
1. Introduction
Macrolactams isolated from actinobacteria have become a large family of natural products, which showed lots of different biological activities and received more and more attention [1,2,3,4]. Some molecules with novel macrolactam frameworks and significant activities have been isolated from various actinobacterial strains, such as dracolactams [5], bombyxamycins [6], macrotermycins [7], verticilactam [8], sceliphrolactam [9], tripartilactam [10], and niizalactam C (4) [11]. The structure of tripartilactam has been revised to be the same as niizalactam C (4) [12]. It possesses a fused [18,6,6]-tricyclic system, which was proposed to be formed from sceliphrolactam via a spontaneous intramolecular [4 + 2] cycloaddition [12].
We have been working on the active metabolites of marine-derived actinobacteria, especially on macrolactams [13,14,15,16]. Some interesting natural products with cytotoxic activity, represented by cyclamenols A−F [15,16], have been identified. To further discover new macrolactams for biological study from marine-derived actinobacteria, the deep-sea sediment-derived Streptomyces sp. OUCMDZ-3159 was selected. This strain was found to produce streptolactam A (1) as the main secondary metabolite. In addition, some trace analogues could be detected by LC-MS. In order to identify these structures, it was fermented on a 30 L scale. As a result, three novel macrolactams, streptolactams A–C (1–3), together with the reported niizalactam C (4) [11] (Figure 1), were isolated and identified. Structurally, streptolactam A (1) is a 26-membered cyclic polyene macrolactam [9], while streptolactam B (2) possesses a novel [20,4,6]-fused tricyclic skeleton, and streptolactam C (3) has an 11,23-oxgen bridged [14,16] bicyclic system. Herein, we reported the isolation, cytotoxic and antimicrobial activity, as well as the complete structural elucidation of compounds 1–4.
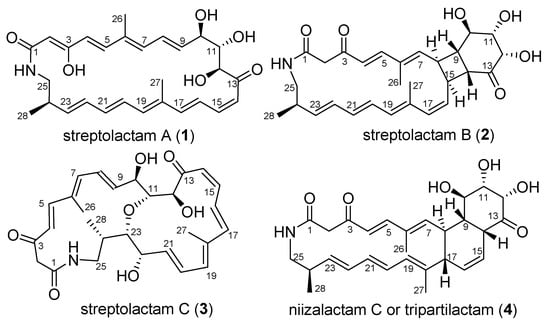
Figure 1.
Chemical structures of compounds 1–4.
2. Results and Discussion
2.1. Structural Elucidation
Compound 1 was obtained as a yellow powder. Its molecular formula could be assigned as C28H35NO6 from the HRESIMS peak at m/z 482.2529 [M + H]+ (calcd 482.2537) (Figure S6 in supporting information). The similarity of 1H and 13C NMR data (Table 1) between compound 1 and sceliphrolactam [9] indicated that they share the same constitution, planar structure, which was confirmed by COSY of H-4/H-5, H-7/H-8/H-9/H-10/H-11/H-12, H-14/H-15/H-16/H-17, H-19/H-20/H-21/H-22/H-23/H-24/H2-25/NH, and H-24/H3-28 (Figure 2 and Figure S4), along with the key HMBC of NH to C-1 and C-2, H-2 to C-1, C-3 and C-4, H3-26 to C-5, C-6 and C-7, H-12 and H-15 to C-13, H3-27 to C-17 and C-19, and H-19 to C-17 (Figure 2 and Figure S5). However, absolute configuration of sceliphrolactam was not determined for its extreme sensitivity under light or heat conditions [9]. In the present study, we tried to solve this issue and first tried to elucidate the configuration of C-12 of compound 1 by preparing its acetonide (1a) followed by Mosher’s method [17] (Figure 2). During the process, the dimethylation derivative (1b) of 1a was obtained (Figure 2). The acetonide 1a was prepared by treating compound 1 with 2,2-dimethoxypropane (2,2-DMP) and pyridinium p-toluenesulfonate (PPTS) in acetone/DMF (3:1) (Figure 2). In the preparation of 3-O-methyl derivative of 1a, we virtually obtained the 2,2-dimethyl derivative (1b). However, the Δδ values between S-(1ba) and R-(1bb) Mosher esters of 1b were some inconsistent (Figure 2 and Figures S41–S44), indicating that Mosher’s method cannot be used to determine the absolute configuration of this compound. Thus, we tried to elucidate the configuration of compound 1 by X-ray diffraction and luckily obtained the single-crystal of 1a. A single-crystal X-ray diffraction pattern of 1a was obtained using the anomalous scattering of Cu Kα radiation, allowing an explicit assignment of its absolute configurations as 10R, 11S, 12S, and 24R (Figure 2). Moreover, it should be noted that compound 1 could be isomerized to the corresponding 3-keto tautomer (1c) which reached a dynamic equilibrium with its 3-enol tautomer (1) in DMSO solution. The 1H and 13C NMR spectra showed some separate signals for 1 and 1c with the approximate ratio of 3:1 in DMSO-d6 (Figures S1–S5). The diagnostic methylene signals (CH2-2) in 1c were observed at δH/C (3.17, 3.44/50.8) (Table S2 and Figures S1–S5).

Table 1.
1H (600 MHz) and 13C (150 MHz) NMR Data for Streptolactams A–C (1–3).
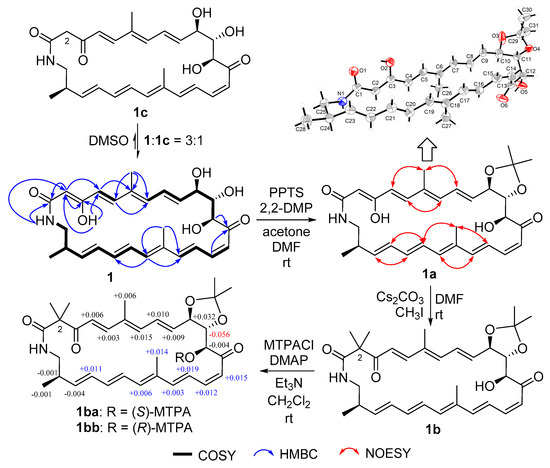
Figure 2.
Structural elucidation of compound 1.
Careful comparison indicated that 13C data of compound 1 (Table 1) were obviously different with those reported for sceliphrolactam [9]. By removal of the calibration of the chemical shifts, we also noted that the most difference is for C-6, C-13 and C-14 which have –1.8, –1.9 and –1.3 ppm difference, respectively. In addition, the value of the specific rotation for compound 1 in the same methanol solution (−392.0 (c 0.05, MeOH)) is different from that of sceliphrolactam (−213 (c 0.09, MeOH)) [9]. Considering no identification of configuration for sceliphrolactam [9], it is reasonable to identify compound 1 as a stereoisomer of sceliphrolactam and a new compound.
Compound 2 was very unstable at room temperature (rt). So, we purified it at a relatively low temperature (18 °C) and measured its NMR spectra at 0 °C. The molecular formula of compound 2 was determined to be C28H35NO6 by HRESIMS (Figure S13). Comparison of its 1H and 13C NMR spectra (Table 1, Figures S7 and S8) with those of compound 4 revealed that they have the similar skeleton. The 1H and 13C NMR data (Table 1), assigned by HSQC (Figure S9), indicated the presence of twelve olefinic carbons, three carbonyl groups (δC 166.3 for amide carbonyl signal; δC 194.8 and 213.1 for keto carbonyl signals), eight sp3-methine groups including three oxygenated carbons, two methylene groups, and three methyl groups. Analysis of its COSY correlations of H-8/H-9/H-14/H-15 revealed the presence of a four-membered ring system that was fused with a six-membered ring, which was confirmed by the COSY correlations of H-10/H-11/H-12 and the key HMBC correlations of H-9 to C-10/C-11/C-13, H-12 to C-14, and H-14 to C-13/C-15/C-16 (Figure 3). The fused [20,4,6] tricyclic framework was determined by the key COSY correlations of H-4/H-5, H-7/H-8, H-15/H-16/H-17, H-24/H3-28, and extending from H-19 to H2-25, and the key HMBC correlations of NH to C-1, H-2 to C-1/C-3, H-4 to C-3/C-6, H-5 to C-3/C-7, H3-26 to C-5/C-6/C-7, and H3-27 to C-17/C-18/C-19 (Figure 3). This structure was deduced to form from the intramolecular [2 + 2] cycloaddition of compound 1.
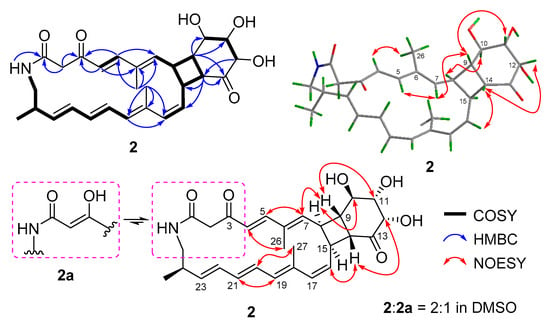
Figure 3.
Key 2D NMR correlations of compound 2.
The geometries of double bonds at Δ4, Δ20, and Δ22 were assigned as E- by the ortho coupling constants (3J) of 15.0, 14.0, and 15.0 Hz, while the 3J value between H-16 and H-17 (11.4 Hz) indicated the Z-geometry of Δ16 double bond (Table 1). The E- geometries of Δ6 and Δ18 double bonds were determined by NOESY correlations of H-5/H-7 and H-20/H3-27 (Figure 3 and Figure S12). The relative configuration of [4,6]-bicyclic system was determined as (8S*,9S*, 10R*,11S*, 12S*,14R*, 15S*)-by the key NOESY correlations of H-7/H-9, H-14/H-16, H-9/H-11, H-12/H-14, and H-8/H-10 (Figure 3 and Figure S12). The fact that compound 2 could be formed from compound 1 (Figure 2) indicated the same (10R,11S,12S,24R)- configurations. Thus, the absolute configuration of compound 2 was determined as shown.
Compound 2, a 3-keto tautomer, can also be reached a dynamic equilibrium with its 3-enol tautomer (2a) in pyridine solution (Figure 3). The 1H and 13C NMR spectra showed some separate signals for 2 and 2a with the approximate ratio of 2:1 in pyridine-d5 (Figures S7–S11). The diagnostic NMR signals of sp2 methine at δH/C 5.53/95.3 and enol hydroxyl at δH 14.7 in 2a could be observed (Table S2 and Figures S7–S11). It is interesting that the tautomerization between 3-enol and 3-keto in DMSO or pyridine solution was only observed for compounds 1 and 2, but not for compounds 3 and 4. This may be largely due to the size of ring. The 3-enol form could increase the ring tension, so that 3-keto form only exists in the small rings while 3-enol and 3-keto forms can coexist in the larger rings.
Streptolactam B (3) was obtained as a yellow powder. Its molecular formula was determined to be C28H35NO7 by HRESIMS (Figure S20). Comparison of its 1D NMR (Table 1, Figures S14 and S15) with those of compound 1 showed that the signals (δC/H 131.4/5.81 and δC/H 137.4/5.36) of a double bond in compound 1 were replaced by two oxygenated methine signals (δC/H 67.9/3.93 and 82.4/3.46) in compound 3. The HMBC correlation of H-11 to C-23 (Figure 3, Figures S18 and S19) indicated that the carbons C-11 and C-23 were connected through an oxygen bridge. The COSY correlations extending from H-19 to H2-25 and the key HMBC correlations of H2-2 to C-1/C-3 (Figure 4 and Figure S18) further supported the structural difference between 1 and 3.
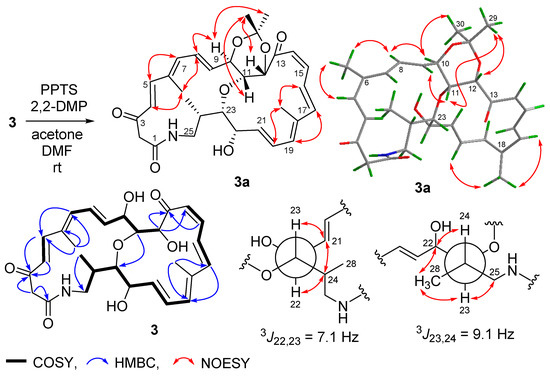
Figure 4.
Preparation and determination of relative configuration of 3a.
The geometries of the Δ4, Δ8, Δ16, and Δ20 double bonds were determined as E- by the coupling constants of 15.4, 14.5, 14.7, and 14.4 Hz, respectively, while the Δ14 double bond was assigned as Z-geometry by the coupling constant of 11.5 Hz (Table 1). In order to verify the relative configuration of compound 3, the acetonide derivative 3a was prepared (Figure 4). The correlations of H-8/H3-26 and H-20/H3-27 could be observed in the NOESY spectrum of compound 3a (Figure 4 and Figure S47), which indicated the E-geometries for the Δ6 and Δ18 double bonds. The (10R*, 11R*, 12S*)- relative configuration was determined by the NOESY correlations of H-9/H3-29, H-9/H-11, H-11/H3-29, H-10/H3-30, and H-12/H3-29 (Figure 4 and Figure S47). J-based configuration analysis (JBCA) method [18] was used to determine the relative configuration of C-22/C-23/C-24. The 1H NMR data of 3a revealed the large coupling constants of H-22/H-23 (J = 7.1 Hz) and H-23/H-24 (J = 9.1 Hz) (Table 1). The NOESY correlations of H-21/H-23, H-21/H-24, and H-22/H-24 (Figure 4 and Figure S47) indicated threo-configuration between C-22 and C-23. The threo-configuration between C-23 and C-24 was concluded from the NOESY correlations of H-22/H-24, H-22/H3-28, H-23/H3-28, and H-23/H-25 (Figure 4 and Figure S47). In addition, compounds 1 and 3 might be biosynthetically formed from the same epoxide precursor, 1p, which subjected to a dihydroxylation of Δ22 double bond followed by an etherification between HO-11 and 9,10-epoxide via an intramolecular nucleophilic ring opening reaction (Figure 5). Furthermore, compounds 1 and 3 showed the similar ECD Cotton effects from long wavelength (420 nm) to short wavelength (250 nm), that is negative first and then two positive effects, indicating they shared the same configurations at C-10 and C-12 which were nearest to the two conjugated enone chromophores, C-3–C-9 and C-13–C-21, and thus contributed most to the ECD Cotton effect. The absolute configuration of C-11 could be determined by comparing its relative configuration with C-10 and C-12 in compound 3a, which is opposite to that of 1. Thus, the absolute configurations of compound 3 were determined as shown.
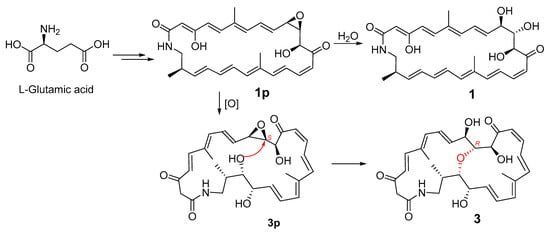
Figure 5.
Postulated biosynthesis of compounds 1 and 3.
Compound 4 was further identical as niizalactam C or tripartilactam by spectroscopic and specific rotation data [10,11,12]. It is reasonable to strongly suggest that 1 and 4 shared a similar absolute configuration based on the fact that compound 4 could be formed from compound 1.
During the isolation and structural elucidation of compound 1, we noticed that compound 1 becomes unstable under light and heat and easy to form compounds 2 and 4. In order to further understand the transformations among compounds 1, 2 and 4, a series of reactions were carried out under different conditions (Figure 6). Compound 1 could exist as a stable structure without light at the temperatures below 30 °C but exhaust and was transformed into compound 4 by the heat Diels-Alder reaction at 50 °C for 6 h, while compound 1 could be transformed into compound 2 via [2 + 2] cycloaddition by light (LED) at low temperature (−15 °C). In the latter reaction, only a little compound 4 was yielded. During the formation of compound 2, only one product with a specific fusing mode, that is a [20,4,6]-fused tricyclic system, was generated, which might be caused by the geometries of double bonds and their relative position in the macrocycle. When compound 2 was placed at 30 °C, it could be transformed into compound 4. At the low temperature (−15 °C) with or without light (LED), compound 2 was stable. These results demonstrated that compound 2 might be an important intermediate during the formation of 4 from 1 stored at rt without protection from light. So, light was the key factor causing compound 1 to change at rt. Avoiding light operation is an effective means to keep the polyene macrolactams stable.
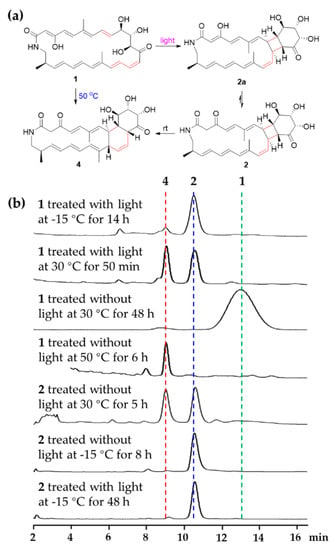
Figure 6.
(a) Abiotic formation of compounds 2 and 4 from compound 1. (b) HPLC profiles (320 nm) of different chemical transformations in MeOH.
2.2. The Bioactivities of Compounds 1–4 from Streptomyces sp. OUCMDZ-3159
Compounds 1, 3, and 4 were evaluated for cytotoxicity against MCF-7, A549, K562, and HL-60 cell lines. No prominent cytotoxic activity against these cell lines was observed (IC50 > 50 μM). Their antimicrobial activity against pathogenic bacteria, Escherichia coli ATCC 11775, Staphylococcus aureus ATCC 6538, Pseudomonas aeruginosa ATCC10145, Clostridium perfringens CGMCC 1.0876 and Bacillus subtilis CGMCC 1.3376, as well as the pathogenic fungus, Candida albicans ATCC 10231 were also tested without light. It is interesting that only compounds 1 and 3 showed a selective antifungal activity against C. albicans with the MIC values of 10.4 and 16.1 μM, respectively. The result of compound 1 further corroborated the reported antifungal activity of the stereoisomer, sceliphrolactam [9]. The biological activity of compound 2 was not tested, because it is very fragile at rt.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were recorded with a JASCO P-1020 digital polarimeter (JASCO Corporation, Tokyo, Japan). UV spectra were recorded on a Beckman DU 640 spectrophotometer (Global Medical Instrumentation, Inc., Ramsey, MN, USA). IR spectra were obtained on a Nicolet Nexus 470 spectrophotometer in KBr discs (Thermo Fisher Scientific, Madison, WI, USA). NMR spectra were recorded on a Bruker Avance 600 spectrometer (Bruker BioSpin AG, Fällanden, Switzerland). ECD spectra were measured on JASCO J-815 spectrometer (JASCO Corporation, Tokyo, Japan). HRESIMS were measured on a Q-TOF Ultima Global GAA076 LC mass spectrometer (Waters Corporation, Milford, MA, USA). Semipreparative HPLC was performed using an ODS column (YMC-pack ODS-A, 10 × 250 mm, 5 μm, 4.0 mL/min). TLC and column chromatography (CC) were performed on plates pre-coated with silica gel GF254 (10–40 μm) and over silica gel (200–300 mesh, Qingdao Marine Chemical Factory, Qingdao, China), and Sephadex LH-20 (Amersham Biosciences, Uppsala, Sweden), respectively. Vacuum-liquid chromatography (VLC) was carried out over silica gel H (Qingdao Marine Chemical Factory).
3.2. Collection and Phylogenetic Analysis
The actinobacterial strain, Streptomyces sp. OUCMDZ-3159, was isolated from a deep-sea sediment collected at depth of 2782 m from the South Mid-Atlantic Ridge (15°9.972′ S, 13°21.348′ W) on October 31, 2012. The sample (2 g) was dried over 24 h in an incubator at 35 °C. The dried sample was diluted to 10–3 g/mL, 100 μL of which was dispersed across a solid-phase agar plate (10 g raffinose, 1 g L-histide, 0.5 g MgSO4.7H2O, 0.01 g FeSO4.7H2O, 0.1 g K2HPO4, 1.2 g bacto-agar, in 1 L seawater, pH 7.0) and incubated at 28 °C for 10 days. A single colony was transferred to Gause’s synthetic agar media. Analysis of the 16S rRNA gene sequence of OUCMDZ-3159 revealed 100% identity to Streptomyces pratensis. The sequence is deposited in GenBank under accession no. MT703834.
3.3. Cultivation and Extraction
The spores of Streptomyces sp. OUCMDZ-3159 were directly transferred to 150 mL of a liquid medium (20 g glucose, 4 g yeast extract, 2 g peptone, 2 g CaCO3, 0.5 g MgSO4, 0.5 g K2HPO4, 0.5 g NH4SO4, in 1 L seawater) in Erlenmeyer flasks (500 mL) and shaken for 14 days (28 ± 0.5 °C, 180 rpm). The whole culture (30 L) was extracted with an equal volume of ethyl acetate (EtOAc) for three times and concentrated in vacuo to yield 20.5 g of EtOAc extract.
3.4. Purification
The EtOAc extract (20.5 g) was separated into nine fractions (Fr.1–Fr.9) on a silica gel VLC column using step gradient elution with CH2Cl2−petroleum ether (0–50%) and then MeOH−CH2Cl2 (0–50%). Fraction 6 was separated into six fractions (Fr.6.1–Fr.6.6) by Sephadex LH-20 eluting with MeOH−CH2Cl2 (1:1) without light. Fr.6.3 was purified by semipreparative HPLC on an ODS column using the solvent system of 40% MeOH aqueous solution to yield compound 3 (16.0 mg, tR 12.5 min). Fr.6.4 was purified by semipreparative HPLC on an ODS column using the solvent system of 65% MeOH aqueous solution to give compounds 4 (5.5 mg, tR 9.1 min), 2 (1.2 mg, tR 10.2 min), and 1 (10.1 mg, tR 12.3 min). Fraction 7 was fractionated into five subfractions (Fr.7.1−Fr.7.5) on a reversed-phase silica gel column, eluting with a step gradient of MeOH−H2O (10–100%) without light. Fr.7.3 and Fr.7.4 were purified by semipreparative HPLC on an ODS column using the solvent system of 65% MeOH and 40% MeOH aqueous solutions to yield compounds 1 (63 mg, tR 12.3 min) and 3 (3.0 mg, tR 12.5 min) without light, respectively.
3.5. Preparation of Compounds 1a and 3a
Compound 1 (25.0 mg) was dissolved in the mixture of DMF (6 mL) and acetone (2 mL), and then pyridinium p-toluenesulfonate (PPTS, 3.0 mg) and 2,2-dimethoxypropane (DMP, 200 μL) were added at 0 °C. The reaction mixture was stirred for 10 h at rt. Then 5 mL of H2O was added, and the solution was extracted three times with EtOAc (5 mL for each). The organic layer was combined and evaporated under reduced pressure to give a yellow gum that was subjected to HPLC purification eluting with 75% MeOH aqueous solution to give compound 1a (15.3 mg, tR 10.4 min, 56% yield). The same reaction of compound 3 (5.0 mg) was carried out and the product 3a (2.0 mg, tR 12.5 min, 37% yield) was purified by HPLC on an ODS column using 55% MeOH aqueous solution.
3.6. Preparation of Compound 1b
Compound 1a (5.0 mg) was dissolved in 1 mL of dimethylformamide (DMF), then Cs2CO3 (2.0 mg) and CH3I (1 μL) were added at 0 °C. The reaction mixture was stirred for 2 h at 28 °C. Then 2 mL of H2O was added, and the solution was extracted three times with ethyl acetate (5 mL for each). The organic layer was combined and evaporated under reduced pressure to give a yellow gum that was purified by HPLC eluting with 80% MeOH aqueous solution to yield compound 1b (2.0 mg, tR 7.9 min, 38% yield).
3.7. Preparation of S-MTPA Ester (1ba) and R-MTPA Ester (1bb) of Compound 1b
Compound 1b (1.0 mg) was dissolved in CH2Cl2 (1 mL), and then triethylamine (10 μL), dimethylaminopyridine (DMAP, 3.0 mg) and (R)-MTPACl (10 μL) were added. The reaction mixture was stirred for 6 h at rt. Then 1 mL of H2O was added, and the solution was extracted three times with CH2Cl2 (5 mL for each). The residue after removal of CH2Cl2 under reduced pressure was purified by semipreparative HPLC (90% MeOH) to yield (S)-MTPA ester 1ba (1.0 mg, tR 9.02 min). With the same method, (R)-MTPA ester 1bb (1.0 mg, tR 8.54 min) was obtained from the reaction of 1b (1.0 mg) with (S)-MTPACl.
3.8. Characterization of the Compounds
Streptolactam A (1): yellow powder; −392.0 (c 0.05, MeOH); ECD (1.00 mM, MeOH) λmax (Δε) 216 (−8.8), 281 (+12.6), 330 (+4.0), 418 (−6.7) nm; UV (MeOH) λmax (log ε) 279 (3.20), 332 (3.39), 421 (2.77) nm; IR (KBr) νmax 3549, 3474, 3415, 3239, 2925, 2853, 1637, 1618, 1571, 1427, 1385, 1058, 619, 477 cm−1; 1H and 13C NMR, see Table S1; HRESIMS m/z 482.2529 [M + H]+ (calcd for C28H36NO6, 482.2537).
Streptolactam B (2): yellowed powder; 1H and 13C NMR at 0 °C, see Table 1; HRESIMS m/z 482.2548 [M + H]+ (calcd for C28H36NO6, 482.2537).
Streptolactam C (3): yellow solid; −792.9 (c 0.05, MeOH); ECD (0.10 mM, MeOH) λmax (Δε) 256 (+18.8), 319 (+4.1), 394 (−12.3) nm; UV (MeOH) λmax (log ε) 401 (4.92), 319 (4.92), 296 (4.90) nm; IR (KBr) νmax 3357, 2961, 2922, 1680, 1618, 1589, 1455, 1384, 1329, 1263, 1089, 1056, 976 cm−1; 1H and 13C NMR, see Table 1; HRESIMS m/z 498.2491 [M + H]+ (calcd for C28H36NO7, 498.2486).
Niizalactam C or Tripartilactam (4): yellow powder; +30.0 (c 0.1, DMSO), −75.0 (c 0.1, MeOH); ECD (1.04 mM, MeOH) λmax 201 (+5.24), 296 (−7.2) nm; 1H and 13C NMR, see Table S1; HRESIMS m/z 482.2544 [M + H]+ (calcd for C28H36NO6, 482.2537).
Compound 1a: orange solid; 1H and 13C NMR, see Table S2; HRESIMS m/z 522.2864 [M + H]+ (calcd for C31H40NO6, 522.2850).
Compound 1b: orange solid; 1H and 13C NMR, see Table S2; HRESIMS m/z 550.3167 [M + H]+ (calcd for C33H44NO6, 550.3163).
Compound 3a: yellow solid; 1H NMR (600 MHz, DMSO-d6): δ 7.17 (1H, dd, J = 15.5, 11.5 Hz, H-16), 6.77 (1H, d, J = 15.5 Hz, H-5), 6.61 (1H, t, J = 11.5 Hz, H-15), 6.60 (1H, d, J = 15.5 Hz, H-17), 6.49 (1H, dd, J = 14.8, 11.5 Hz, H-8), 6.41 (1H, dd, J = 14.3, 11.5 Hz, H-20), 6.21 (1H, d, J = 11.5 Hz, H-14), 6.14 (1H, d, J = 15.5 Hz, H-4), 6.13 (1H, d, J = 14.8 Hz, H-7), 6.12 (1H, d, J = 11.5 Hz, H-19), 5.52 (1H, dd, J = 14.7, 9.0 Hz, H-9), 5.46 (1H, dd, J = 15.0, 10.3 Hz, H-21), 4.64 (1H, d, J = 1.8 Hz, H-12), 4.37 (1H, d, J = 9.0 Hz, H-10), 4.15 (1H, dd, J = 9.0, 1.8 Hz, H-11), 3.91 (1H, d, J = 18.1 Hz, H-2a), 3.88 (1H, dd, J = 10.3, 7.1 Hz, H-22), 3.86 (1H, dd, J = 11.2, 7.7 Hz, H-25a), 3.47 (1H, d, J = 18.1 Hz, H-2b), 3.44 (1H, dd, J = 9.1, 7.1 Hz, H-23), 2.77 (1H, t, J = 11.2 Hz, H-25b), 1.85 (1H, m, H-24), 1.81 (3H, s, CH3-26), 1.61 (3H, s, CH3-27), 1.01 (3H, d, J = 6.7 Hz, CH3-28), 1.33 (3H, s, CH3-29), 1.35 (3H, s, CH3-30); HRESIMS m/z 538.2809 [M + H]+ (calcd for C31H40NO7, 538.2799), 560.2628 [M + Na]+ (calcd for C31H39NO7Na, 560.2619).
S-MTPA ester (1ba) of 1b: 1H NMR (600 MHz, CDCl3): δ 7.58 (1H, m, NH), 7.31 (1H, dd, J = 15.5, 12.0 Hz, H-16), 7.02 (1H, d, J = 14.8 Hz, H-4), 6.66 (1H, t, J = 11.5 Hz, H-15), 6.61 (1H, d, J = 15.1 Hz, H-17), 6.59 (1H, dd, J = 14.3, 11.4 Hz, H-8&21), 6.54 (1H, d, J = 15.1 Hz, H-5), 6.33 (1H, d, J = 12.5 Hz, H-19), 6.06 (1H, overlapped, H-7&14), 6.04 (2H, overlapped, H-20&22), 5.73 (1H, d, J = 2.3 Hz, H-12), 5.46 (1H, overlapped, H-9), 5.44 (1H, overlapped, H-23), 4.54 (1H, t, J = 8.1 Hz, H-10), 4.16 (1H, dd, J = 8.4, 2.3 Hz, H-11), 3.66 (1H, dd, J = 8.0, 4.6 Hz, H-25a), 2.60 (1H, ddd, J = 13.0, 10.7, 3.9 Hz, H-25b), 2.51 (1H, m, H-24), 1.87 (3H, s, H-26), 1.79 (3H, s, H3-27), 1.54 (3H, s), 1.42 (3H, s), 1.34 (3H, s), 1.05 (1H, d, J = 6.6 Hz, H3-28), 1.02 (3H, s); ESIMS m/z 766.5 [M + H]+.
R-MTPA ester (1bb) of 1b: 1H NMR (600 MHz, CDCl3): δ 7.57 (1H, m, NH), 7.29 (1H, dd, J = 14.9, 11.0 Hz, H-16), 7.02 (1H, d, J = 15.1 Hz, H-4), 6.65 (1H, t, J = 11.2 Hz, H-15), 6.60 (1H, d, J = 15.0 Hz, H-17), 6.56 (1H, overlapped, H-8&21), 6.53 (1H, d, J = 14.9 Hz, H-5), 6.33 (1H, d, J = 11.3 Hz, H-19), 6.05 (1H, overlapped, H-7&14), 6.03 (2H, overlapped, H-20&22), 5.74 (1H, d, J = 2.2 Hz, H-12), 5.46 (1H, overlapped, H-9), 5.44 (1H, overlapped, H-23), 4.51 (1H, t, J = 8.1 Hz, H-10), 4.22 (1H, dd, J = 8.5, 2.2 Hz, H-11), 3.66 (1H, m, H-25a), 2.61 (1H, ddd, J = 18.2, 14.4, 4.0 Hz, H-25b), 2.51 (1H, m, H-24), 1.87 (3H, s, H3-26), 1.77 (3H, s, H3-27), 1.54 (3H, s), 1.43 (3H, s), 1.42 (3H, s), 1.35 (3H, s), 1.05 (1H, d, J = 6.6 Hz, H3-28); ESIMS m/z 766.4 [M + H]+.
3.9. X-Ray Crystallographic Analysis
Compound 1a was obtained as an orange crystal with molecular formula of C31H39NO6 from CH2Cl2/MeOH. Further, the crystal data were got on a Bruker Smart APEXDUO area detector diffractometer with graphite monochromated Cu-Kα radiation (λ = 1.54178 Å) (Table S4). The structure was solved by direct methods (SHELXS-97) and expanded using Fourier techniques (SHELXL-97). Crystallographic data (excluding structure factors) for structure 1a in this paper have been deposited in the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 996760.
3.10. Chemical Interconversion of Compounds 1,2 and 4
Streptolactam A (1), isolated from an EtOAc extract from Streptomyces sp. OUCMDZ-3159 without light, was dissolved in MeOH at a concentration of 2.5 mM and stirred with or without light at rt (30 °C), −15 °C and 50 °C, respectively. Compound 2 was dissolved in MeOH at a concentration of 1 mM with or without light at rt (30 °C) and −15 °C, respectively. The reaction mixtures were analyzed every 30 min by use of HPLC on an ODS column eluting with 65% MeOH aqueous solution at a flow rate of 1 mL/min. The isolated compounds 1, 2 and 4 were used as the standards (Figure 6).
Compound 2 was very fragile at rt. In order to identify its structure, we tried to obtain more materials through chemical transformation. Compound 1 was not stable under light, which could be converted into compounds 2 and 4. So, we treated compound 1 (35.0 mg, 25mM in MeOH) with sunlight for 1.5 h, and then the mixture was separated by HPLC on an ODS column at 18 °C using 65% MeOH aqueous solution to yield compounds 4 (4.0 mg, tR 9.1 min, 11.4% yield) and 2 (2.0 mg, tR 10.2 min, 5.7% yield). The NMR spectra of compound 2 were measured at 0 °C in pyridine-d5.
3.11. Cytotoxicity Assay
Cytotoxicity was assayed against A549 and MCF-7 cell lines by the MTT [19], and K562 and HL-60 cell lines CCK-8 [20] methods. Adriamycin was used as the positive control with the IC50 values of 1.00, 0.63, 0.73 and 0.58 for the cell lines MCF-7, A549, K562, and HL-60 respectively.
3.12. Antimicrobial Assay
The antimicrobial activities against pathogenic bacteria, Escherichia coli ATCC 11775, Staphylococcus aureus ATCC 6538, Pseudomonas aeruginosa ATCC10145, Clostridium perfringens CGMCC 1.0876 and Bacillus subtilis CGMCC 1.3376, as well as the pathogenic fungus, Candida albicans ATCC 10231 were evaluated by an agar dilution method [21]. The tested strains were cultivated in LB agar plates for bacteria and YPD agar plates for C. albicans at 37 °C. Compounds 1, 3 and 4 and positive controls were dissolved in MeOH at different concentrations from 100 to 0.05 μg/mL by the continuous 2-fold dilution methods. A 10 μL quantity of test solution was absorbed by a paper disk (5 mm diameter) and placed on the assay plates. After 12 h incubation, zones of inhibition (mm in diameter) were recorded. The minimum inhibitory concentrations (MICs) were defined as the lowest concentration at which no microbial growth could be observed. Ciprofloxacin lactate (for bacteria) and ketoconazole (for fungus) were used as positive control for E. coli, S. aureus, P. aeruginosa, C. perfringens, B. subtilis, C. albicans with MIC values of 1.9, 1.9, 3.8, 1.9, 0.94, and 0.02 μM, respectively.
4. Conclusions
In summary, we identified four polyene macrolactams (1–4) from a deep-sea sediment-derived Streptomyces strain, OUCMDZ-3159. Compounds 1 and 2 with 20-membered or larger ring moieties existed a keto-enol tautomerism in the DMSO solution or pyridine solution. The abiotic formation of 2 and 4 from 1 was clarified through a package of heat and light induced intramolecular pericyclic reactions. This study indicated that light was a key factor that made the polyene macrolactams unstable at rt. The results provide ideas for the research on the non-enzymatic formation of polycyclic macrolactams.
Supplementary Materials
The following are available online at https://www.mdpi.com/1660-3397/19/1/13/s1. Tables S1–S3: NMR data for compounds 4, 1a, 1b, 1c and 2a and X-ray crystallographic analysis. Figure S1–S48: NMR and HRESIMS spectra.
Author Contributions
P.W. performed the experiments for the isolation, structure elucidation, reactions under different conditions, and biological activity assay and prepared the draft manuscript; D.W. jointly contributed to isolation, preparation of compounds and performed HRESIMS and ECD experiments. R.Z. performed the fermentation of the strain; Y.W. and F.K. participated in the structure elucidation; P.F. and W.Z. supervised the research work and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (Nos. U1906213, 41876172, 41806086), the National Key R&D Program of China (No. 2018YFC1406705) and the Fundamental Research Funds for the Central Universities (No. 201841006).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Skellam, E.J.; Stewart, A.K.; Strangman, W.K.; Wright, J.L.C. Identification of micromonolactam, a new polyene macrocyclic lactam from two marine Micromonospora strains using chemical and molecular methods: Clarification of the biosynthetic pathway from a glutamate starter unit. J. Antibiot. 2013, 66, 431–441. [Google Scholar]
- Sugiyama, R.; Nishimura, S.; Matsumori, N.; Tsunematsu, Y.; Hattori, A.; Kakeya, H. Structure and biological activity of 8-deoxyheronamide C from a marine-derived Streptomyces sp.: Heronamides target saturated hydrocarbon chains in lipid membranes. J. Am. Chem. Soc. 2014, 136, 5209–5212. [Google Scholar] [PubMed]
- Schulze, C.J.; Donia, M.S.; Siqueira-Neto, J.L.; Ray, D.; Raskatov, J.A.; Green, R.E.; McKerrow, J.H.; Fischbach, M.A.; Linington, R.G. Genome-directed lead discovery: Biosynthesis, structure elucidation, and biological evaluation of two families of polyene macrolactams against Trypanosoma brucei. ACS Chem. Biol. 2015, 10, 2373–2381. [Google Scholar] [PubMed]
- Genilloud, O. Actinomycetes: Still a source of novel antibiotics. Nat. Prod. Rep. 2017, 34, 1203–1232. [Google Scholar]
- Hoshino, S.; Okada, M.; Awakawa, T.; Asamizu, S.; Onaka, H.; Abe, I. Mycolic acid containing bacterium stimulates tandem cyclization of polyene macrolactam in a lake sediment derived rare actinomycete. Org. Lett. 2017, 19, 4992–4995. [Google Scholar]
- Shin, Y.H.; Beom, J.Y.; Chung, B.; Shin, Y.; Byun, W.S.; Moon, K.; Bae, M.; Lee, S.K.; Oh, K.B.; Shin, J.; et al. Bombyxamycins A and B, cytotoxic macrocyclic lactams from an intestinal bacterium of the silkworm Bombyx mori. Org. Lett. 2019, 21, 1804–1808. [Google Scholar]
- Beemelmanns, C.; Ramadhar, T.R.; Kim, K.H.; Klassen, J.L.; Cao, S.; Wyche, T.P.; Hou, Y.; Poulsen, M.; Bugni, T.S.; Currie, C.R.; et al. Macrotermycins A–D, glycosylated macrolactams from a termite-associated Amycolatopsis sp. M39. Org. Lett. 2017, 19, 1000–1003. [Google Scholar]
- Nogawa, T.; Okano, A.; Takahashi, S.; Uramoto, M.; Konno, H.; Saito, T.; Osada, H. Verticilactam, a new macrolactam isolated from a microbial metabolite fraction library. Org. Lett. 2010, 12, 4564–4567. [Google Scholar]
- Oh, D.C.; Poulsen, M.; Currie, C.R.; Clardy, J. Sceliphrolactam, a polyene macrocyclic lactam from a wasp-associated Streptomyces sp. Org. Lett. 2011, 13, 752–755. [Google Scholar]
- Park, S.H.; Moon, K.; Bang, H.S.; Kim, S.H.; Kim, D.G.; Oh, K.B.; Shin, J.; Oh, D.C. Tripartilactam, a cyclobutane-bearing tricyclic lactam from a Streptomyces sp. in a dung beetle’s brood ball. Org. Lett. 2012, 14, 1258–1261. [Google Scholar]
- Hoshino, S.; Okada, M.; Wakimoto, T.; Zhang, H.P.; Hayashi, F.; Onaka, H.; Abe, I. Niizalactams A–C, multicyclic macrolactams isolated from combined culture of Streptomyces with mycolic acid-containing bacterium. J. Nat. Prod. 2015, 78, 3011–3017. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Kim, E.; Lee, J.; Shin, J.; Yoon, Y.J.; Oh, D.C. Structure revision and the biosynthetic pathway of tripartilactam. J. Nat. Prod. 2020, 83, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Liu, P.; Li, X.; Wang, Y.; Wang, S.; Hong, K.; Zhu, W. Cyclic bipyridine glycosides from the marine-derived actinomycete Actinoalloteichus cyanogriseus WH1-2216-6. Org. Lett. 2011, 13, 5948–5951. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Zhu, Y.; Mei, X.; Wang, Y.; Jia, H.; Zhang, C.; Zhu, W. Acyclic congeners from Actinoalloteichus cyanogriseus provide insights into cyclic bipyridine glycoside formation. Org. Lett. 2014, 16, 4264–4267. [Google Scholar] [CrossRef]
- Shen, J.; Fan, Y.; Zhu, G.; Chen, H.; Zhu, W.; Fu, P. Polycyclic macrolactams generated via intramolecular diels–alder reactions from an Antarctic Streptomyces species. Org. Lett. 2019, 21, 4816–4820. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wang, J.; Chen, H.; Wang, Y.; Zhu, W.; Fu, P. Cyclamenols E and F, two diastereoisomeric bicyclic macrolactams with a cyclopentane moiety from an Antarctic Streptomyces species. Org. Chem. Front. 2020, 7, 310–317. [Google Scholar] [CrossRef]
- Seco, J.M.; Quiñoá, E.; Riguera, R. A practical guide for the assignment of the absolute configuration of alcohols, amines and carboxylic acids by NMR. Tetrahedron Asymmetry 2001, 12, 2915–2925. [Google Scholar] [CrossRef]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon-proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Xiong, T.; Chen, X.; Wei, H.; Xiao, H. Influence of PJ34 on the genotoxicity induced by melphalan in human multiple myeloma cells. Arch. Med. Sci. 2015, 11, 301–306. [Google Scholar] [CrossRef]
- Zaika, L.L.J. Spices and herbs: Their antimicrobial activity and its determination. J. Food Safety 1988, 9, 97–118. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).