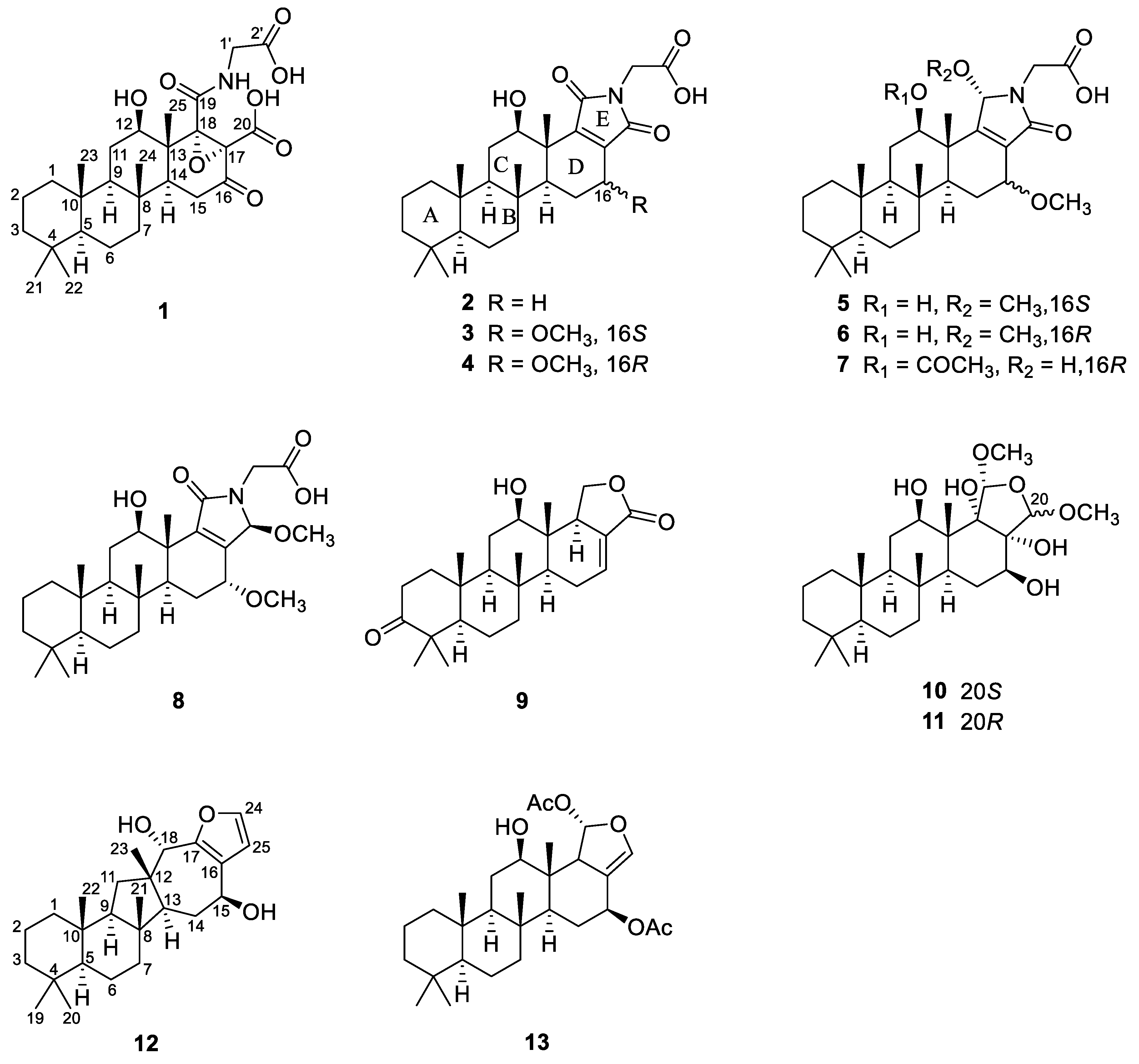
2.1. Structure Elucidation
The molecular formula of compound
1 was deduced to be C
27H
39NO
8, corresponding to 9 degrees of unsaturation, by HRFABMS analysis ([M-H
2O-H]
− m/z 486.2491, calcd for C
27H
36NO
7, 486.2492). The
13C NMR data of this compound showed signals indicative of four carbonyl carbons (δ
C 204.2, 177.8, 170.4, and 168.7), two oxygenated and non-protonated carbons (δ
C 73.3 and 68.7) and one oxymethine carbon (δ
C 74.7) (
Table 1). The remaining 20 carbons were all aliphatic (four non-protonated, three methine, eight methylene, and five methyl carbons). Therefore,
1 was thought to be a pentacyclic compound. The
1H NMR spectra also showed five singlet methyl signals, revealing a terpene or related structure. In conjunction with the mass data and inherent degrees of unsaturation, our preliminary interpretation of the 1-D NMR data suggested that
1 was a highly oxygenated pentacyclic sesterterpene with a nitrogen-containing functionality.
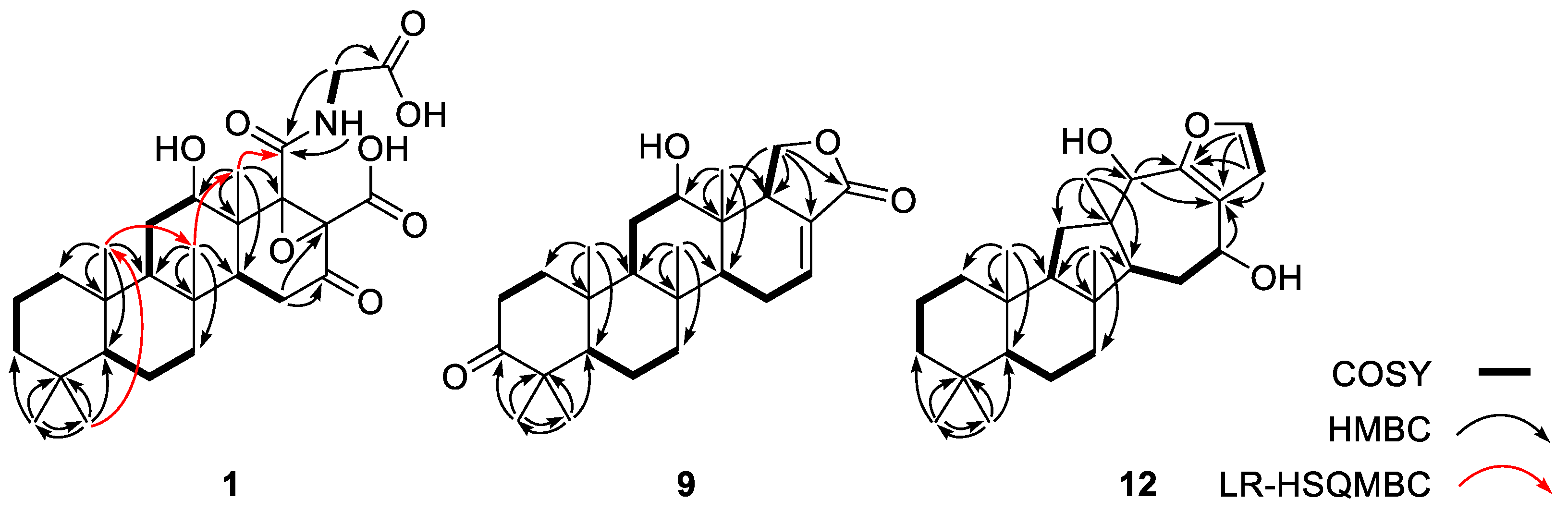
The planar structure of
1 was determined by combined 2-D NMR analyses (
Figure 1). After the initial matching of the protons with their bearing carbons by the HSQC data, three aliphatic proton spin systems were deduced from the
1H-
1H COSY data. These were consisted of three methylenes (C-1-C-3), one methine and two methylenes (C-5-C-7), and two methines and one methylene (C-9, C-11 and C-12), respectively. Subsequently, these linear systems were assembled by using the HMBC correlations with bridgehead singlet methyl protons (H
3-21-H
3-25): H
3-21/C-3, C-4 and C-5, H
3-22/C-3, C-4 and C-5, H
3-23/C-1,C-5, C-9, and C-10, H
3-24/C-7, C-8, C-9, and C-14, and H
3-25/ C-12, C-13 and C-14. Then these HMBC-based assemblies were confirmed by the LR-HSQMBC correlations between the methyl groups: H
3-22/C-23, H
3-23/C-24 and H
3-24/C-25. Overall
1 was found to possess rings A-C as a 6/6/6 system frequently seen in scalaranes [
1,
3].
Based on the COSY data, the C-14 methine group was directly connected to a methylene (δ
C/δ
H 35.4/2.35 and 2.29, C-15) that showed no further proton–proton couplings (
Figure 1). The HMBC cross peaks of these protons placed a ketone (δ
C 204.2, C-16) and a non-protonated carbon (δ
C 68.7, C-17) at the neighboring positions: H-14/C-16 and H
2-15/C-16 and C-17. Similarly, another non-protonated carbon (δ
C 73.3, C-18) was placed based on its HMBC cross peaks to previously assigned protons: H-12/C-18 and H
3-25/C-18. The assignments of C-17 and C-18 were supported by the LR-HSQMBC data [
21], a variant of an HMBC experiment, thus constructing a cyclohexanone (ring D): H-14/C-18 and H
2-15/C-17 (
Supporting Information Figure S8). The chemical shifts of these non-protonated carbons at δ
C 73.3 and 68.7 indicated not only the attachments of oxygen but also the presence of an epoxide moiety involving these carbons, accounting for the last degree of unsaturation required by the mass data.
The remaining NMR signals were those of an exchangeable NH (δ
H 7.88), a methylene (δ
C/δ
H 43.9/4.15 and 3.33) and three carbonyl carbons (δ
C 177.8, 170.4 and 168.7). The NH and methylene were directly connected to each other based on their vicinal coupling constants (
J = 7.4, 3.2 Hz) and COSY data. This spin system was expanded to an
N-carboglycine moiety by the HMBC cross peaks with the carbonyl carbons at δ
C 177.8 and 168.7. A crucial long-range correlation of the latter carbon with H
3-25 by LR-HSQMBC data linked the
N-carboglycine moiety to C-18, assigning the carbonyl carbons at C-2’ and C-19, respectively (
Figure 1). Although no NMR correlations were observed to the carbonyl carbon at δ
C 170.4, this carbon (C-20) must be attached at C-18, the only open position in the main framework. Of the eight oxygens indicated by the mass data of
1, the four carbonyl groups at C-16, C-19, C-20, and C-2’ and the epoxide at C-17-C-18 only accounted for five. Therefore, the three remaining oxygens must be OH groups at C-12, C-19 and C-20, forming a hydroxyl moiety and two carboxylic acids. Thus, the planar structure of
1 was determined to be a glycine- and epoxide-bearing scalarane sesterterpene.
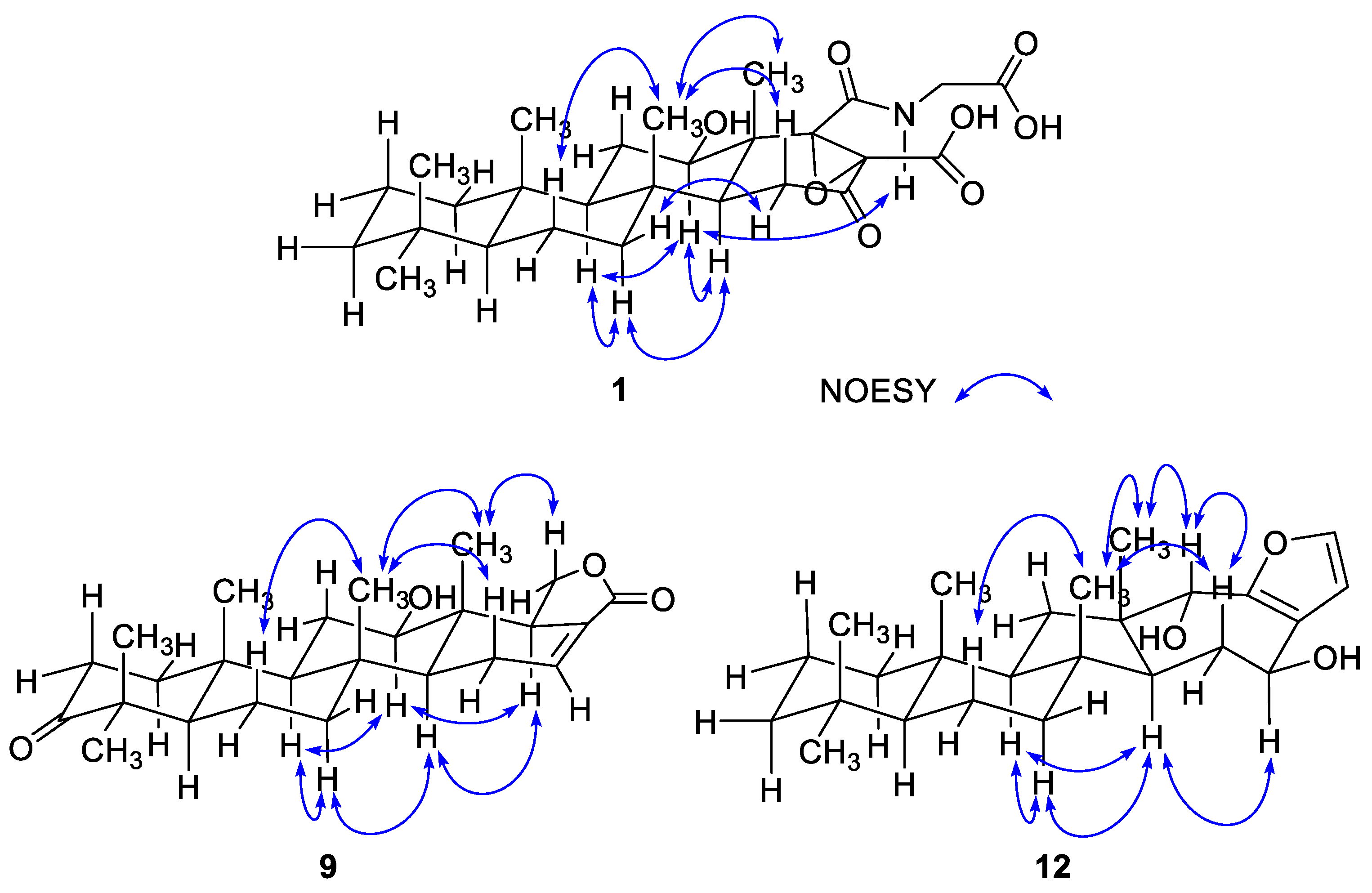
The configurations at the stereogenic centers of
1 were assigned based on NOESY data, which readily showed all
trans junctures for rings A–D, which are typical of scalaranes and similar sesterterpenes (
Figure 2). This interpretation was also supported by the characteristic carbon chemical shifts of the bridgehead methines and methyl groups. The β-orientation (12
R* configuration) was assigned based on the NOESY cross peak for H-12/H-14 and its vicinal coupling constants (
J = 11.0, 4.4 Hz) with H
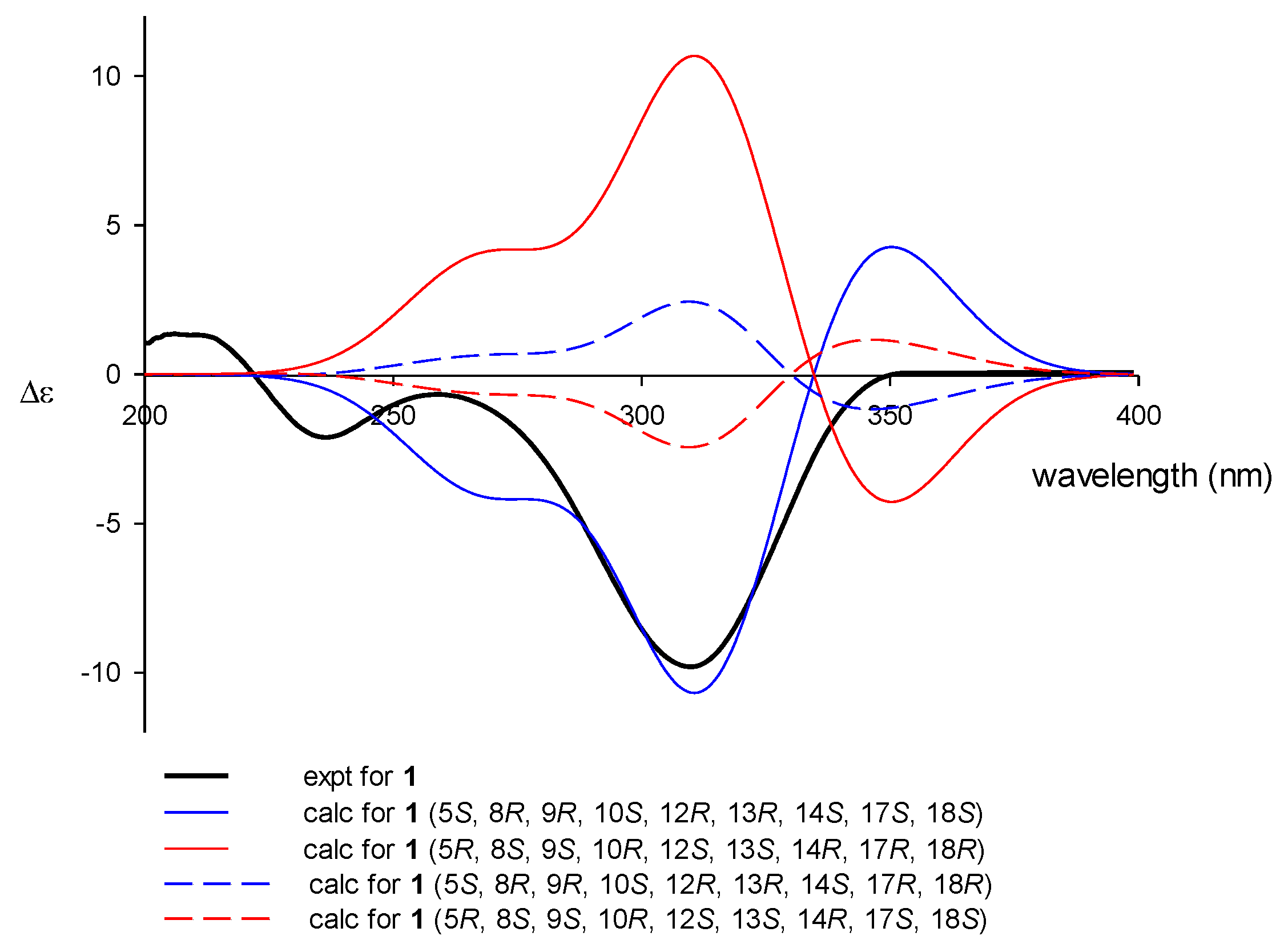
2-11. For the C-17-C-18 epoxide, which does not have any bound hydrogens, severe steric crowding with the neighboring C-25 methyl group indicated that the olefinic precursor underwent α-oriented attack by the oxygen. This interpretation was confirmed by ECD calculations (
Figure 3). Given the all
trans ring junctures and 12
R* configuration, the calculated ECD profile for the 17
S and 18
S configurations matched well with the observed profile in both the intensity and wavelength of the signals. In this way, the absolute configurations of the ring junctures and C-12 were also satisfactorily assigned as (5
S, 8
R, 9
R, 10
S, 12
R, 13
R, 14
S, 17
S, 18
S). Thus, compound
1, designated hyrtioscalarin A, was determined to be a new glycine-bearing scalarane.
The molecular formula of compound
2 was deduced to be C
27H
39NO
5 by HRFABMS analysis ([M-H]
− m/z 456.2760, calcd for C
27H
38NO
5, 456.2750), which corresponds to 9 degrees of unsaturation. The spectroscopic data of this compound were highly reminiscent of those of
1, suggesting they shared the same glycine-bearing scalarane core. Detailed examination of the
1H and
13C NMR and HSQC data, however, revealed remarkable differences, the most noticeable of which were the replacement of the C-16 ketone and C-17-C-18 epoxide of
1 with a methylene group (δ
C/δ
H 23.4/2.53 and 2.26) and two non-protonated carbons (δ
C 150.8 and 144.1) in
2 (
Table 1).
The structural differences were defined by a combination of 2-D NMR data (
Supporting Information Figure S1). First, the A–C rings of
1 were found to be intact in
2 based on their COSY, HSQC and HMBC data. Then, tracing the proton spin system using the COSY data revealed that the C-14 methine group (δ
C/δ
H 56.4/1.18) was connected to the methylene at C-15 (δ
C/δ
H 17.8/1.98 and 1.67), which was connected in turn to the methylene at C-16 (δ
C/δ
H 23.4/2.53 and 2.26). Subsequently, two non-protonated carbons were placed at C-17 (δ
C 144.1) and C-18 (δ
C 150.8), constructing ring D as a cyclohexene moiety, based on the HMBC cross peaks of H
2-15/C-17, H
2-16/C-17, and H
3-25/C-18.
The partial formula of the A-D ring portion of
2 was C
23H
36O, leaving a C
4H
3NO
4 unit for the remainder of the molecule. The
1H NMR and HSQC data of
2 revealed the remaining fragment was an isolated methylene (δ
C/δ
H 40.2/4.19 (2H), C-1’) with HMBC cross peaks with three carbonyl carbons (δ
C 175.1, 171.8 and 171.5) (
Supporting Information Figure S1). Among these, the carbon at δ
C 171.5 was placed at C-20 based on its additional HMBC cross peak with H
2-16. Therefore, one of the remaining carbonyl carbons must be located at C-19 and attached at C-18, the last open end of the ring system, while the other was a free carboxylic acid carbon at C-2’. The noticeably small and broad shape of the carbon signal at δ
C 171.8 in the
13C NMR data secured the assignment of this group at C-2’, leaving the signal at δ
C 175.1 as C-19. Therefore,
2 must possess a glycine-derived succinimide group (C-17-C-20), fulfilling the remaining 4 degrees of unsaturation required by the mass data. The configurations of stereogenic centers in the A–C rings were also assigned to be identical to those of
1 by the NOESY data (
Supporting Information Figure S2) and biogenetic consideration with
1. Thus, hyrtioscalarin B (
2) was determined to be a scalarane sesterterpene bearing a succinimide group.
Compounds
3 and
4 were isolated as pale-yellow amorphous solids, and they were found to have the same molecular formula (C
28H
41NO
6) by HRFABMS analyses. The spectroscopic data of these compounds were very similar to each other and highly reminiscent of those of
2, suggesting they shared the same succinimide-bearing scalarane structure. The most noticeable differences in the
13C,
1H and HSQC NMR data of
3 and
4 from those of
2 were the replacement of a methylene with a methine (δ
C/δ
H 3: 73.6/4.18,
4: 68.4/4.10) and a methoxy group (δ
C/δ
H 3: 58.1/3.55,
4: 57.8/3.45) (
Supporting Information Tables S1 and S2).
These compounds were defined as 16-methoxy derivatives of
2 based on their combined 1-D and 2-D NMR data, including the crucial HMBC H
3-OCH
3/C-16 cross peaks in the spectra of both compounds (
Supporting Information Figure S1). The relative configurations at the epimeric C-16 stereogenic center were also determined from the NOESY data. In addition to the same all
trans ring junctures and 12
R configurations as seen in
1 and
2 derived from the NOESY data and biogenetic consideration, additional NOESY cross peaks of H-14/H-16, H-15α (δ
H 2.32)/H-16, H-15β (δ
H 1.65)/H
3-25, and H-15β/H
3-24 assigned the 16
S configuration for
3 (
Supporting Information Figure S2). In contrast, the opposite 16
R configuration was assigned for
4 based on the cross peaks of H-14/H
3-16OCH
3, H-15α (δ
H 2.12)/H
3-16OCH
3 and H-15β/H
3-25. The absolute configurations of this compound were further supported the ECD calculations (
Supporting Information Figure S79). Thus, compounds
3 and
4, designated hyrtioscalarin C and D, respectively, were defined as epimeric 16-methoxy derivatives of hyrtioscalarin B (
2).
Compounds
5 and
6 were obtained as another pair of epimeric pale-yellow solids and were found to have the formula C
29H
45NO
6 by HRFABMS analyses. The
13C and
1H NMR data of these compounds were similar to those of
3 and
4, indicating they shared the same glycine-incorporated scalarane structures. Aided by the HSQC data, the most conspicuous differences in the
13C and
1H NMR data were the replacement of a carbonyl carbon with a methoxy and a methine group (δ
C/δ
H 5: 84.8/5.75 and 50.3/3.09,
6: 84.5/5.69 and 50.1/3.12) (
Supporting Information Tables S1 and S2). The remarkable differentiation of the C-17 and C-18 olefinic carbons between compounds
3/
4 and
5/
6 suggested that structural differences were present in compounds
5/
6 around the amide carbons at C-19 and C-20. This interpretation was confirmed by the COSY and HMBC data of
5, which assigned a methoxy-methine group at C-19 based on the proton-carbon correlations of H
2-15/C-17; H-16/C-17; H-19/C-17, C-18 and C-20; H
3-25/C-18; H
2-1’/C-19, C-20, and C-2’; H
3-16OCH
3/C-16; and H
3-19OCH
3/C-19 (
Supporting Information Figure S1). Possibly due to the small amounts of materials isolated, the HMBC data of
6 were less informative than those of
5 (
Supporting Information Table S4). However, in conjunction with a comparison of their
13C and
1H NMR data, the planar structure of
6 was defined to be identical to that of
5 based on the key HMBC cross peaks of H
2-1’/C-19, C-20, and C-2’; H
3-16OCH
3/C-16; and H
3-19OCH
3/C-19. Overall, these compounds possessed γ-methoxy-α,β-unsaturated butyrolactam rings as a common structural motif (ring E).
Compounds
5 and
6 each possessed two methoxy-bearing stereogenic centers, C-16 and C-19. The relative configurations at these centers were determined from their NOESY data as well as the biogenetic consideration with other scalaranes. For
5, the 16
S and 19
S configurations were assigned based on the cross peaks of H-12/H-14, H-12/H
3-19OCH
3, H-14/H-16, H-15β (δ
H 1.65)/H
3-25, H-19/H
3-25, and H-19/H-1’ (δ
H 3.65) (
Supporting Information Figure S2). Similarly, the 16
R and 19
S configurations were assigned for
6 based on the cross peaks of H-12/H-14, H-12/H
3-19OCH
3, H-15α (δ
H 2.04)/H
3-16OCH
3, H-15β (δ
H 1.53)/H
3-25, H-19/H
3-25, and H-19/H-1’ (δ
H 3.69). These results clearly indicated that
5 and
6 were epimeric at the methoxy-bearing C-16 position. Overall, hyrtioscalarins E (
5) and F (
6) are epimeric scalaranes bearing a glycine-derived γ-methoxy-α,β-unsaturated butyrolactam moiety.
The molecular formula of compound
7 was established as C
30H
45NO
7 based on its HRFABMS data ([M-H]
− m/z 530.3112, calcd for C
30H
44NO
7, 530.3118). The spectroscopic data of this compound were similar to those of
5 and
6, suggesting an analogous structure. The most noticeable differences in the
13C and
1H NMR data were the appearance of an acetyl group (δ
C/δ
H 173.4 and 22.2/2.03) and the lack of a methoxy group (
Supporting Information Tables S1 and S2). Based upon the extensive 2-D NMR analyses, the structural differences were readily found to be the 12-acetyl and 19-hydroxy groups (
Supporting Information Figure S1). After assigning the same ring junctures and 12
R configurations using the NOESY data and biogenetic consideration, the 16
R and 19
S configurations were also assigned in a similar manner based on the cross peaks of H-12/H-14, H-14/H
3-16OCH
3, H-15α (δ
H 2.09)/H
3-16OCH
3, H-15β (δ
H 1.71)/H
3-25, H-19/H
3-25, H-19/H-1’ (δ
H 3.88), and H-19/H
3-12OCOCH
3 (
Supporting Information Figure S2). Thus, hyrtioscalarin G (
7) was defined as a new scalarane bearing a γ-hydroxy-α,β-unsaturated butyrolactam group.
The molecular formula of compound
8 was established to be C
29H
45NO
6, identical to that of
5 and
6, by HRFABMS analysis ([M-H]
− m/z 502.3169, calcd for C
29H
44NO
6, 502.3169). Although the spectroscopic data of this compound were also very similar to those of
5 and
6, a detailed examination of its
13C and
1H NMR data revealed significant differences among the carbons and protons of the butyrolactam (ring E) and the adjacent positions (
Supporting Information Tables S1 and S2). Although the 2-D NMR data indicated the same A–D rings as in the other compounds, the butyrolactam moiety of
8 was revealed to have a distinct substitution mode. That is, key HMBC cross peaks were found for H
2-15/C-17; H-16/C-17, C-18 and C-19; H
2-1’/C-19, C-20 and C-2’; and H
3-20OCH
3/C-19 (
Supporting Information Figure S1). Therefore, methylation must have occurred at the C-20 lactam carbon instead of C-19. After assigning the all
trans ring junctures and 12
R configurations from the NOESY data and biogenetic consideration, the 16
R and 20
S configurations of the methoxy-bearing stereogenic centers were also determined from the NOESY cross peaks of H-15α (δ
H 2.16)/H
3-16OCH
3, H-15β (δ
H 1.71)/H
3-25, H-16/H
3-20OCH
3, H-20/H
3-16OCH
3, and H
2-1’ (δ
H 3.87)/H
3-20OCH
3 (
Supporting Information Figure S2). This interpretation was further supported by the ECD calculation (
Supporting Information Figure S79). In this way, compound
8, designated hyrtioscalarin H, was determined to be a new butyrolactam-bearing scalarane.
Hyrtioscalarins A-H (
1–
8) all possess glycine-derived nitrogenous functionalities. A literature study showed that among the numerous scalarane sesterterpenes reported to date, few are nitrogenous. To the best of our knowledge, the only examples are molliorins A–C from
Cacospongia mollior [
11,
12,
13], an unnamed compound (
14, a congener in this work) from
Hyatella sp. [
14], petrosaspongiolactams A–C from
Petrosaspongia sp. [
15], scalalactam A–D from
Spongia sp. [
16], a pyrrole containing scalarane from
Scalarispongia sp. [
17], and 24-methoxypetrosaspongia C from
Hyrtios erectus [
18]. Moreover, only the one compound from
Hyatella sp. possesses the same glycine-derived moiety as seen in
1–
8, emphasizing the unusual structural features of these new compounds. Indeed, our finding of diverse nitrogenous derivatives suggests that these compounds are important structural variants of scalaranes.
In addition to glycine-bearing scalaranes
1–
8, a number of non-nitrogenous analogs were also isolated. The molecular formula of compound
9 was deduced to be C
25H
36O
4, corresponding to 8 degrees of unsaturation, from its HRFABMS data ([M-H]
− m/z 399.2531, calcd for C
25H
35O
4, 399.2535). The combined
13C,
1H and HSQC NMR data of this compound showed the presence of two carbonyl carbons (δ
C 220.3 and 172.4) and two olefinic carbons (δ
C/δ
H 137.2/6.81 and 128.6) in the deshielded region, which together account for 3 degrees of unsaturation (
Table 2). Signals for an oxymethine (δ
C/δ
H 81.4/3.41) and an oxymethylene (δ
C/δ
H 70.8/4.50 and 4.20) were also conspicuous in the NMR data. Other signals in the shielded region of the
13C NMR spectra were those of four non-protonated, four methine, six methylene, and five methyl carbons, which matched well with the corresponding protons based on the HSQC data. Altogether, these data indicated that
9 is pentacyclic and likely to be a typical scalarane sesterterpene.
The planar structure of
9 was elucidated from its 2-D NMR data (
Figure 1). First, the COSY data revealed the presence of a spin system involving two methylenes (δ
C/δ
H 40.2/1.98 and 1.58 and 34.8/2.53 and 2.47, C-1 and C-2) and requiring a non-protonated carbon at C-3. Based on the HMBC data, a ketone carbon (δ
C 220.3) was placed at this position based on its correlations with neighboring protons at H
2-1, H
2-2, H
3-21, and H
3-22. Similarly, the A–C ring portion was defined as the same 6/6/6 tricyclic moiety seen in
1-
8 based on the COSY and HMBC data, including crucial long-range correlations between the five bridgehead methyl protons and their neighboring carbons. Similar to other scalaranes, the chemical shifts (δ
C/δ
H 81.4/3.41), vicinal coupling constants (
J = 11.4, 3.4 Hz) and HMBC cross peaks to the C-25 methyl group (H-12/C-25, H
3-25/C-12, H-9/C-12, and H
2-11/C-12) placed a β-hydroxy group at C-12.
The COSY data located a double bond at C-16 based on the proton–proton correlations of H-14 with H-16 via H2-15 (δH 1.34, 2.39 and 2.27, and 6.81, respectively). Due to the small amount of material obtained, no HMBC data were available to directly support this interpretation. However, the chemical shifts of the C-18 methine group (δC/δH 51.6/2.92), which showed long-range coupling with H3-25, were definitive enough to place an olefinic carbon at C-17 (δC 128.6). Then, the placement of an oxymethylene (δC/δH 70.8/4.50 and 4.20) at C-19 was accomplished based on direct proton–proton couplings with H-18. Finally, the HMBC cross peaks of H2-19 with the neighboring carbons (H2-19/C-13, C-17, C-18, and C-20) not only confirmed its location but also constructed a five-membered lactone as ring E.
The configuration at the newly constructed C-18 center was assigned as
R based on the NOESY cross peaks of H-18 with the α-oriented H-12 and H-14 protons as well as the all
trans ring junctures derived from the NOESY data and biogenetic relation with other scalaranes (
Figure 2). Thus, the structure of
9, designated 12-deacetyl-3-oxoscalarin (following the previously reported analog scalarin), was determined to be a scalarane lactone bearing a 3-keto group. A literature study showed that scalarane sesterterpenes possessing a 3-keto group are quite rare. To the best of our knowledge, the only four reported examples are two scalaranes from
H. erectus [
22] and
H. erecta [
23], a norscalarane from the mollusk
Dorisprismatica (=
Glossodoris)
atromarginata [
24] and a scalarane with acetoxy group at C-3 position from
H. erecta [
25].
An oxidized-furan-bearing scalarane (
10) was isolated as a pale-yellow solid, and it was found to have the molecular formula C
27H
46O
7, corresponding to 5 degrees of unsaturation, by HRFABMS ([M-H]
− m/z 481.3168, calcd for C
27H
45O
7, 481.3165). With the aid of the HSQC data, the
13C and
1H NMR spectra of this compound showed signals for two oxygen-bearing non-protonated carbons (δ
C 83.1 and 81.0), four methines (δ
C/δ
H 106.1/5.22, 103.2/4.99, 74.4/4.34, and 74.2/3.72) and two methyl groups (δ
C/δ
H 56.0/3.56 and 55.6/3.40) (
Supporting Information Tables S1 and S2). Since all the other carbon and proton signals were observed in the shielded regions and showed no evidence of unsaturation,
10 was thought to be a pentacyclic sesterterpene.
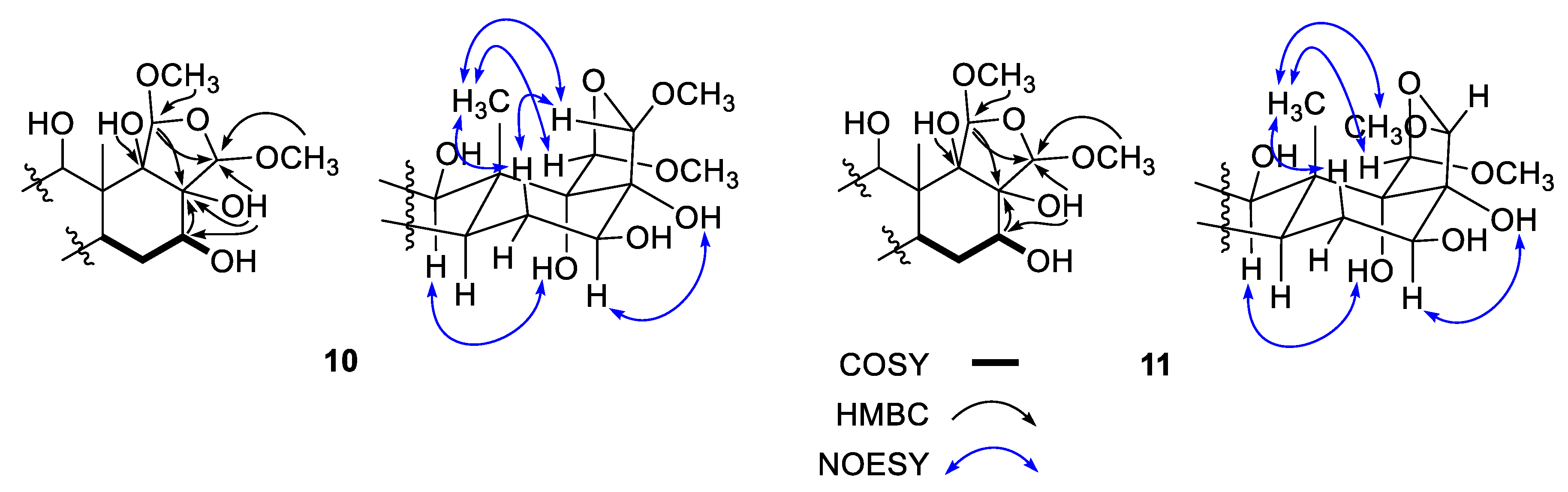
Based upon the results of 2-D NMR analyses and comparison with other scalaranes,
10 was readily found to possess the same A-C rings, including the 12-βOH group seen in the compounds described above, and most of the oxygenated functionalities of
10 were located on rings D and E. The HMBC data, including the correlations of hydroxy protons, were crucial to defining the remaining structure (
Figure 4). That is, starting at H-14 (δ
H 1.32), the COSY correlations placed a hydroxy methine at C-16 (δ
C/δ
H 74.2/3.72), which was confirmed by the HMBC cross peaks of this methine carbon with H-14 and H
2-15 (δ
H 1.79 and 1.44). The neighboring carbon, C-17 (δ
C 81.0), was similarly assigned as being a hydroxy-bearing non-protonated carbon based on the correlations of H
2-15/C-17, H-16/C-17, and 17-OH (δ
H 2.32)/C-16 and C-17. Another hydroxy group was also placed at C-18 (δ
C 83.1) by the HMBC correlations of H
3-25/C-18 and of 18-OH (δ
H 3.87)/C-13 and C-18.
In addition, two methoxy groups (δC/δH 55.6/3.40 and 56.0/3.56) were directly attached to oxymethines (δC/δH 106.1/5.22 and 103.2/4.99, C-19 and C-20) by diagnostic 3-bond HMBC interactions. The deshielded chemical shifts of these oxymethines in conjunction with the remaining oxygen from the mass data indicated these were part of an acetal. This interpretation was confirmed, and a tetrahydrofuran moiety was constructed by a series of HMBC cross peaks, namely, H-19/C-13, C-17 and C-20, H-20/C-17, and 18-OH (δH 3.47)/C-20. Thus, the planar structure of 10 was defined as a polyoxygenated scalarane.
Compound
10 possessed stereogenic centers at the D/E ring juncture and adjacent sites, and their relative configurations were assigned based on the NOESY data (
Figure 4). The H-14/H-16 cross peak defined a β-orientation for the 16-OH group, which was in good agreement with the vicinal coupling constants (
J = 12.0, 5.7 Hz) of H-16. In addition, the H
3-25 protons showed cross peaks with H-15β (δ
H 1.44), H-19 and H-20. To achieve sufficient spatial proximity with the bridgehead H
3-25 protons, not only a
cis D/E ring juncture but also β-orientations of both H-19 and H-20 in ring E are required. Overall, aided by the biogenetic consideration, the configurations of the stereogenic centers were assigned as 12
R, 16
S, 17
R, 18
S, 19
R, and 20
S. Thus, 17(
R),18(
S)-dihydroxy-19(
R),20(
S)-dimethoxysesterstatin 5 (
10), was determined to be a tetrahydrofuran-bearing scalarane sesterterpene.
The molecular formula of compound
11 was deduced to be C
27H
46O
7, identical to that of
10, based on HRFABMS analysis ([M-H]
− m/z 481.3172, calcd for C
27H
45O
7, 481.3165). The spectroscopic data of this compound were also very similar to those of
10, implying the same planar structure, which was confirmed by 1-D and 2-D NMR analyses. However, a detailed comparison of its
13C and
1H NMR data revealed remarkable differences among the carbons and protons of ring E and the adjacent positions, suggesting that
10 and
11 are epimers (
Supporting Information Tables S1 and S2). To confirm this, the NOESY data of
11 showed a cross peak for H-14/H-16, indicating the presence of the same 16β-OH group as in
10. Additionally, cross peaks of H
3-25 with H-15β (δ
H 1.65), H-19 and H
3-20OCH
3 were observed. To generate these signals, the D/E ring juncture must be
cis, identical to that in
10, but an opposite orientation was required at the C-20 stereogenic center, indicating a 20
R configuration (
Figure 4). Thus, 17(
R),18(
S)-dihydroxy-19(
R),20(
R)-dimethoxysesterstatin 5 (
11) was defined as another tetrahydrofuran-bearing scalarane.
In addition to scalaranes, the
H. erectus extract contained a sesterterpene with a different skeleton. The molecular formula of compound
12 was established as C
25H
38O
3, corresponding to 7 degrees of unsaturation, by HRFABMS analysis ([M-H]
− m/z 385.2738, calcd for C
25H
37O
3, 385.2743). The
13C NMR data of this compound showed four olefinic carbons (δ
C 150.2, 141.4, 124.2, and 111.5) (
Table 2). The chemical shifts (δ
H 7.30 and 6.50) and coupling constants (
J = 1.7 Hz for both) of the corresponding protons in the
1H NMR data were indicative of a disubstituted furan moiety. Aided by the HSQC data, two oxymethines (δ
C/δ
H 76.1/4.46 and 69.8/4.66) were also identified from the NMR data. The other signals were those of four non-protonated, three methine, seven methylene and five methyl groups. Accordingly,
12 must be a pentacyclic sesterterpene.
A detailed comparison of the 1-D NMR data with those of other compounds revealed remarkable differences in the chemical shifts of several carbons and protons, prompting extensive 2-D NMR analyses (
Figure 1). First, the COSY data revealed the presence of two linear chains of three protonated carbons (C-1-C-3 and C-5-C-7). Subsequently, several long-range correlations from these carbons to the protons of four singlet methyl groups (H
3-19, H
3-20, H
3-21 and H
3-22) secured a 6/6 bicyclic moiety (rings A and B), the same as its congeners.
In the process of determining the structures of rings A and B, the C-9 methine group (δ
C/δ
H 61.4/1.30) was confidently assigned based on its HMBC cross peaks with the H
3-21 and H
3-22 protons. In contrast to the scalaranes, however, the proton spin system involving H-9 terminated at the C-11 methylene (δ
C/δ
H 34.9/1.89 and 1.37), placing a non-protonated carbon at neighboring C-12. This was confirmed by the HMBC cross peaks of C-11 and C-12 (δ
C 43.9) with a common methyl proton (δ
H 1.01, H
3-23). This methyl proton showed an additional HMBC cross peak with a methine group (δ
C/δ
H 49.3/1.82, C-13). Then, a crucial 3-bond correlation with H
3-21 confirmed the attachment of C-13 to C-8 of ring B, establishing a five-membered ring (C-8, C-9, C-11-C-13, ring C) (
Figure 1).
The COSY data revealed a linear spin system of H-13 (δH 1.82), H2-14 (δH 1.90 and 1.70) and H-15 (δH 4.66), analogous to the H-14-H-16 spin system in the scalarane congeners. The assignment of an oxygenated methine at C-15 (δC 69.8) is also reminiscent of several previously reported compounds. This three-carbon unit was then extended by two additional non-protonated carbons (δC 124.2 and 150.2, C-16 and C-17, respectively) by the HMBC cross peaks of H2-14/C-16 and of H-15/C-16 and C-17. Similarly, another hydroxy-bearing methine (δC/δH 76.1/4.46, C-18) was attached to C-12 by the HMBC cross peaks of H-18/C-12 and C-14 and of H3-23/C-18. Then, C-18 was linked to C-17 by the similar correlations of H-18 with C-16 and C-17, establishing a seven-membered ring (C-12-C-18, ring D).
The preliminary examination of the
13C and
1H NMR data revealed the presence of a disubstituted furan moiety that required the linkage of C-16 and C-17 to the remaining carbons at C-24 (δ
C/δ
H 141.4/7.30) and C-25 (δ
C/δ
H 111.5/6.50). This confirmed by the small proton–proton coupling constants (
J = 1.7 Hz) between H-24 and H-25 as well as their HMBC cross peaks with C-16 and C-17 (
Figure 1). Thus, a 1,2-disubstituted furan moiety was deduced as the final structural motif (ring E). Overall, the planar structure of
12 was determined to be that of a salmahyrtisane-type sesterterpene [
25,
26,
27].
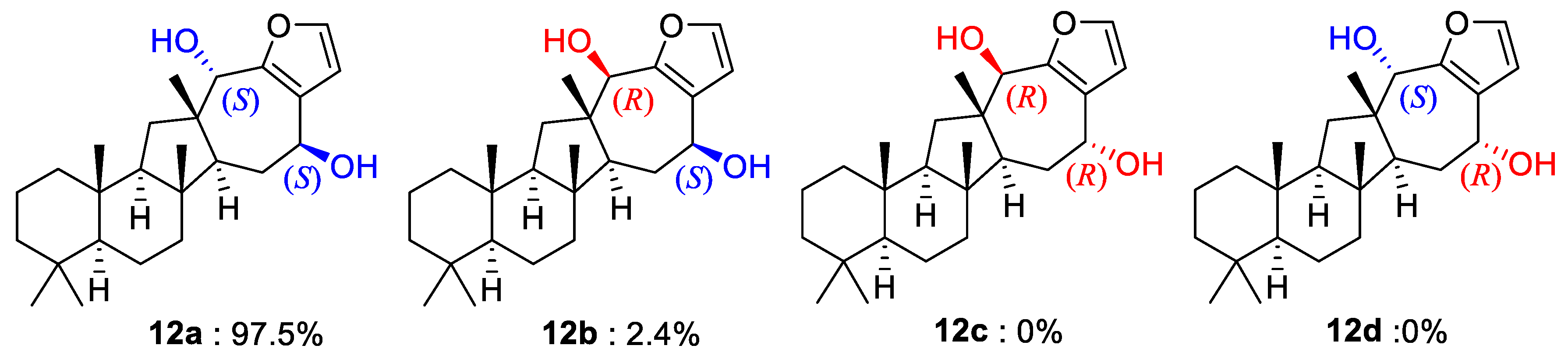
In addition to the ring junctures, compound
12 possessed two hydroxy-bearing stereogenic centers at C-15 and C-18. The NOESY data showed several diagnostic 1,3-diaxial cross peaks between the bridgehead methyl protons and spatially adjacent protons, assigning all
trans ring junctures (
Figure 2). The orientations of the hydroxy groups were assigned as 15β and 18α by a number of NOESY cross peaks for protons on ring D and in adjacent positions (H-13/H-15, H-14α (δ
H 1.90)/H-15, H-14β (δ
H 1.70)/H
3-21 and 23, and H-18/H
3-23). Therefore, the 15
S and 18
S configurations were assigned. The NOESY-based configurations were further supported by the DP4 calculations in which these configurations (
12a) matched the NMR data better than did any other configurational combination (
12b–
12d) (
Figure 5 and
Supporting Information Figure S77). Thus, compound
12, designated sarmahyrtisol B, was elucidated as a new sesterterpene.
The salmahyrtisane skeleton of
12 is rare, and to date, only three compounds with this skeleton have been reported: salmahyrtisol A from the Red Sea sponge
H. erecta (=
H. erectus) [
25], similan A from a Thai specimen of
H. gumininae [
26], and hippospongide A from a Taiwanese
Hippospongia sp [
27]. The structures of these compounds were closely related to each other by the possession of oxygenated functionalities at C-15 and C-18 on an identical furan-bearing carbon framework. Despite the weak or no cytotoxicity of these compounds, the unusual carbon framework and co-isolation with scalaranes and other sesterterpenes makes the biosynthesis of sarmahyrtisanes of great interest. Accordingly, salmahyrsitol A and hippospongide A were recently prepared by biomimetic synthesis [
28]. Notably,
12 has been proposed as a biosynthetic intermediate but was isolated for the first time as a natural product in this work. Both our finding of a sarmahyrtisane from the
Hyrtios sponge and its co-isolation with scalaranes coincides well with previous works [
25,
26,
27].
Several new compounds, such as
3/
4,
5/
6, and
10/
11, were isolated as configurational pairs at their methoxy-bearing centers. In addition,
10 and
11 showed typical oxidized furans for ring E. These findings suggest that these compounds are abiotic products derived from natural precursors, the results of solvolysis and Michael additions, either during prolonged storage or the isolation process [
29]. Even considering this, however, the quantities, diversity and unusual structural features of these new compounds demonstrate both the immense structural variation in sponge sesterterpenes and the chemical prosperity of
Hyrtios sponges.
In addition to
1-
12, the extract of
H. erectus contained several known scalaranes (
Supporting Information Figure S78). Based upon the results of 1-D and 2-D NMR and mass spectroscopy, these were identified as heteronemin (
13, known from various sponges) [
30,
31,
32], a glycine-bearing derivative (
14, known from a
Hyatella sp.) [
14], sesterstatins 5 and 6 (
15 and
16, known from
H. erecta (=
H. erectus)) [
33,
34], 16-hydroxyscalarolide (
17, known from
H. erectus) [
35], 12-
O-deacetyl-12-
epi-scalarin (
18, known from a
Spongia sp. [
36] and
H. erectus [
37]), and hyrtiosin A and 16-
O-deacetyl-16-
epi-scalarolbutenolide (
19 and
20, known from
H. erecta) [
38,
39]. The spectroscopic data of these compounds were in good accordance with those in the literature. Compound
13 was the major constituent, far exceeding all the known and new compounds isolated in this work.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}