Uncovering the Core Microbiome and Distribution of Palmerolide in Synoicum adareanum Across the Anvers Island Archipelago, Antarctica

 , , , , , , and
, , , , , , and 
Abstract
1. Introduction
2. Results
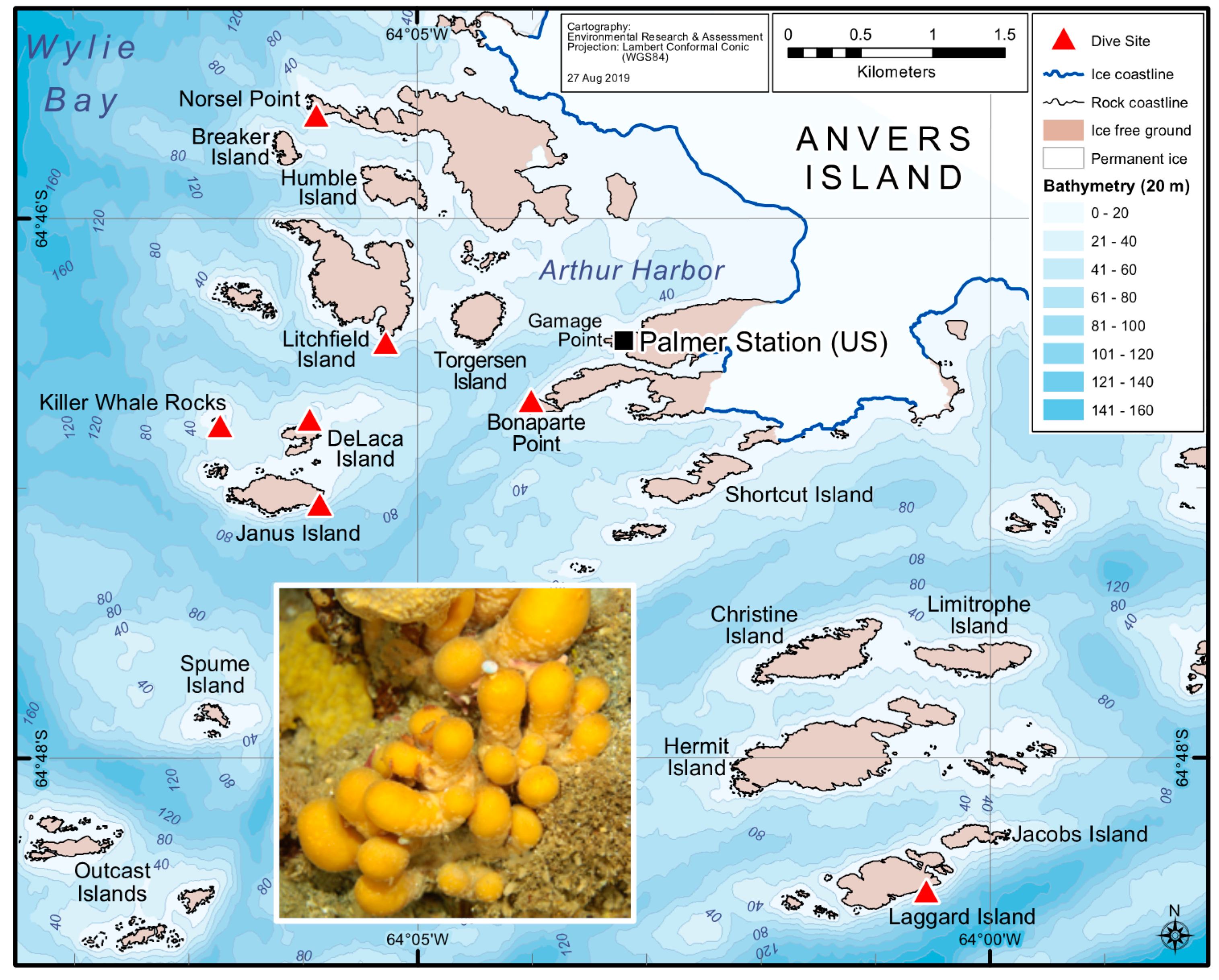
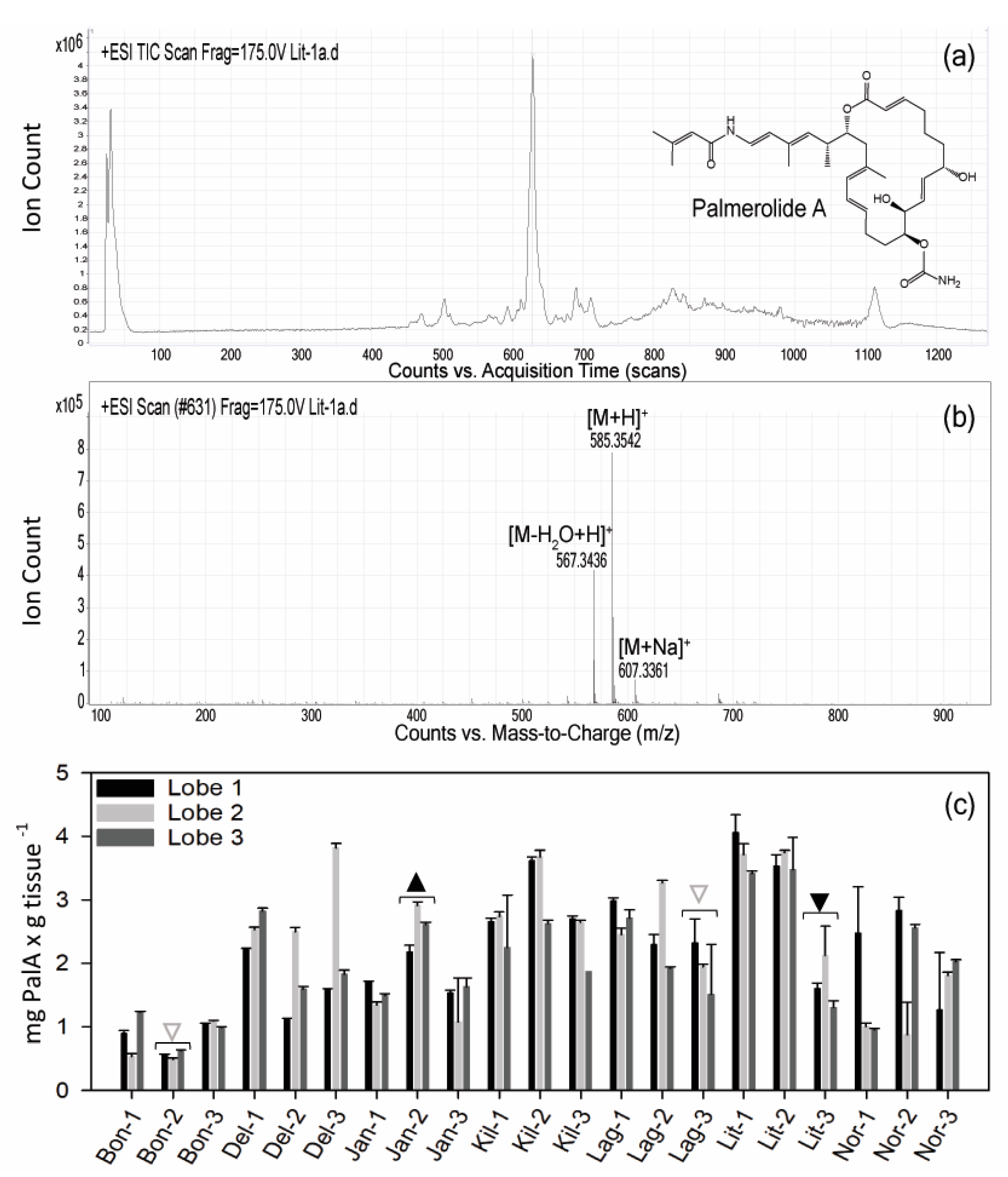
2.1. Variation in Holobiont PalA Levels Across Ascidian Colonies and Collection Sites
2.2. Characterization of Host-Associated Cultivated Bacteria
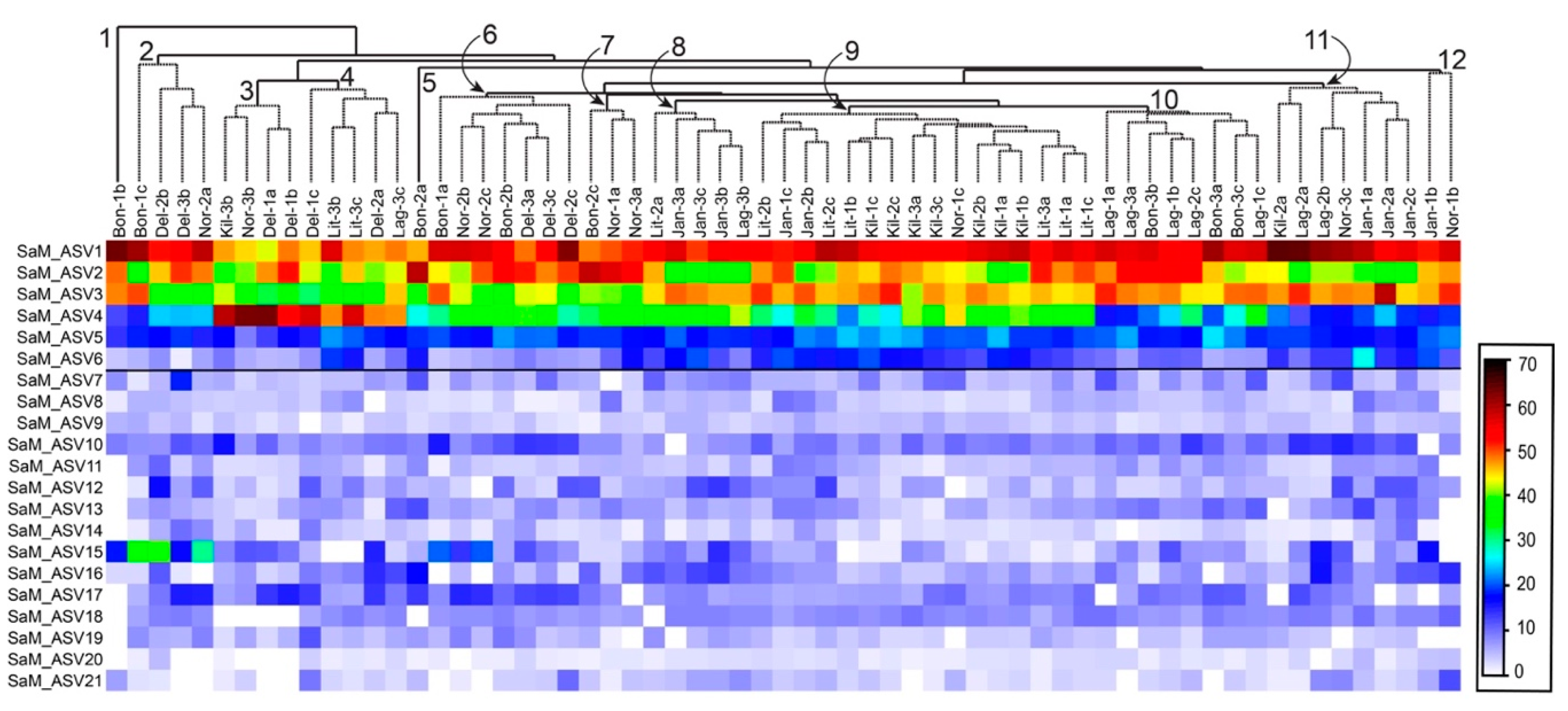
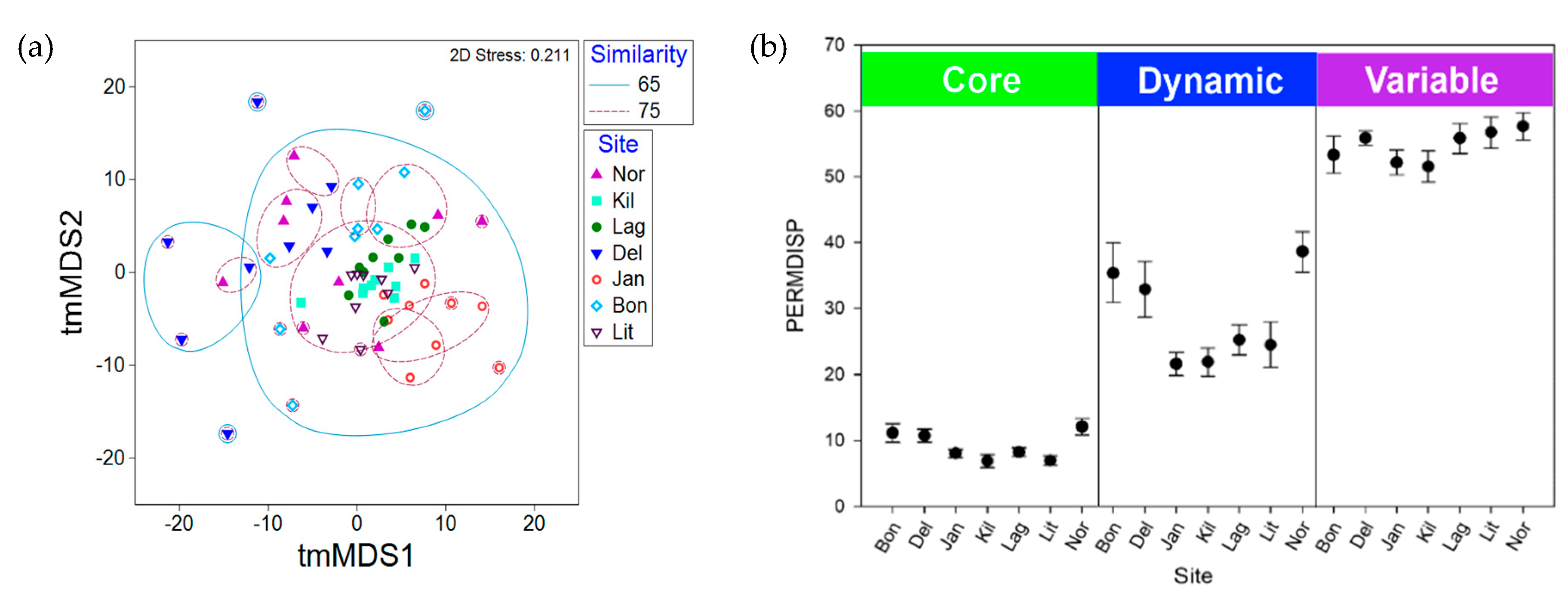
2.3. Synoicum Adareanum Microbiome (SaM)
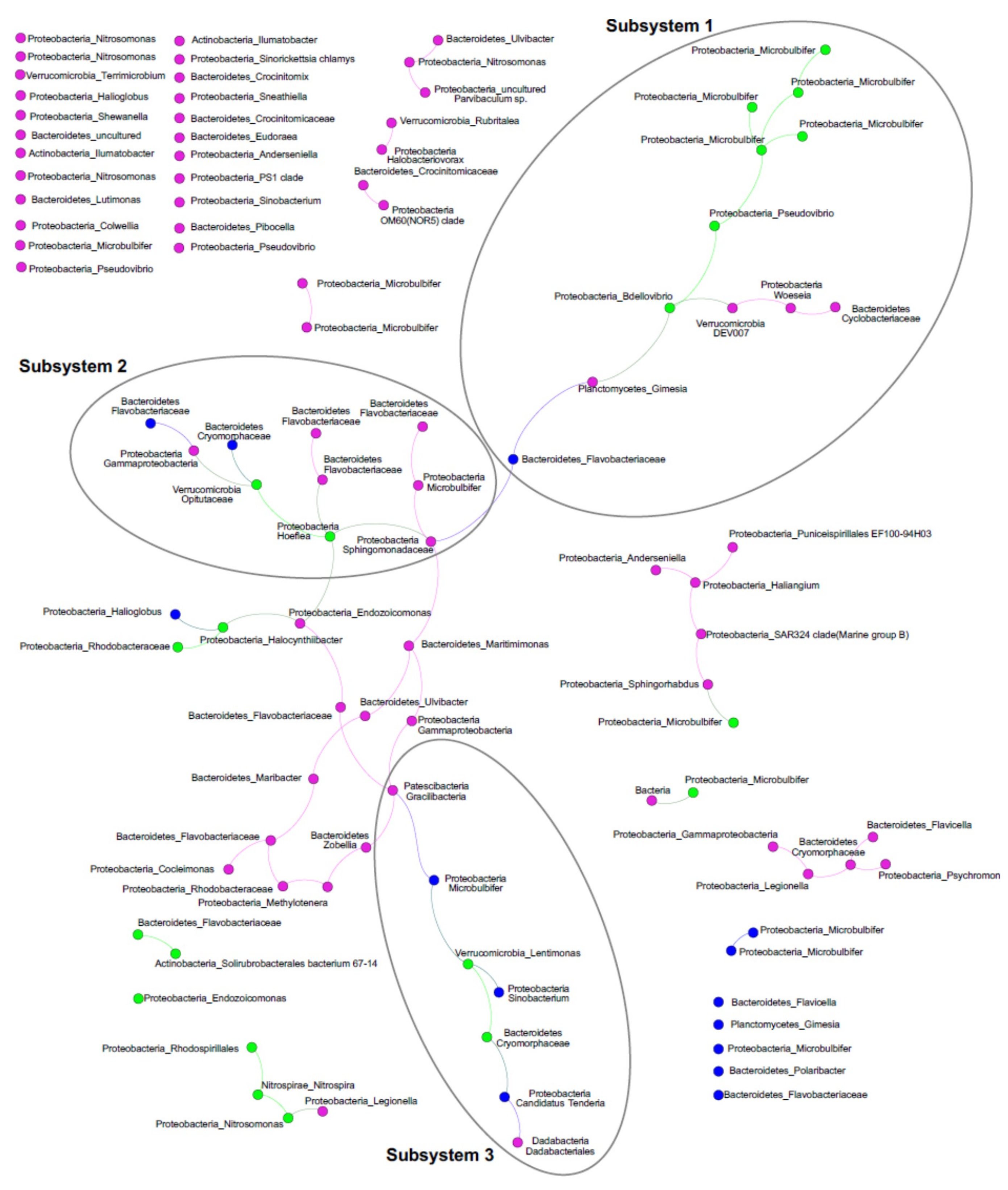
2.4. ASV Co-Occurrences, and Relationship to PalA
2.5. Culture Collection: Microbiome and Bacterioplankton Comparisons
2.6. Microbiome: Bacterioplankton Comparisons
3. Discussion
3.1. Core Microbiome
3.2. Secondary Metabolite Distributions and Bioaccumulation in Marine Biota
3.3. The Biosynthetic Potential of the Core
4. Conclusions
5. Materials and Methods
5.1. Cultivation-Dependent Effort
5.2. Field Sample Collections for Cultivation-Independent Efforts
5.3. Palmerolide A Screening
5.4. S. Adareanum-Associated Microbial Cell Preparation
5.5. DNA Extractions
5.6. The 16S rRNA Gene Sequencing
5.7. Bioinformatic Analysis of 16S rRNA Gene tag Sequences
5.8. Statistical Analyses
5.9. Biosynthetic Gene Cluster Analysis
5.10. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taylor, M.W.; Tsai, P.; Simister, R.L.; Deines, P.; Botte, E.; Ericson, G.; Schmitt, S.; Webster, N.S. ‘Sponge-specific’ bacteria are widespread (but rare) in diverse marine environments. ISME J. 2013, 7, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Ainsworth, T.D.; Krause, L.; Bridge, T.; Torda, G.; Raina, J.B.; Zakrzewski, M.; Gates, R.D.; Padilla-Gamino, J.L.; Spalding, H.L.; Smith, C.; et al. The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 2015, 9, 2261–2274. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Agreda, A.; Leggat, W.; Bongaerts, P.; Ainsworth, T.D. The microbial signature provides insight into the mechanistic basis of coral success across reef habitats. mBio 2016, 7, e00560-16. [Google Scholar] [CrossRef] [PubMed]
- Burgsdorf, I.; Erwin, P.M.; Lopez-Legentil, S.; Cerrano, C.; Haber, M.; Frenk, S.; Steindler, L. Biogeography rather than association with cyanobacteria structures symbiotic microbial communities in the marine sponge Petrosia ficiformis. Front. Microbiol. 2014, 5, 529. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.W.; Williams, G.J.; Barott, K.L.; Carlson, C.A.; Dinsdale, E.A.; Edwards, R.A.; Haas, A.F.; Haynes, M.; Lim, Y.W.; McDole, T.; et al. Local genomic adaptation of coral reef-associated microbiomes to gradients of natural variability and anthropogenic stressors. Proc. Natl. Acad. Sci. USA 2014, 111, 10227–10232. [Google Scholar] [CrossRef]
- Pantos, O.; Bongaerts, P.; Dennis, P.G.; Tyson, G.W.; Hoegh-Guldberg, O. Habitat-specific environmental conditions primarily control the microbiomes of the coral Seriatopora hystrix. ISME J. 2015, 9, 1916–1927. [Google Scholar] [CrossRef]
- Lo Guidice, A.; Azzaro, M.; Schiaparelli, S. Microbial Symbionts of Antarctic Marine Benthic Invertebrates. In The Ecological Role of Microorganisms in the Antarctic Environment; Castro-Sowinski, S., Ed.; Springer Polar Sciences, Springer Nature: Basel, Switzerland, 2019; pp. 277–296. [Google Scholar]
- Cardenas, C.A.; Gonzalez-Aravena, M.; Font, A.; Hestetun, J.T.; Hajdu, E.; Trefault, N.; Malmbergg, M.; Bongcarn-Rudloff, E. High similarity in the microbiota of cold-water sponges of the Genus Mycale from two different geographical areas. PeerJ 2018, 6. [Google Scholar] [CrossRef]
- Webster, N.S.; Negri, A.P.; Munro, M.; Battershill, C.N. Diverse microbial communities inhabit Antarctic sponges. Environ. Microbiol. 2004, 6, 288–300. [Google Scholar] [CrossRef]
- Steinert, G.; Wemheuer, B.; Janussen, D.; Erpenbeck, D.; Daniel, R.; Simon, M.; Brinkhoff, T.; Schupp, P.J. Prokaryotic diversity and community patterns in Antarctic continental shelf sponges. Front. Mar. Sci. 2019, 6, 297. [Google Scholar] [CrossRef]
- Webster, N.S.; Bourne, D. Bacterial community structure associated with the Antarctic soft coral, Alcyonium antarcticum. FEMS Microbiol. Ecol. 2007, 59, 81–94. [Google Scholar] [CrossRef]
- Murray, A.E.; Rack, F.R.; Zook, R.; Williams, M.J.M.; Higham, M.L.; Broe, M.; Kaufmann, R.S.; Daly, M. Microbiome composition and diversity of the ice-dwelling sea anemone, Edwardsiella andrillae. Integr. Comp. Biol. 2016, 56, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Riesenfeld, C.S.; Murray, A.E.; Baker, B.J. Characterization of the microbial community and polyketide biosynthetic potential in the Palmerolide-producing tunicate, Synoicum adareanum. J. Nat. Prod. 2008, 71, 1812–1818. [Google Scholar] [CrossRef] [PubMed]
- Palermo, J.A.; Brasco, M.F.R.; Spagnuolo, C.; Seldes, A.M. Illudalane sesquiterpenoids from the soft coral Alcyonium paessleri: The first natural nitrate esters. J. Org. Chem. 2000, 65, 4482–4486. [Google Scholar] [CrossRef] [PubMed]
- Diyabalanage, T.; Amsler, C.D.; McClintock, J.B.; Baker, B.J. Palmerolide A, a cytotoxic macrolide from the Antarctic tunicate Synoicum adareanum. J. Am. Chem. Soc. 2006, 128, 5630–5631. [Google Scholar] [CrossRef] [PubMed]
- Erwin, P.M.; Pineda, M.C.; Webster, N.; Turon, X.; Lopez-Legentil, S. Down under the tunic: Bacterial biodiversity hotspots and widespread ammonia-oxidizing archaea in coral reef ascidians. ISME J. 2014, 8, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Legentil, S.; Turon, X.; Espluga, R.; Erwin, P.M. Temporal stability of bacterial symbionts in a temperate ascidian. Front. Microbiol. 2015, 6, 1022. [Google Scholar] [CrossRef]
- Evans, J.S.; Erwin, P.M.; Shenkar, N.; Lopez-Legentil, S. Introduced ascidians harbor highly diverse and host-specific symbiotic microbial assemblages. Sci. Rep. 2017, 7, 11033. [Google Scholar] [CrossRef]
- Evans, J.S.; Erwin, P.M.; Shenkar, N.; Lopez-Legentil, S. A comparison of prokaryotic symbiont communities in nonnative and native ascidians from reef and harbor habitats. FEMS Microbiol. Ecol. 2018, 94, fiy139. [Google Scholar] [CrossRef]
- Cahill, P.L.; Fidler, A.E.; Hopkins, G.A.; Wood, S.A. Geographically conserved microbiomes of four temperate water tunicates. Environ. Microbiol. Rep. 2016, 8, 470–478. [Google Scholar] [CrossRef]
- Dror, H.; Novak, L.; Evans, J.S.; Lopez-Legentil, S.; Shenkar, N. Core and dynamic microbial communities of two invasive ascidians: Can host-symbiont dynamics plasticity affect invasion capacity? Microb. Ecol. 2019, 78, 170–184. [Google Scholar] [CrossRef]
- Erwin, P.M.; Pineda, M.C.; Webster, N.; Turon, X.; Lopez-Legentil, S. Small core communities and high variability in bacteria associated with the introduced ascidian Styela plicata. Symbiosis 2013, 59, 35–46. [Google Scholar] [CrossRef]
- Blasiak, L.C.; Zinder, S.H.; Buckley, D.H.; Hill, R.T. Bacterial diversity associated with the tunic of the model chordate Ciona intestinalis. ISME J. 2014, 8, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Tianero, M.D.B.; Kwan, J.C.; Wyche, T.P.; Presson, A.P.; Koch, M.; Barrows, L.R.; Bugni, T.S.; Schmidt, E.W. Species specificity of symbiosis and secondary metabolism in ascidians. ISME J. 2015, 9, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Adachi, K.; Chen, C.Y.; Kasai, H.; Kanoh, K.; Shizuri, Y.; Misawa, N. Discovery of a marine bacterium producing 4-hydroxybenzoate and its alkyl esters, parabens. Appl. Environ. Microbiol. 2006, 72, 5556–5561. [Google Scholar] [CrossRef]
- Fukunaga, Y.; Kurahashi, M.; Tanaka, K.; Yanagi, K.; Yokota, A.; Harayama, S. Pseudovibrio ascidiaceicola sp. nov., isolated from ascidians (sea squirts). Int. J. Syst. Evol. Microbiol. 2006, 56, 343–347. [Google Scholar] [CrossRef]
- Schreiber, L.; Kjeldsen, K.U.; Funch, P.; Jensen, J.; Obst, M.; Lopez-Legentil, S.; Schramm, A. Endozoicomonas are specific, facultative symbionts of sea squirts. Front. Microbiol. 2016, 7, 1042. [Google Scholar] [CrossRef]
- Gonzalez, J.M.; Mayer, F.; Moran, M.A.; Hodson, R.E.; Whitman, W.B. Microbulbifer hydrolyticus gen.nov., sp. nov., and Marinobacterium georgiense gen.nov.sp.nov., two marine bacteria from a lignin-rich pulp mill waste enrichment community. Int. J. Syst. Evol. Microbiol. 1997, 47, 369–376. [Google Scholar]
- Schmidt, E.W.; Donia, M.S. Life in cellulose houses: Symbiotic bacterial biosynthesis of ascidian drugs and drug leads. Curr. Opin. Biotechnol. 2010, 21, 827–833. [Google Scholar] [CrossRef]
- Dou, X.; Dong, B. Origins and bioactivities of natural compounds derived from marine ascidians and their symbionts. Mar. Drugs 2019, 17, 670. [Google Scholar] [CrossRef]
- Little, A.E.; Robinson, C.J.; Peterson, S.B.; Raffa, K.F.; Handelsman, J. Rules of engagement: Interspecies interactions that regulate microbial communities. Annu. Rev. Microbiol. 2008, 62, 375–401. [Google Scholar] [CrossRef]
- Imamura, N.; Nishuma, M.; Takader, T.; Adachi, K.; Sakai, M.; Sano, H. New anticancer antibiotics pelagiomicins, produced by a new marine bacterium Pelagiobacter variabilis. J. Antibiot. 1997, 50, 8–12. [Google Scholar] [CrossRef]
- Quevrain, E.; Domart-Coulon, I.; Pernice, M.; Bourguet-Kondracki, M.L. Novel natural parabens produced by a Microbulbifer bacterium in its calcareous sponge host Leuconia nivea. Environ. Microbiol. 2009, 11, 1527–1539. [Google Scholar] [CrossRef]
- Martínez-García, M.; Stief, P.; Díaz-Valdés, M.; Wanner, G.; Ramos-Esplá, A.; Dubilier, N.; Antón, J. Ammonia-oxidizing Crenarchaeota and nitrification inside the tissue of a colonial ascidian. Environ. Microbiol. 2008, 10, 2991–3001. [Google Scholar]
- Hatzenpichler, R. Diversity, physiology, and niche differentiation of ammonia-oxidizing archaea. Appl. Environ. Microbiol. 2012, 78, 7501–7510. [Google Scholar] [CrossRef]
- Murray, A.E.; Preston, C.M.; Massana, R.; Taylor, L.T.; Blakis, A.; Wu, K.; DeLong, E.F. Seasonal and spatial variability of bacterial and archaeal assemblages in the coastal waters off Anvers Island, Antarctica. Appl. Environ. Microbiol. 1998, 64, 2585–2595. [Google Scholar] [CrossRef]
- Murray, A.E.; Grzymski, J.J. Diversity and genomics of Antarctic marine micro-organisms. Phil. Trans. Roy. Soc. B Biol. Sci. 2007, 362, 2259–2271. [Google Scholar] [CrossRef]
- Grzymski, J.J.; Riesenfeld, C.S.; Williams, T.J.; Dussaq, A.M.; Ducklow, H.; Erickson, M.; Cavicchioli, R.; Murray, A.E. A metagenomic assessment of winter and summer bacterioplankton from Antarctica Peninsula coastal surface waters. ISME J. 2012, 6, 1901–1915. [Google Scholar] [CrossRef]
- Rhoades, D. Evolution of Plant Chemical Defense Against Herbivores. Herbivores: Their Interaction with Secondary Plant Metabolites; Academic Press: New York, NY, USA, 1979. [Google Scholar]
- McPhail, K.L.; Davies-Coleman, M.T.; Starmer, J. Sequestered chemistry of the Arminacean nudibranch Leminda millecra in Algoa Bay, South Africa. J. Nat. Prod. 2001, 64, 1183–1190. [Google Scholar] [CrossRef]
- Carbone, M.; Gavagnin, M.; Haber, M.; Guo, Y.W.; Fontana, A.; Manzo, E.; Genta-Jouve, G.; Tsoukatou, M.; Rudman, W.B.; Cimino, G.; et al. Packaging and delivery of chemical weapons: A defensive trojan horse stratagem in chromodorid nudibranchs. PLoS ONE 2013, 8, e62075. [Google Scholar] [CrossRef]
- Winters, A.E.; White, A.M.; Dewi, A.S.; Mudianta, I.W.; Wilson, N.G.; Forster, L.C.; Garson, M.J.; Cheney, K.L. Distribution of defensive metabolites in nudibranch molluscs. J. Chem. Ecol. 2018, 44, 384–396. [Google Scholar] [CrossRef]
- Schupp, P.; Eder, C.; Paul, V.; Proksch, P. Distribution of secondary metabolites in the sponge Oceanapia sp. and its ecological implications. Mar. Biol. 1999, 135, 573–580. [Google Scholar] [CrossRef]
- Freeman, C.J.; Gleason, D.F. Chemical defenses, nutritional quality, and structural components in three sponge species: Ircinia felix, I. campana, and Aplysina fulva. Mar. Biol. 2010, 157, 1083–1093. [Google Scholar] [CrossRef]
- Roue, M.; Domart-Coulon, I.; Ereskovsky, A.; Dejediat, C.; Pererz, T.; Bourguet-Kondracki, M.-L. Cellular localization of clathridimine, an antimicrobial 2-aminoimidazole alkaloid produced by the Mediterranean calcareous sponge Clathrina clathrus. J. Nat. Prod. 2010, 73, 1277–1282. [Google Scholar] [CrossRef]
- Furrow, F.B.; Amsler, C.D.; McClintock, J.B.; Baker, B.J. Surface sequestration of chemical feeding deterrents in the Antarctic sponge Latrunculia apicalis as an optimal defense against sea star spongivory. Mar. Biol. 2003, 143, 443–449. [Google Scholar] [CrossRef]
- Becerro, M.A.; Paul, V.J.; Starmer, J. Intracolonial variation in chemical defenses of the sponge Cacospongia sp. and its consequences on generalist fish predators and the specialist nudibranch predator Glossodoris pallida. Mar. Ecol. Prog. Ser. 1998, 168, 187–196. [Google Scholar] [CrossRef]
- Siriak, T.; Intaraksa, N.; Kaewsuwan, S.; Yuenyongsawad, S.; Suwanborirux, K.; Plubrukarn, A. Intracolonial allocation of tisoxazole macrolides in the sponge Pachastrissa nux. Chem. Biodivers. 2011, 8, 238–246. [Google Scholar]
- Richardson, A.D.; Aalbersberg, W.; Ireland, C.M. The patellazoles inhibit protein synthesis at nanomolar concentrations in human colon tumor cells. Anti Cancer Drugs 2008, 16, 533–541. [Google Scholar] [CrossRef]
- Kwan, J.C.; Donia, M.S.; Han, A.W.; Hirose, E.; Haygood, M.G.; Schmidt, E.W. Genome streamlining and chemical defense in a coral reef symbiosis. Proc. Natl. Acad. Sci. USA 2012, 109, 20655–20660. [Google Scholar] [CrossRef]
- Gouiffes, D.; Juge, M.; Grimaud, N.; Welin, L.; Sauviat, M.; Barbin, Y.; Laurent, D.; Roussakis, C.; Henichart, J.; Verbist, J. Bistramide A, a new toxin from the urochordata Lissoclinum bistratum Sluiter: Isolation and preliminary characterization. Toxicon 1988, 26, 1129–1136. [Google Scholar] [CrossRef]
- Schmidt, E.W. The secret to a successful relationship: Lasting chemistry between ascidians and their symbiotic bacteria. Invertebr. Biol. 2015, 134, 88–102. [Google Scholar] [CrossRef]
- Subramani, R.; Aalbersberg, W. Culturable rare Actinomycetes: Diversity, isolation and marine natural product discovery. Appl. Microbiol. Biotechnol. 2013, 97, 9291–9321. [Google Scholar] [CrossRef]
- Ziemert, N.; Lechner, A.; Wietz, M.; Millan-Aguinaga, N.; Chavarria, K.L.; Jensen, P.R. Diversity and evolution of secondary metabolism in the marine actinomycete genus Salinispora. Proc. Natl. Acad. Sci. USA 2014, 111, E1130–E1139. [Google Scholar] [CrossRef]
- Lopera, J.; Miller, I.J.; McPhail, K.L.; Kwan, J.C. Increased biosynthetic gene dosage in a genome-reduced defensive bacterial symbiont. mSystems 2017, 2, e00096-17. [Google Scholar] [CrossRef]
- Holmstrom, C.; James, S.; Neilan, B.A.; White, D.C.; Kjelleberg, S. Pseudoalteromonas tunicata sp. nov., a bacterium that produces antifouling agents. Int. J. Syst. Bacteriol. 1998, 48, 1205–1212. [Google Scholar] [CrossRef]
- Baker, B.J.; Dent, B. Synoicum adareanum sampling underwater video March 2011 Palmer Station Antarctica, V3. Dryad Dataset 2020. Available online: https://doi.org/10.5061/dryad.gxd2547gw (accessed on 30 May 2020).
- Massana, R.; Murray, A.E.; Preston, C.M.; DeLong, E.F. Vertical distribution and phylogenetic characterization of marine planktonic Archaea in the Santa Barbara Channel. Appl. Environ. Microbiol. 1997, 63, 50–56. [Google Scholar] [CrossRef]
- Takahashi, S.; Tomita, J.; Nishioka, K.; Hisada, T.; Nishijima, M. Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next-generation sequencing. PLoS ONE 2014, 9, e105592. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.; Walters, W.; Berg-Lyons, D.; Lozupone, C.; Turnbaugh, P.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger data sets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Tackmann, J.; Matias Rodrigues, J.F.; von Mering, C. Rapid inference of direct interactions in large-scale ecological networks from heterogeneous microbial sequencing data. Cell Syst. 2019, 9, 286–296.e8. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the International AAAI ICWSM Conference on weblogs and social media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar]
- Cristóbal, E.; Ayuso, S.V.; Justel, A.; Toro, M. Robust optima and tolerance ranges of biological indicators: A new method to identify sentinels of global warming. Ecol. Res. 2013, 29, 55–68. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]
- Markowitz, V.M.; Chen, I.-M.A.; Palaniappan, K.; Chu, K.; Szeto, E.; Grechkin, Y.; Ratner, A.; Jacob, B.; Huang, J.; Williams, P.; et al. IMG: The integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012, 40, D115–D122. [Google Scholar] [CrossRef]
- Blin, K.; Medema, M.H.; Kottmann, R.; Lee, S.Y.; Weber, T. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2017, 45, D555–D559. [Google Scholar] [CrossRef]
- Murray, A.; Baker, B. Synoicum adareanum Microbiome. SCAR—Microbial Antarctic Resource System Metadata Dataset. , 2020. Available online: https://doi.org/10.15468/aewoib (accessed on 30 May 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phlya or Class | Whole Community | Core80 | Dynamic50 | Variable |
|---|---|---|---|---|
| Proteobacteria | ||||
| Gammaproteobacteria | 71.990 ± 6.640 | 73.280 ± 6.330 | 51.300 ± 23.160 | 43.710 ± 23.630 |
| Alphaproteobacteria | 22.900 ± 5.390 | 23.830 ± 5.930 | 23.830 ± 19.700 | |
| Deltaproteobacteria | 0.170 ± 0.100 | 0.160 ± 0.100 | 1.110 ± 2.000 | |
| Bacteroidetes | 2.830 ± 2.140 | 0.790 ± 0.690 | 46.550 ± 22.910 | 17.40 ± 14.690 |
| Verrucomicrobia | 1.560 ± 2.800 | 1.590 ± 2.930 | 2.830 ± 3.970 | |
| Nitrospirae | 0.270 ± 0.230 | 0.290 ± 0.240 | 0.020 ± 0.170 | |
| Planctomycetes | 0.120 ± 0.130 | 2.150 ± 3.060 | 4.600 ± 12.840 | |
| Actinobacteria | 0.100 ± 0.080 | 0.050 ± 0.050 | 3.150 ± 3.530 | |
| Patescibacteria | 0.020 ± 0.090 | 0.840 ± 2.050 | ||
| Dadabacteria | 0.020 ± 0.003 | 1.220 ± 2.030 | ||
| Uncl. Bacteria | 0.009 ± 0.017 | 0.720 ± 1.530 | ||
| Dependentiae | 0.004 ± 0.018 | 0.340 ± 1.990 | ||
| Chlamydiae | 0.002 ± 0.006 | 0.190 ± 0.830 | ||
| Acidobacteria | 0.000 ± 0.003 | 0.020 ± 0.130 | ||
| Chloroflexi | 0.000 ± 0.003 | 0.020 ± 0.130 | ||
| Epsilonbacteraeota | 0.000 ± 0.001 | 0.010 ± 0.070 |
| ASV_ID | Phylum, Highest Taxonomic Assignment | Average Relative Abundance (%) | Rank | Nearest Neighbor % Identity | NRP BGC | PKS BGC | Combined NRP-PKS |
|---|---|---|---|---|---|---|---|
| SaM_ASV1, 2, 4, 5, 10, 17, 18 | Proteobacteria, Microbulbifer | 77.54 ± 21.86 | 1, 2, 4, 5, 8, 9, 10 | 97.42 | + | + | - |
| SaM_ASV7 | Proteobacteria, Endozoicomonas | 0.47 ± 0.51 | 13 | 96.71 | + | + | - |
| SaM_ASV13 | Proteobacteria, Nitrosomonas | 0.46 ± 0.35 | 14 | 99.77 | + | + | + |
| SaM_ASV3 | Proteobacteria, Pseudovibrio | 19.92 ± 4.74 | 3 | 98.75 | + | + | + |
| SaM_ASV6 | Proteobacteria, Hoeflea | 1.59 ± 1.36 | 6 | 99.25 | + | + | + |
| SaM_ASV16 | Proteobacteria, Halocynthiibacter | 0.63 ± 0.64 | 11 | 99.75 | + | - | - |
| SaM_ASV11 | Nitrospirota/Nitrospirae, Nitrospira | 0.27 ± 0.23 | 15 | 98.32 | + | + | + |
| SaM_ASV12 | Bacteroidota/Bacteroidetes, Lutibacter | 0.50 ± 0.55 | 12 | 94.54 | + | - | + |
| SaM_ASV14 | Verrucomicrobiota/Verrucomicrobia, Lentimonas | 0.16 ± 0.22 | 19 | 99.77 | + | - | + |
| Sam_ASV21 | Proteobacteria, Rhodobacteraceae | 0.18 ± 0.26 | 18 | 98.75 | - | - | - |
| SaM_ASV8 | Proteobacteria, Rhodospirillales | 0.22 ± 0.22 | 17 | 86.60 | - | - | - |
| SaM_ASV9 | Bdellovibrionota/Proteobacteria, Bdellovibrionaceae | 0.15 ± 0.09 | 20 | 90.35 | - | - | - |
| SaM_ASV19 | Bacteroidetes, Cryomorphaceae | 0.24 ± 0.23 | 16 | 89.10 | - | - | - |
| SaM_ASV15 | Verrucomicrobiota/Verrucomicrobia, Opitutaceae | 1.34 ± 2.77 | 7 | 90.14 | - | - | - |
| SaM_ASV20 | Actinobacteria, Solirubrobacterales | 0.05 ± 0.05 | 21 | 91.80 | - | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murray, A.E.; Avalon, N.E.; Bishop, L.; Davenport, K.W.; Delage, E.; Dichosa, A.E.K.; Eveillard, D.; Higham, M.L.; Kokkaliari, S.; Lo, C.-C.; et al. Uncovering the Core Microbiome and Distribution of Palmerolide in Synoicum adareanum Across the Anvers Island Archipelago, Antarctica. Mar. Drugs 2020, 18, 298. https://doi.org/10.3390/md18060298
Murray AE, Avalon NE, Bishop L, Davenport KW, Delage E, Dichosa AEK, Eveillard D, Higham ML, Kokkaliari S, Lo C-C, et al. Uncovering the Core Microbiome and Distribution of Palmerolide in Synoicum adareanum Across the Anvers Island Archipelago, Antarctica. Marine Drugs. 2020; 18(6):298. https://doi.org/10.3390/md18060298
Chicago/Turabian StyleMurray, Alison E., Nicole E. Avalon, Lucas Bishop, Karen W. Davenport, Erwan Delage, Armand E.K. Dichosa, Damien Eveillard, Mary L. Higham, Sofia Kokkaliari, Chien-Chi Lo, and et al. 2020. "Uncovering the Core Microbiome and Distribution of Palmerolide in Synoicum adareanum Across the Anvers Island Archipelago, Antarctica" Marine Drugs 18, no. 6: 298. https://doi.org/10.3390/md18060298
APA StyleMurray, A. E., Avalon, N. E., Bishop, L., Davenport, K. W., Delage, E., Dichosa, A. E. K., Eveillard, D., Higham, M. L., Kokkaliari, S., Lo, C.-C., Riesenfeld, C. S., Young, R. M., Chain, P. S. G., & Baker, B. J. (2020). Uncovering the Core Microbiome and Distribution of Palmerolide in Synoicum adareanum Across the Anvers Island Archipelago, Antarctica. Marine Drugs, 18(6), 298. https://doi.org/10.3390/md18060298