Bioactive Metabolites from the Deep-Sea-Derived Fungus Diaporthe longicolla FS429


Abstract
1. Introduction
2. Results and Discussion
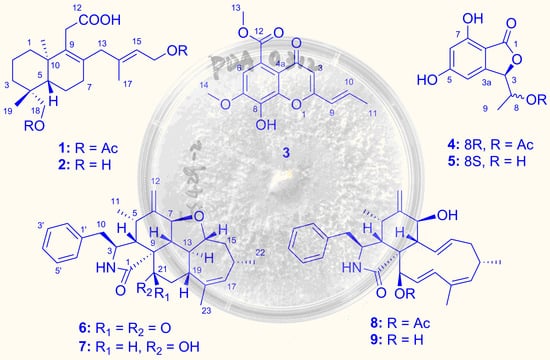
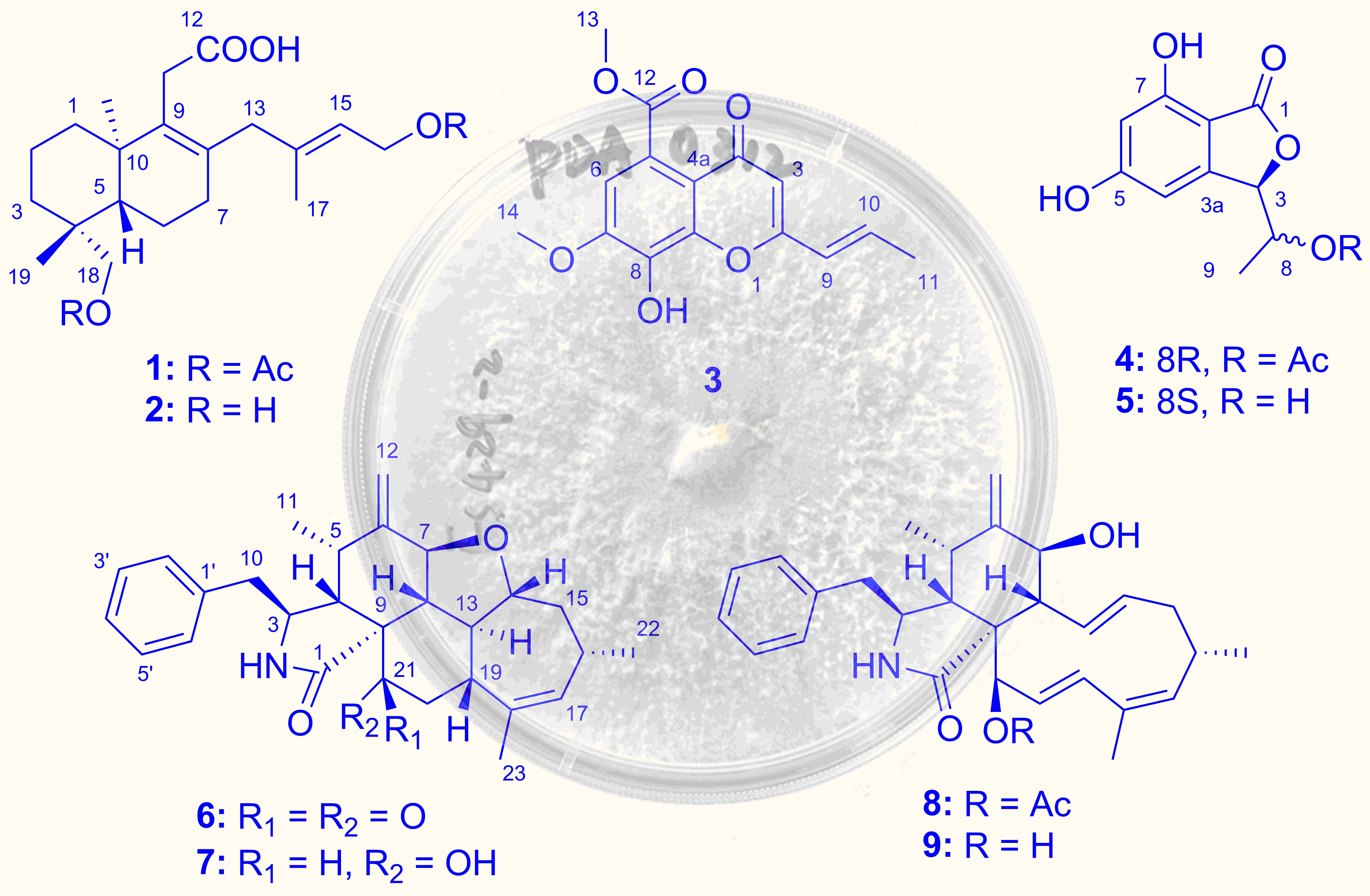
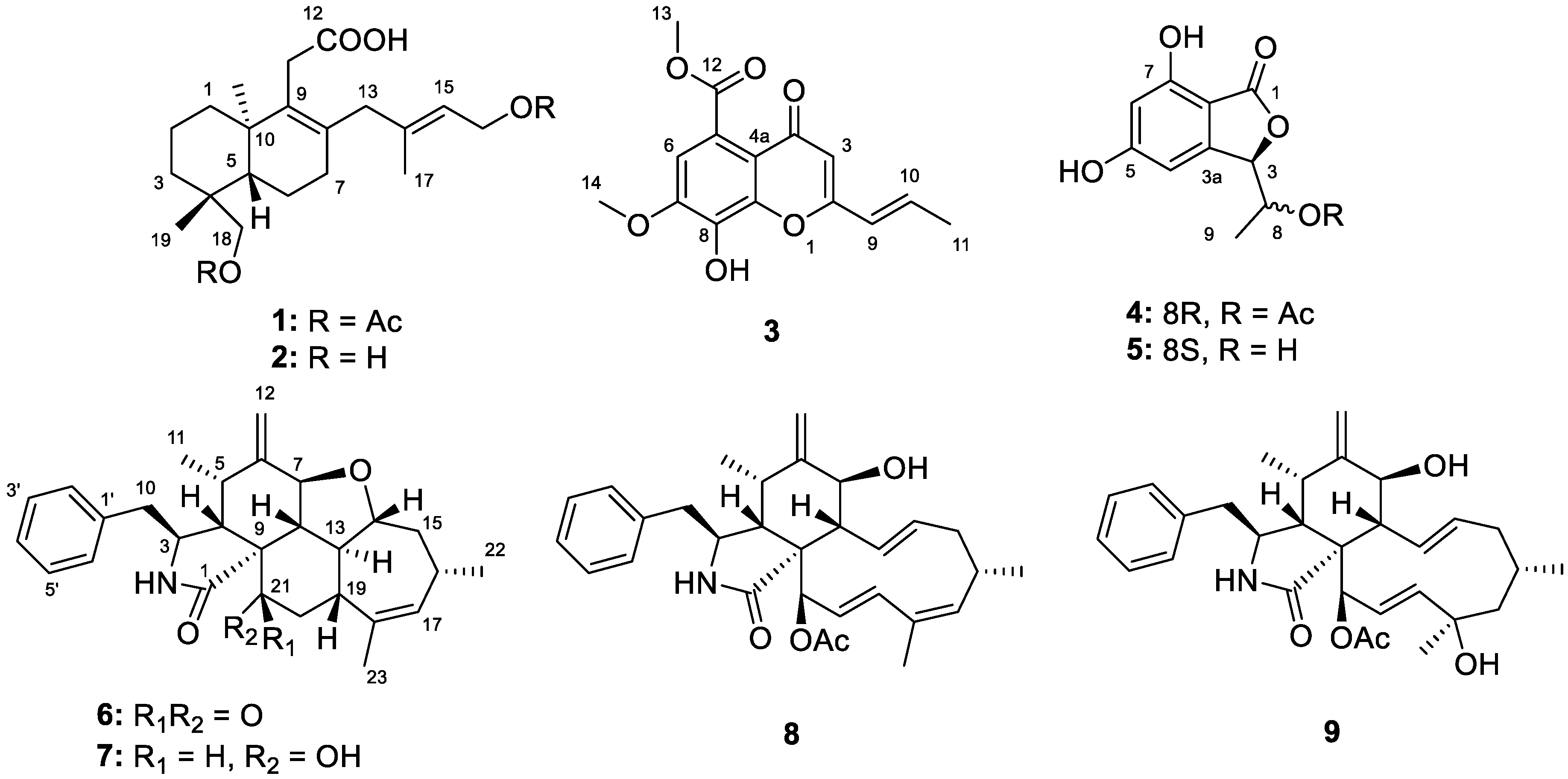
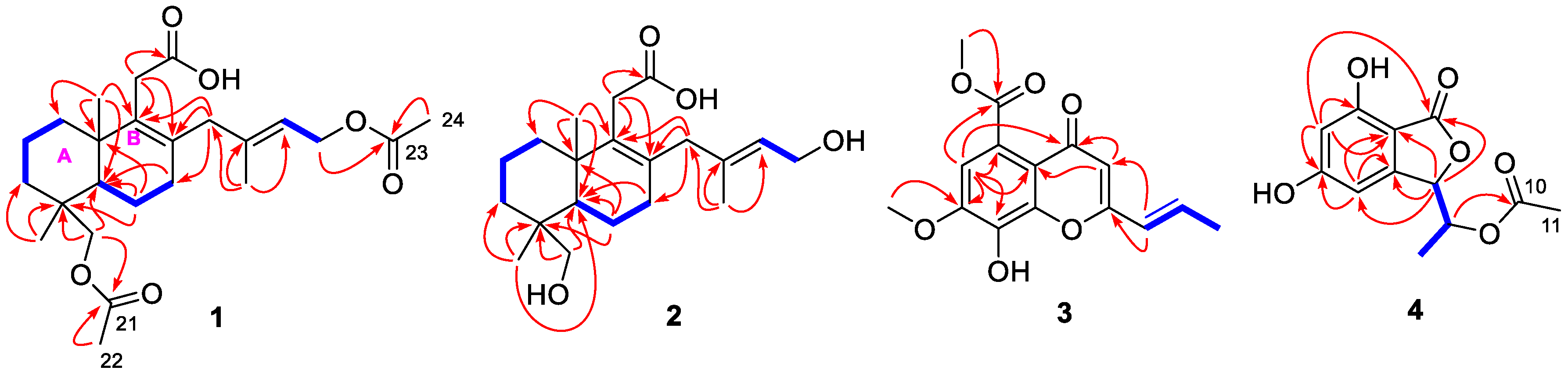
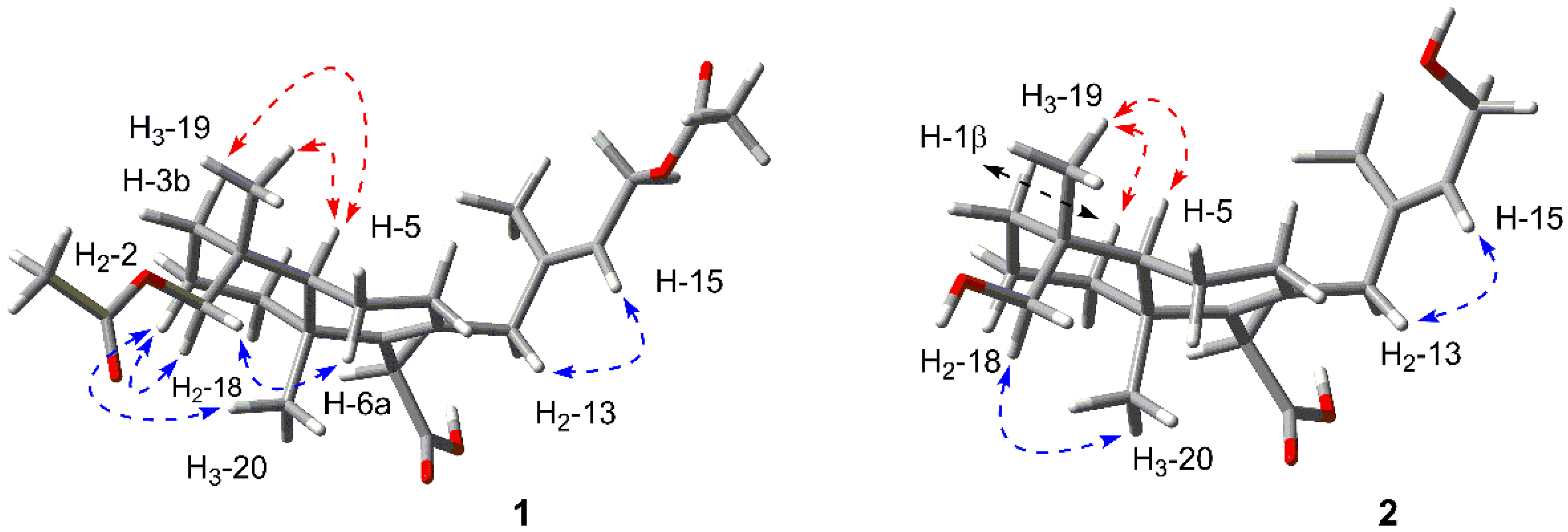
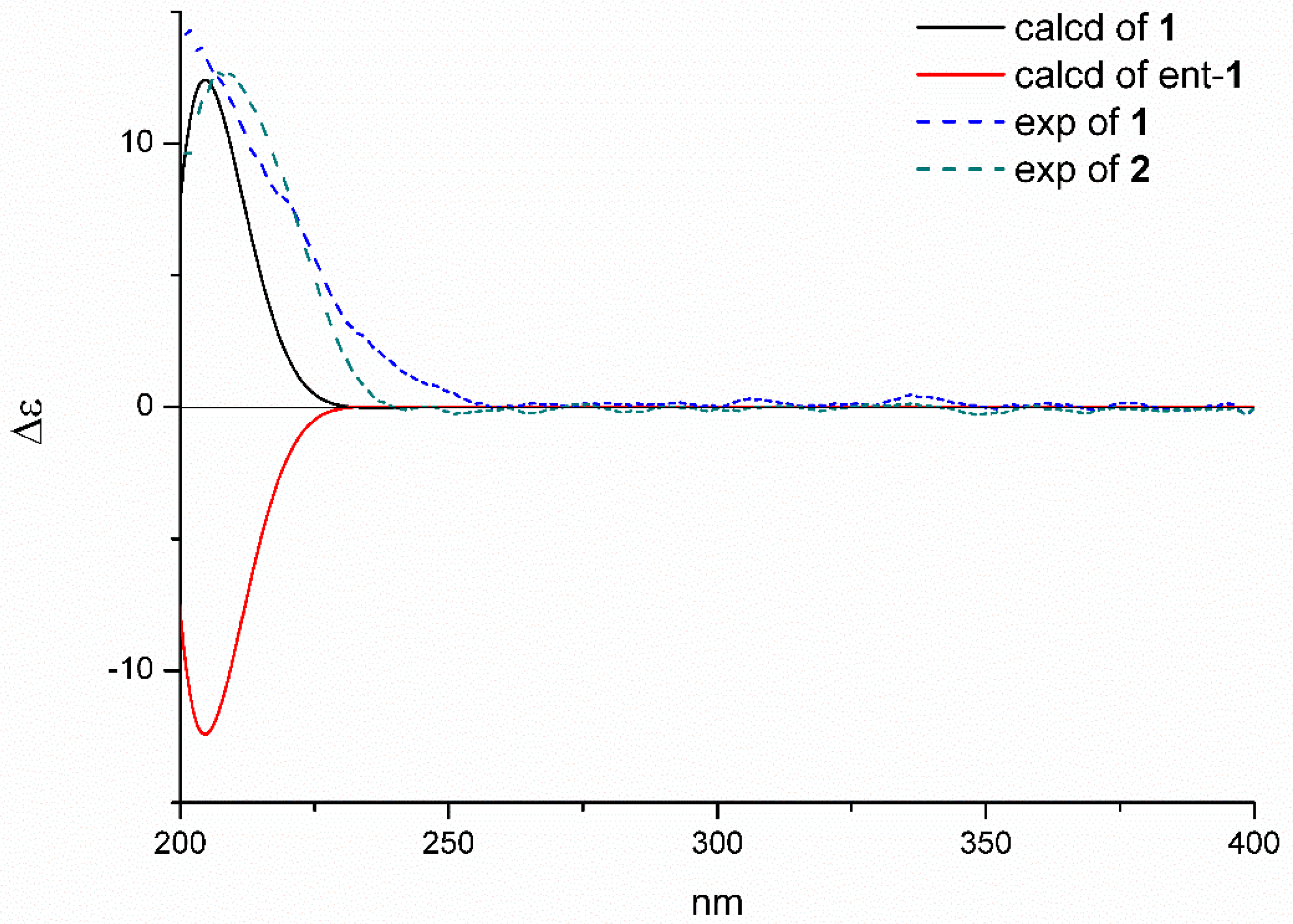
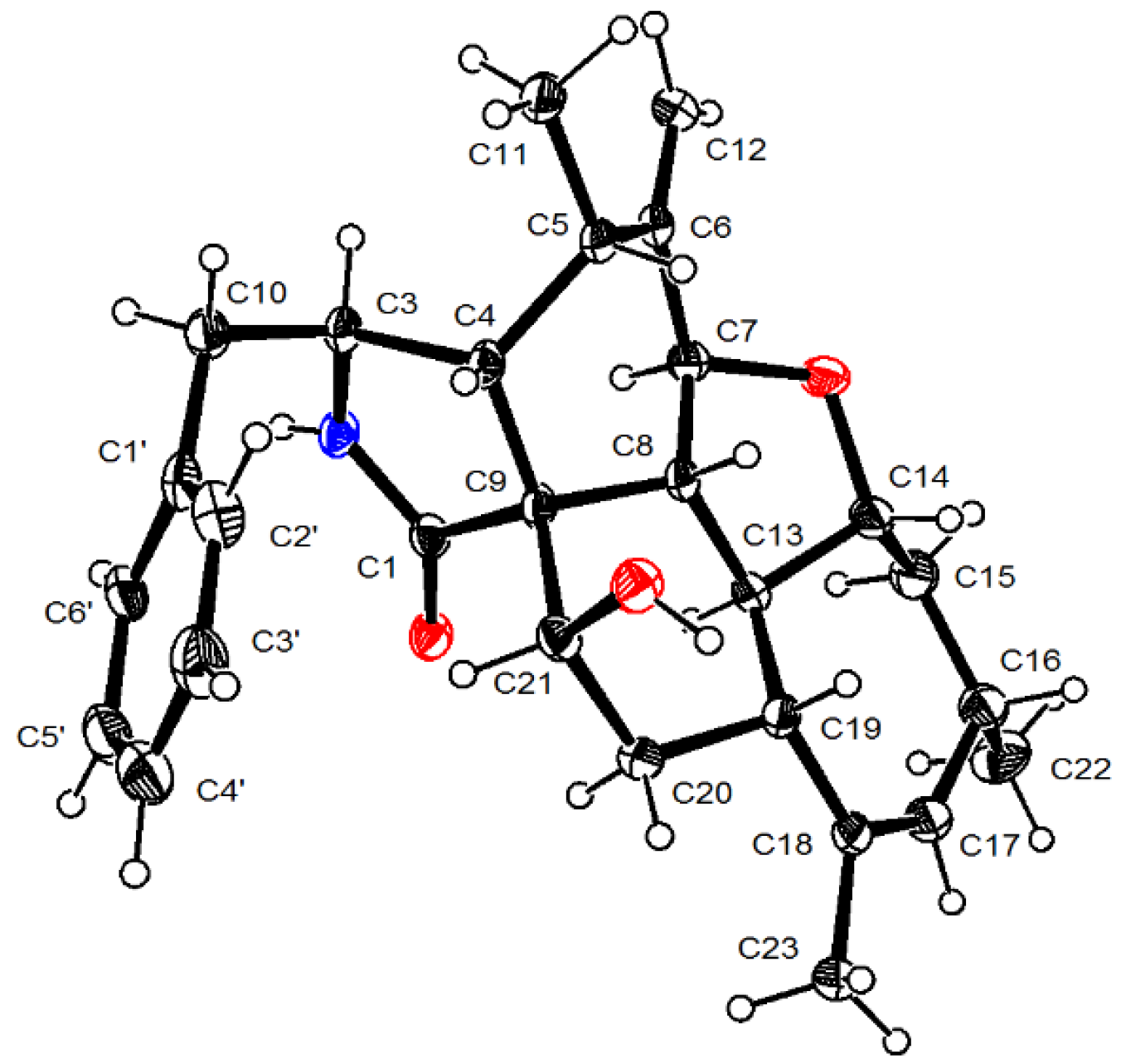
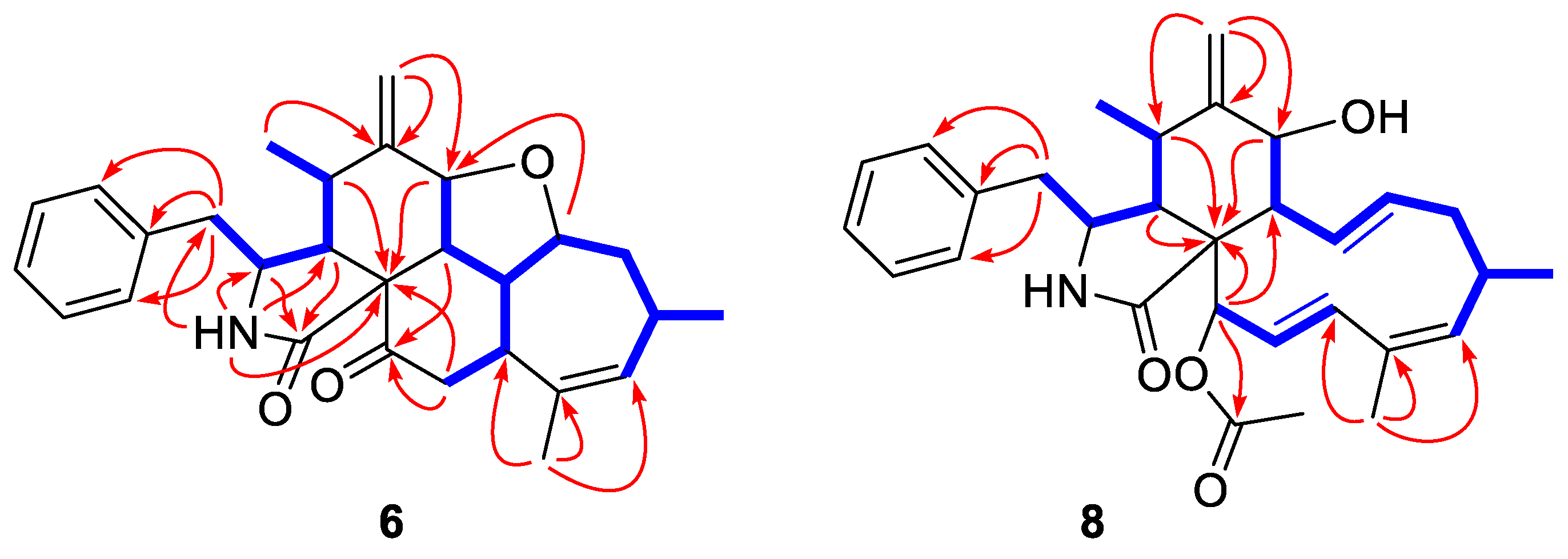
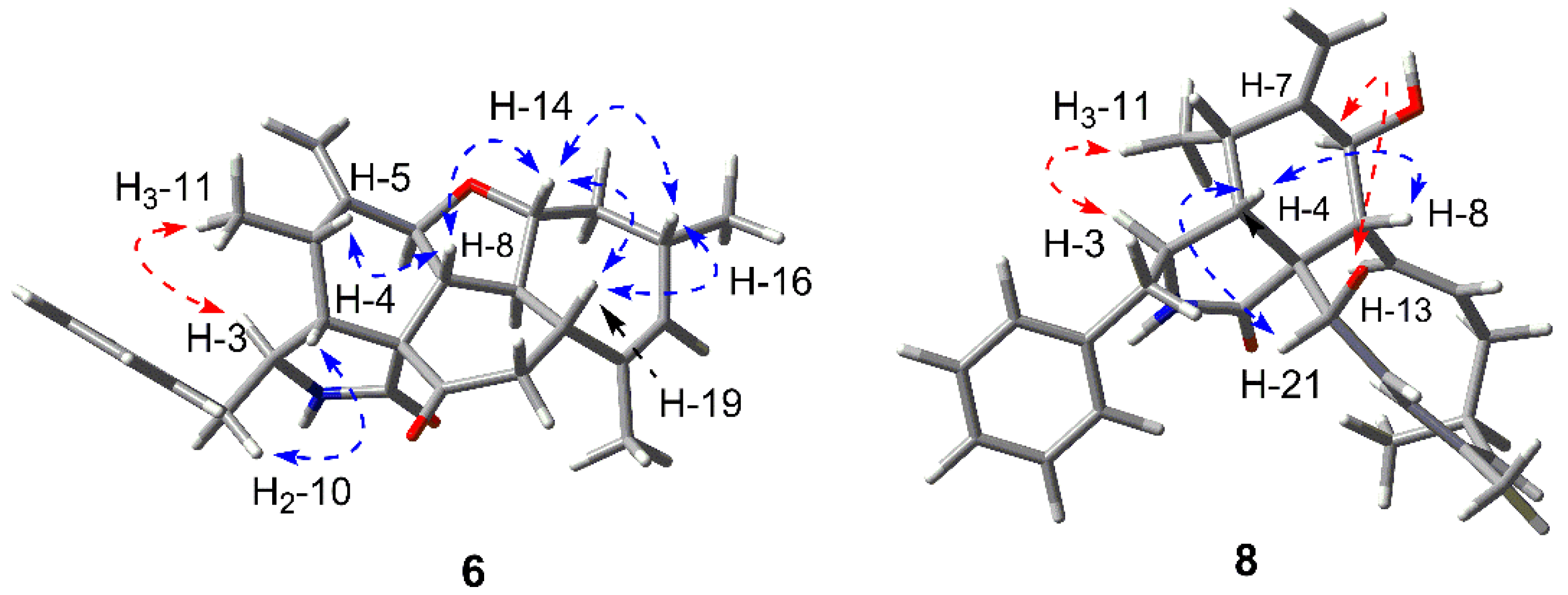
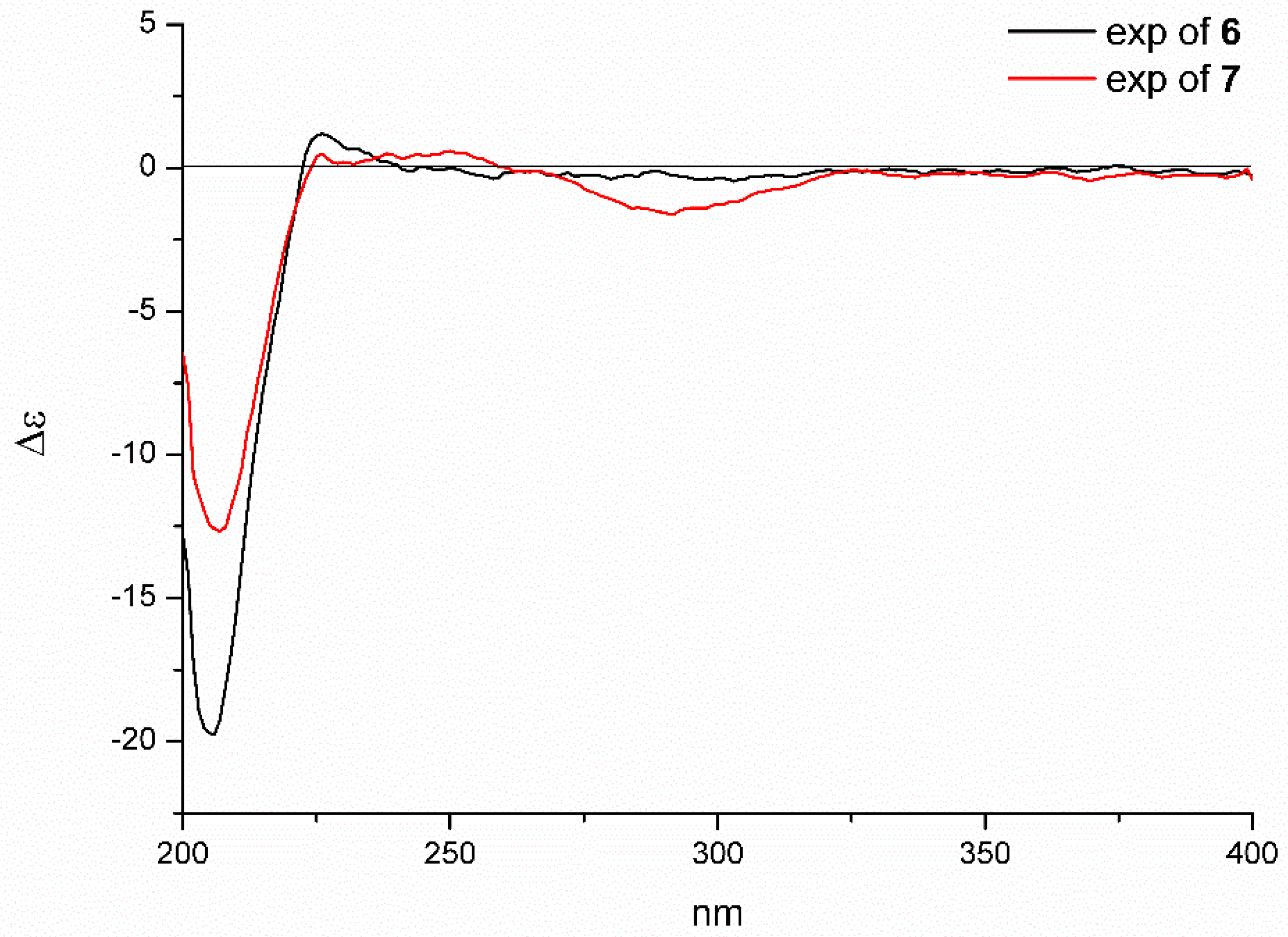
2.1. Structure Elucidation of the New Compounds
2.2. Bioactivities
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation, Extraction, and Isolation
3.3.1. Longidiacid A (1)
3.3.2. Longidiacid B (2)
3.3.3. Longichromone A (3)
3.3.4. Longiphthalidin A (4)
3.3.5. Longichalasin A (6)
3.3.6. Longichalasin B (8)
3.4. Details of Quantum Chemical Calculations
3.5. Cytotoxicity Assay
3.6. MptpB Inhibitory Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mahé, S.; Rédou, V.; Calvez, T.L.; Vandenkoornhuyse, P.; Burgaud, G. Fungi in deep-sea environments and metagenomics. In The Ecological Genomics of Fungi; Martin, F., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 325–354. [Google Scholar]
- Wang, Y.-T.; Xue, Y.-R.; Liu, C.-H. A brief review of bioactive metabolites derived from deep-sea fungi. Mar. Drugs 2015, 13, 4594–4616. [Google Scholar] [CrossRef]
- Roth, F.J.; Orpurt, P.A.; Ahearn, D.G. Occurrence and distribution of fungi in a subtropical marine environment. Can. J. Bot. 1964, 42, 375–383. [Google Scholar] [CrossRef]
- Cui, C.-B.; Kakeya, H.; Osada, H. Novel mammalian cell cycle inhibitors, spirotryprostatins A and B, produced by Aspergillus fumigatus, which inhibit mammalian cell cycle at G2/M phase. Tetrahedron 1996, 52, 12651–12666. [Google Scholar] [CrossRef]
- Niu, S.; Xia, J.-M.; Li, Z.; Yang, L.-H.; Yi, Z.-W.; Xie, C.-L.; Peng, G.; Luo, Z.-H.; Shao, Z.; Yang, X.-W. Aphidicolin chemistry of the deep-sea-derived fungus Botryotinia fuckeliana MCCC 3A00494. J. Nat. Prod. 2019, 82, 2307–2331. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, J.; Liao, Y.-Y.; Cheng, G.; Chen, J.; Mo, S.; Yuan, L.; Cheng, X.-D.; Qin, J.-J.; Shao, Z. Aspeterreurone A, a cytotoxic dihydrobenzofuran-phenyl acrylate hybrid from the deep-sea-derived fungus Asperillus terreus CC-S06-18. J. Nat. Prod. 2020, 83, 1998–2003. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Xie, C.-L.; Xia, J.-M.; Liu, Q.-M.; Peng, G.; Liu, G.-M.; Yang, X.-W. Botryotins A–H, tetracyclic diterpenoids representing three carbon skeletons from a deep-sea-derived Botryotinia fuckeliana. Org. Lett. 2020, 22, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Sun, Z.; Peng, J.; Zhu, M.; Che, Q.; Zhang, G.; Zhu, T.; Gu, Q.; Li, D. Secondary metabolites produced by combined culture of Penicillium crustosum and a Xylaria sp. J. Nat. Prod. 2019, 82, 2013–2017. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Tan, H.; Chen, Y.; Li, S.; Huang, Z.; Guo, H.; Li, H.; Gao, X.; Liu, H.; Zhang, W. Lithocarpins A–D: Four tenellone-macrolide conjugated [4 + 2] hetero-adducts from the deep-sea derived fungus Phomopsis lithocarpus FS508. Org. Chem. Front. 2018, 5, 1792–1797. [Google Scholar] [CrossRef]
- Chen, S.; Liu, Z.; Tan, H.; Chen, Y.; Li, S.; Li, H.; Zhuang, S.; Liu, H.; Zhang, W. Phomeroids A and B: Two novel cytotoxic meroterpenoids from the deep-sea-derived fungus Phomopsis tersa FS441. Org. Chem. Front. 2020, 7, 557–562. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, X.-P.; Ju, Z.-R.; Liao, X.-J.; Huang, X.-J.; Zhang, C.; Zhao, B.-X.; Xu, S.-H. Aspergchromones A and B, two new polyketides from the marine sponge-associated fungus Aspergillus sp. SCSIO XWS03F03. J. Asian Nat. Prod. Res. 2017, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Watanabe, H.; Usui, T.; Osada, H.; Kitahara, T. Synthesis and bioactivities of acetophthalidin and its derivatives. Heterocycles 1998, 48, 2049–2060. [Google Scholar]
- Takenaka, Y.; Morimoto, N.; Hamada, N.; Tanahashi, T. Phenolic compounds from the cultured mycobionts of Graphis proserpens. Phytochemistry 2011, 72, 1431–1435. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.; Raju, R.; Salim, A.A.; Khalil, Z.G.; Capon, R.J. Cytochalasins from an Australian marine sediment-derived Phomopsis sp. (CMB-M0042F): Acid-mediated intramolecular cycloadditions enhance chemical diversity. J. Org. Chem. 2017, 82, 9704–9709. [Google Scholar] [CrossRef]
- Izawa, Y.; Hirose, T.; Shimizu, T.; Koyama, K.; Natori, S. Six new 10-phenyl-[11]-cytochalasans, cytochalasins N–S from Phomopsis sp. Tetrahedron 1989, 45, 2322–2335. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.K.K.N.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013.
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef]
- Liu, Z.; Fan, Z.; Sun, Z.; Liu, H.; Zhang, W. Dechdigliotoxins A–C, three novel disulfide-bridged gliotoxin dimers from deep-sea sediment derived fungus Dichotomomyces cejpii. Mar. Drugs. 2019, 17, 596. [Google Scholar] [CrossRef]
- Liu, Z.; Dong, Z.; Qiu, P.; Wang, Q.; Yan, J.; Lu, Y.; Wasu, P.; Hong, K.; She, Z. Two new bioactive steroids from a mangrove-derived fungus Aspergillus sp. Steroids 2018, 140, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, Q.; Li, S.; Cui, H.; Sun, Z.; Chen, D.; Lu, Y.; Liu, H.; Zhang, W. Polypropionate derivatives with Mycobacterium tuberculosis protein tyrosine phosphatase B inhibitory activities from the deep-sea-Derived fungus Aspergillus fischeri FS452. J. Nat. Prod. 2019, 82, 3440–3449. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 2 a | ||
|---|---|---|---|---|
| δC, Type. | δH (J in Hz) | δC, Type. | δH (J in Hz) | |
| 1 | 35.8, CH2 | 1.71, m | 35.4, CH2 | 1.83, m |
| 1.74, m | 1.80, m | |||
| 2 | 18.4, CH2 | 1.49, m | 18.2, CH2 | 1.58, m |
| 1.41, m | ||||
| 3 | 35.8, CH2 | 1.28, m | 34.9, CH2 | 1.33, m |
| 1.05, dd (13.5, 4.8) | 0.93, m | |||
| 4 | 37.0, C | 38.4, C | ||
| 5 | 51.9, CH | 1.40, m | 51.5, CH | 1.50, m |
| 6 | 19.0, CH2 | 1.82, dd (12.2, 5.9) | 18.8, CH2 | 1.76, m |
| 1.46, m | 1.44, m | |||
| 7 | 31.5, CH2 | 2.02, m | 31.1, CH2 | 2.02, m |
| 8 | 131.8, C | 128.7, C | ||
| 9 | 135.8, C | 138.8, C | ||
| 10 | 38.7, C | 38.6, C | ||
| 11 | 32.8, CH2 | 3.11, d (17.4) | 36.2, CH2 | 2.96, d (16.9) |
| 2.97, d (17.4) | 2.80, d (16.9) | |||
| 12 | 178.3, C | 179.9, C | ||
| 13 | 42.9, CH2 | 2.82, d (16.0) | 42.4, CH2 | 2.94, d (16.1) |
| 2.51, d (16.0) | 2.35, d (16.1) | |||
| 14 | 139.6, C | 136.5, C | ||
| 15 | 118.6, CH | 5.24, t (6.9) | 122.7, CH | 5.28, t (6.8) |
| 16 | 61.3, CH2 | 4.58, d (6.9) | 58.1, CH2 | 4.07, d (6.8) |
| 17 | 16.7, CH3 | 1.64, s | 15.4, CH3 | 1.60, s |
| 18 | 67.1, CH2 | 4.24, d (11.0) | 63.7, CH2 | 3.77, d (11.9) |
| 3.92, d (11.0) | 3.31, (overlap) | |||
| 19 | 27.1, CH3 | 0.99, s | 26.0, CH3 | 0.96, s |
| 20 | 20.5, CH3 | 0.99, s | 20.0, CH3 | 0.98, s |
| 21 | 171.5, C | |||
| 22 | 21.1, CH3 | 2.06, s | ||
| 23 | 171.3, C | |||
| 24 | 21.1, CH3 | 2.06, s | ||
| Position | 3 a | Position | 4 b | ||
|---|---|---|---|---|---|
| δC, Type. | δH (J in Hz) | δC, Type. | δH (J in Hz) | ||
| 1 | 37.0, C | 1 | 170.2, C | ||
| 2 | 161.3, C | 3 | 81.0, CH | 5.43, brd (3.1) | |
| 3 | 108.4, CH | 6.06, s | 3a | 150.6, C | |
| 4 | 176.9, C | 4 | 100.9, CH | 6.42, m | |
| 4a | 116.1, C | 5 | 165.4, C | ||
| 5 | 124.0, C | 6 | 102.5, CH | 6.32, d (1.8) | |
| 6 | 107.4, CH | 6.94, s | 7 | 158.2, C | |
| 7 | 148.9, C | 7a | 103.8, C | ||
| 8 | 134.9, C | 8 | 69.2, CH | 5.38, dq (3.1, 6.5) | |
| 8a | 144.3, C | 9 | 14.6, CH3 | 1.33, d (6.5) | |
| 9 | 123.8, CH | 6.18, dq (15.5, 1.7) | 10 | 170.1, C | |
| 10 | 137.2, CH | 6.93, dq (15.5, 6.9) | 11 | 19.3, CH3 | 1.89, s |
| 11 | 18.7, CH3 | 1.98, dd (6.9, 1.7) | |||
| 12 | 170.0, C | ||||
| 13 | 53.1, CH3 | 3.97, s | |||
| 14 | 56.8, CH3 | 4.01, s | - | ||
| Position | 6 a | 8 a | ||
|---|---|---|---|---|
| δC, Type. | δH (J in Hz) | δC, Type. | δH (J in Hz) | |
| 1 | 170.9, C | 174.1, C | ||
| 2 | - | 5.45, brs | - | - |
| 3 | 53.9, CH | 3.34, dt (9.8, 4.1) | 53.6, CH | 3.25, m |
| 4 | 43.3, CH | 3.07, t (4.1) | 50.6, CH | 2.13, d (5.3, 3.2) |
| 5 | 35.1, CH | 2.87, m | 32.6, CH | 2.80, m |
| 6 | 147.4, C | 147.2, C | ||
| 7 | 76.3, CH | 4.26, dd (12.4, 2.6) | 69.6, CH | 3.72, d (10.6) |
| 8 | 52.1, CH | 2.22, t (12.4) | 47.7, CH | 3.04, brt (10.1) |
| 9 | 59.7, C | 48.2, C | ||
| 10 | 45.2, CH2 | 2.95, dd (13.6, 4.1) | 45.5, CH2 | 2.84, dd (13.5, 4.9) |
| 2.58, dd (13.6, 9.8) | 2.70, dd (13.5, 9.6) | |||
| 11 | 14.6, CH3 | 1.18, d (6.7) | 13.5, CH3 | 0.98, d (6.7) |
| 12 | 114.5, CH2 | 5.39, brt (2.4) | 114.6, CH2 | 5.29, brs |
| 5.23, brt (2.5) | 5.10, brs | |||
| 13 | 44.4, CH | 2.43, dt (12.4, 9.8) | 130.4, CH | 6.02, dd (15.6, 9.6) |
| 14 | 87.3, CH | 3.70, ddd (12.4, 11.8, 3.1) | 137.2, CH | 5.72, ddd (15.6, 10.8, 4.8) |
| 15 | 39.8, CH2 | 2.01, dt (11.8, 3.1) | 42.9, CH2 | 2.24, m |
| 1.47, q (11.8) | 2.00, m | |||
| 16 | 30.3, CH | 2.13, m | 31.7, CH | 2.70, m |
| 17 | 133.3, CH | 5.27, brs | 136.0, CH | 5.26, brd (7.6) |
| 18 | 137.3, C | 132.5, C | ||
| 19 | 41.7, CH | 2.60, m | 136.1, CH | 6.67, d (16.4) |
| 20 | 43.3, CH2 | 3.47, dd (14.0, 12.4) | 120.4, CH | 5.61, dd (16.4, 3.7) |
| 2.73, dd (14.0, 5.1) | ||||
| 21 | 204.9, C | 78.1, CH | 5.48, m | |
| 22 | 24.5, CH3 | 1.13, d (7.2) | 24.0, CH3 | 1.05, d (6.9) |
| 23 | 23.4, CH3 | 1.76, s | 21.1, CH3 | 1.82, s |
| 1′ | 137.4, C | 137.4, C | ||
| 2′/6′ | 129.0, CH | 7.15, brd (6.80) | 129.1, CH | 7.15, m |
| 3′/5′ | 129.0, CH | 7.32, m | 128.9, CH | 7.32, m |
| 4′ | 127.1, CH | 7.25, m | 127.1, CH | 7.26, m |
| Compounds | IC50 (µM) a | |||
|---|---|---|---|---|
| SF-268 | MCF-7 | HepG-2 | A549 | |
| 1 | >150 | >150 | >150 | >150 |
| 2 | >150 | >150 | >150 | >150 |
| 3 | >150 | >150 | >150 | >150 |
| 4 | 33.83 ± 2.43 | 88.95 ± 3.35 | 91.86 ± 8.74 | 88.25 ± 5.87 |
| 5 | >150 | >150 | >150 | >150 |
| 6 | 65.33 ± 1.59 | 73.48 ± 0.42 | 63.84 ± 2.73 | 64.00 ± 0.50 |
| 7 | 74.38 ± 6.24 | 79.55 ± 2.82 | 63.67 ± 1.25 | 70.29 ± 2.55 |
| 8 | 16.44 ± 0.75 | 36.45 ± 1.97 | 59.09 ± 1.30 | 33.34 ± 1.24 |
| 9 | 68.94 ± 2.15 | 91.91 ± 4.86 | 94.04 ± 2.56 | 84.52 ± 4.57 |
| Cisplatin b | 3.18 ± 0.04 | 2.78 ± 0.15 | 2.21 ± 0.02 | 1.49 ± 0.02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Chen, Y.; Li, S.; Wang, Q.; Hu, C.; Liu, H.; Zhang, W. Bioactive Metabolites from the Deep-Sea-Derived Fungus Diaporthe longicolla FS429. Mar. Drugs 2020, 18, 381. https://doi.org/10.3390/md18080381
Liu Z, Chen Y, Li S, Wang Q, Hu C, Liu H, Zhang W. Bioactive Metabolites from the Deep-Sea-Derived Fungus Diaporthe longicolla FS429. Marine Drugs. 2020; 18(8):381. https://doi.org/10.3390/md18080381
Chicago/Turabian StyleLiu, Zhaoming, Yuchan Chen, Saini Li, Qinglin Wang, Caiyun Hu, Hongxin Liu, and Weimin Zhang. 2020. "Bioactive Metabolites from the Deep-Sea-Derived Fungus Diaporthe longicolla FS429" Marine Drugs 18, no. 8: 381. https://doi.org/10.3390/md18080381
APA StyleLiu, Z., Chen, Y., Li, S., Wang, Q., Hu, C., Liu, H., & Zhang, W. (2020). Bioactive Metabolites from the Deep-Sea-Derived Fungus Diaporthe longicolla FS429. Marine Drugs, 18(8), 381. https://doi.org/10.3390/md18080381