Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study

 , ,
, ,  ,
,  ,
,  and
and 
Abstract
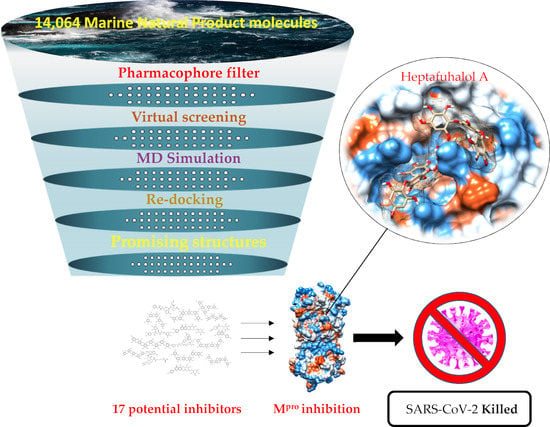
1. Introduction
2. Results and Discussion
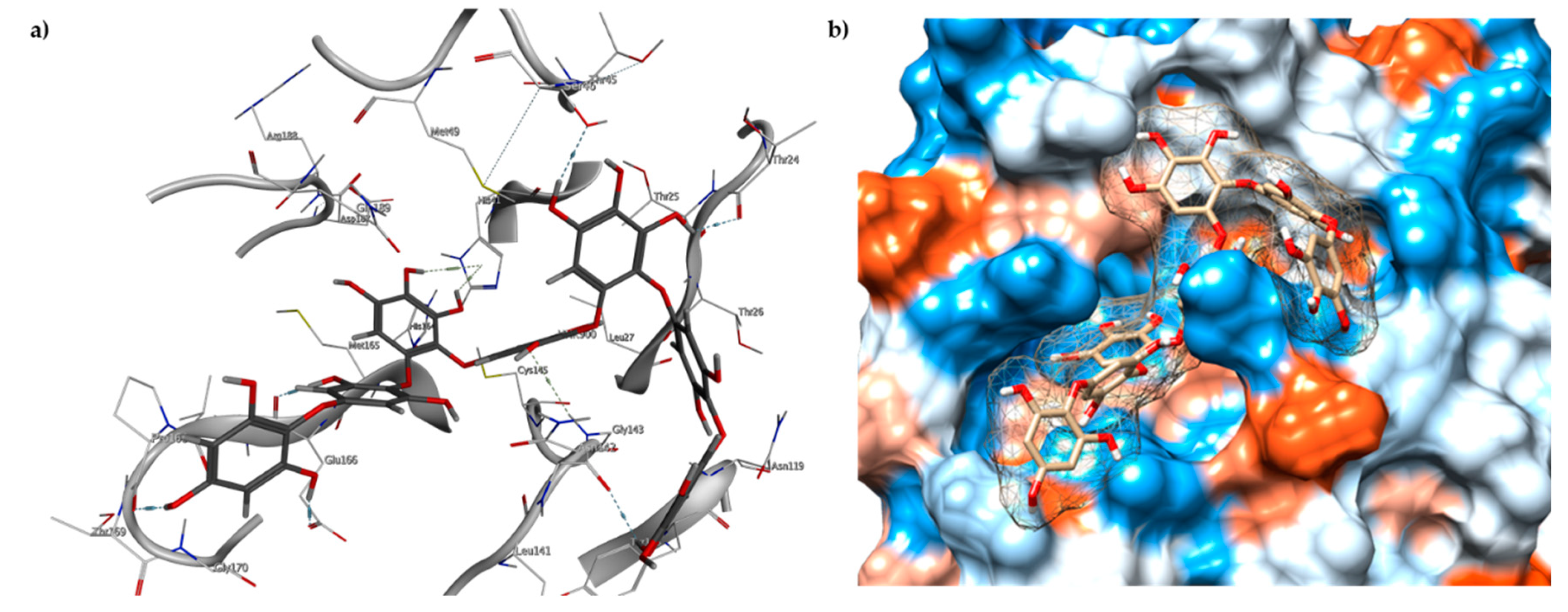
2.1. Catalytic Site of the SARS-CoV-2 Mpro
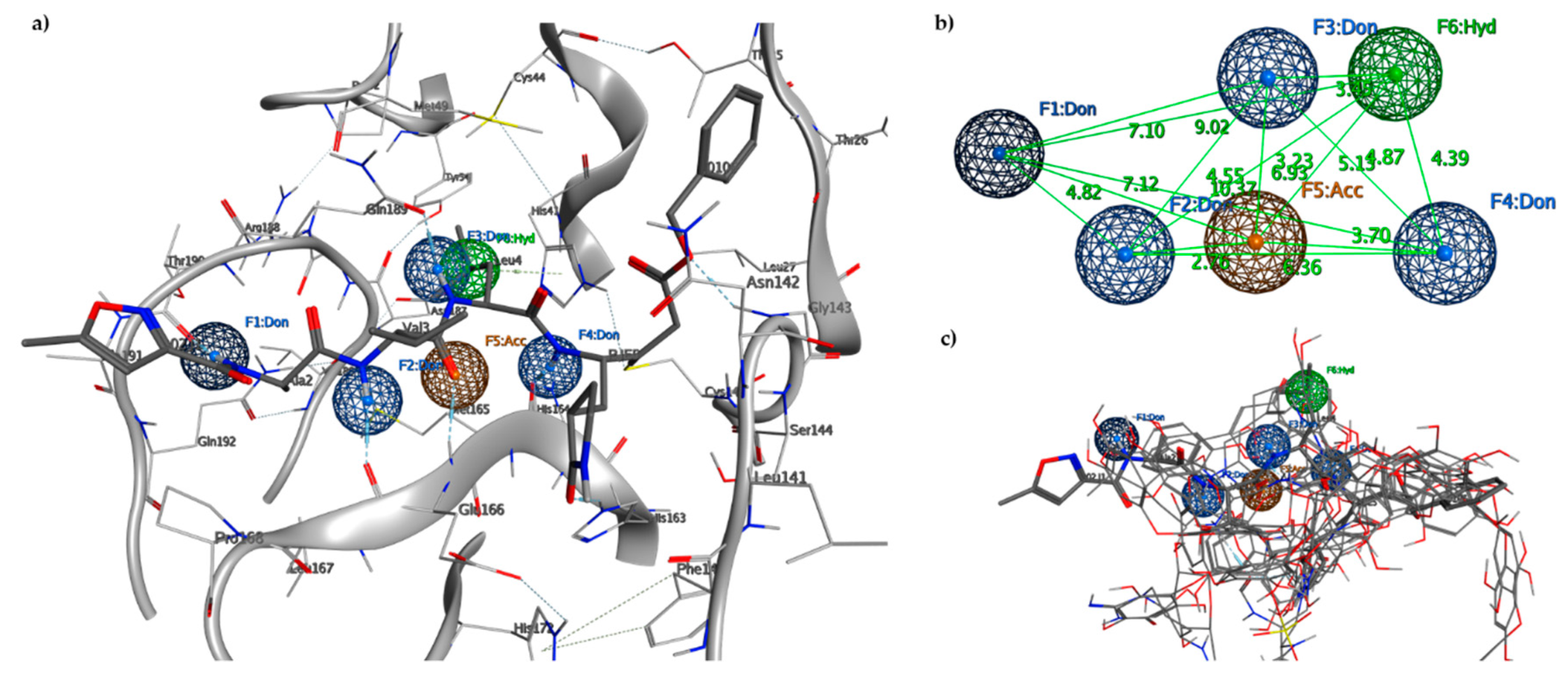
2.2. Pharmacophore Model
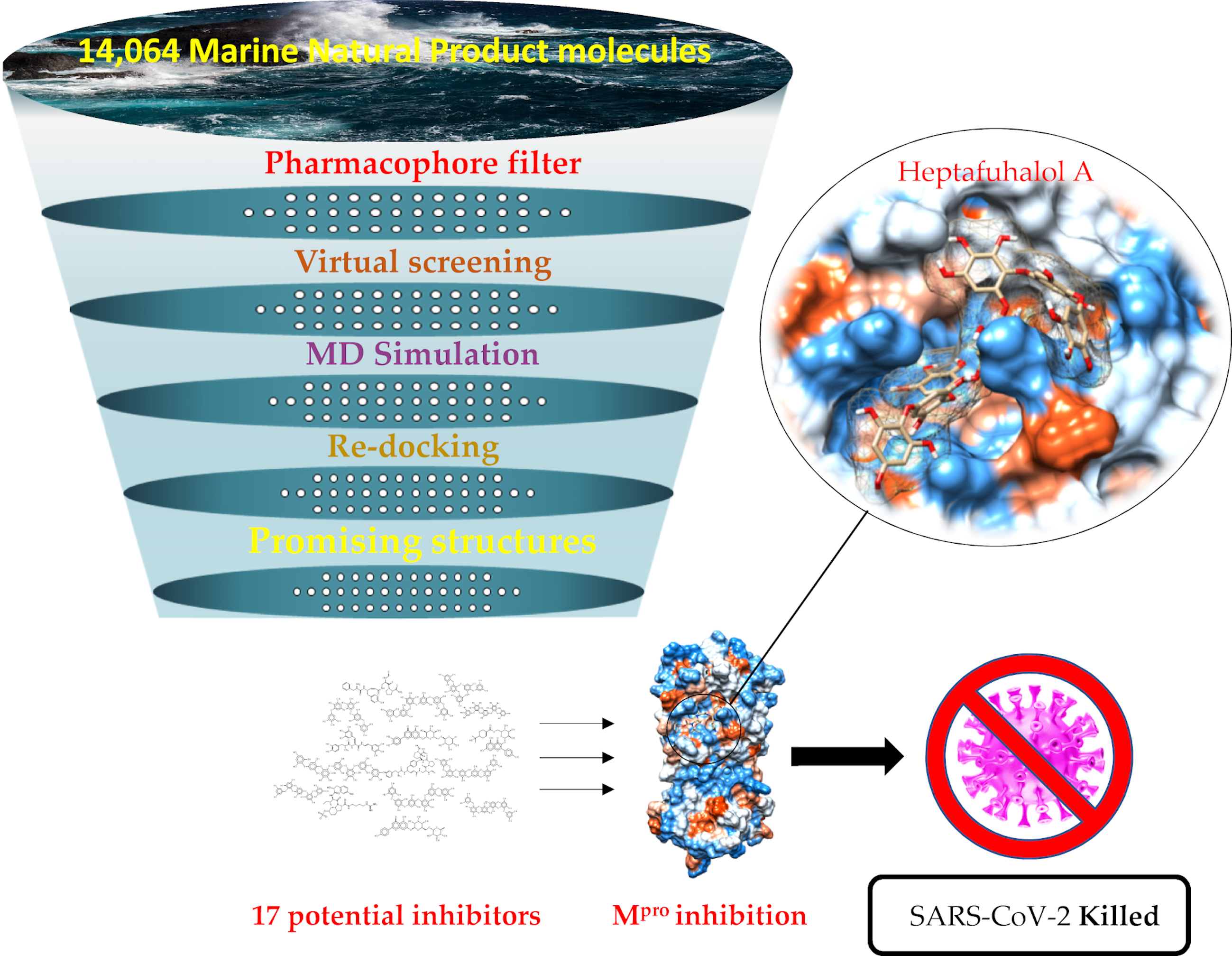
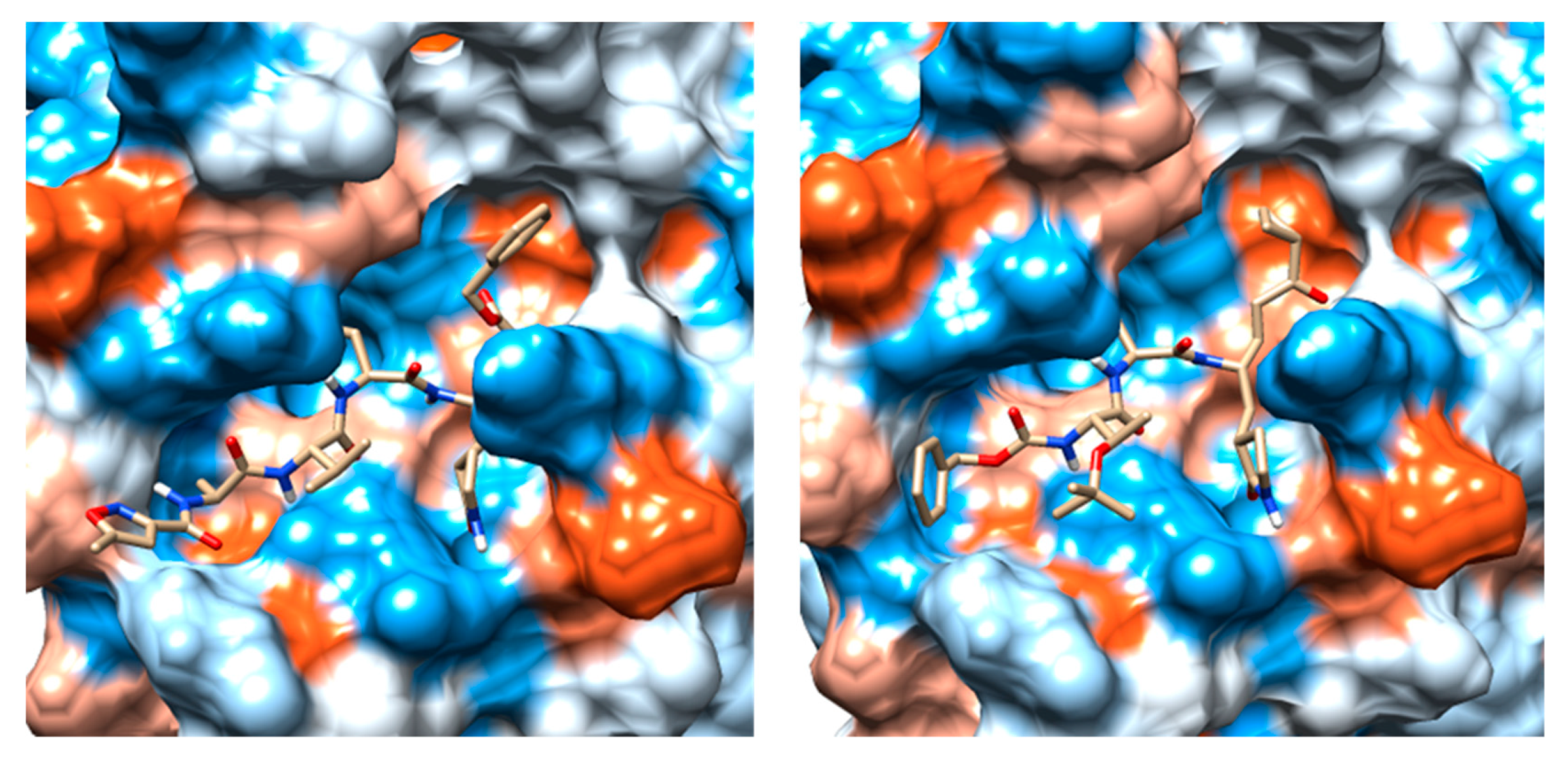
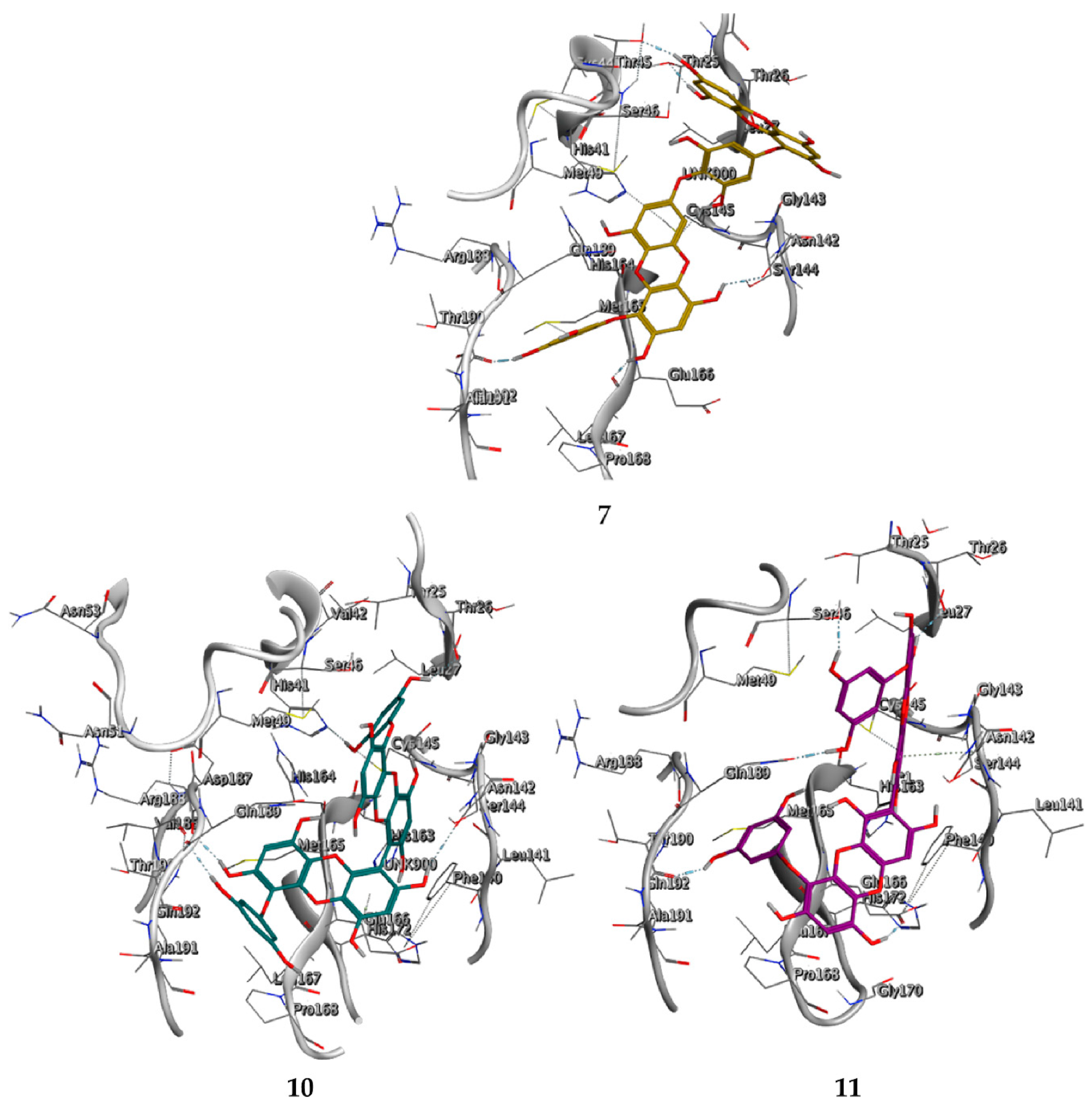
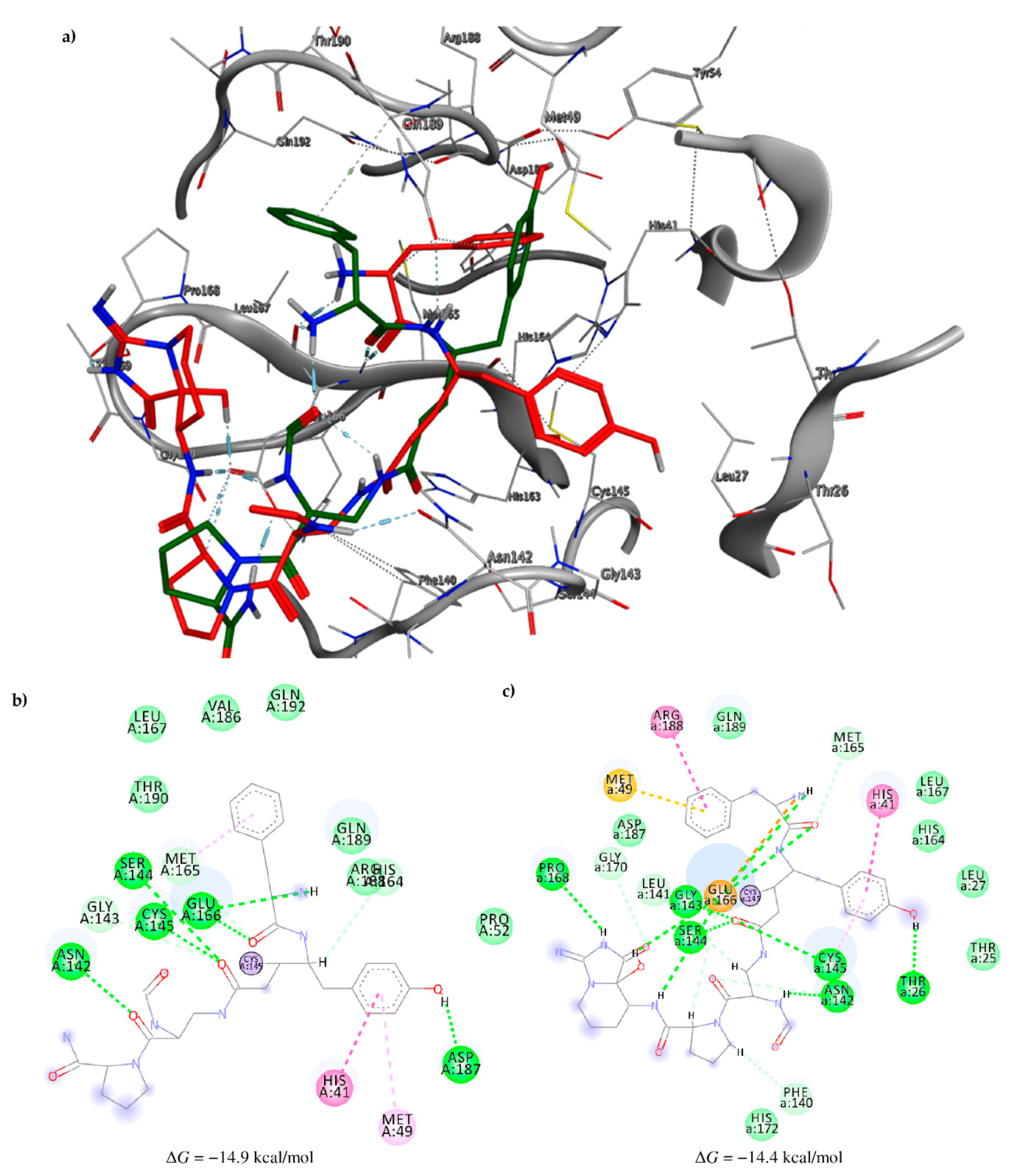
2.3. Molecular Docking and MD Simulation
3. Materials and Methods
3.1. Dataset of Compounds
3.2. Pharmacophore-based Virtual Screening and Database Preparation
3.3. Structures Preparation and Minimization
3.4. Molecular Docking
3.5. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Hui, D.S.; Azhar, E.I.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; McHugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health - The latest 2019 novel coronavirus outbreak in Wuhan, China. Int J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.Y. Coronaviruses - drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef]
- Song, Z.; Xu, Y.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Munster, V.J.; Koopmans, M.; van Doremalen, N.; van Riel, D.; de Wit, E. A Novel Coronavirus Emerging in China - Key Questions for Impact Assessment. N. Engl. J. Med. 2020, 382, 692–694. [Google Scholar] [CrossRef] [PubMed]
- Morse, J.S.; Lalonde, T.; Xu, S.; Liu, W.R. Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-nCoV. Chembiochemistry 2020, 21, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Peng, C.; Shi, Y.; Zhu, Z.; Mu, K.; Wang, X.; Zhu, W. Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bacha, U.; Barrila, J.; Velazquez-Campoy, A.; Leavitt, S.A.; Freire, E. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry-Us 2004, 43, 4906–4912. [Google Scholar] [CrossRef]
- Bzówka, M.; Mitusińska, K.; Raczyńska, A.; Samol, A.; Tuszyński, J.; Góra, A. Molecular Dynamics Simulations Indicate the COVID-19 Mpro Is Not a Viable Target for Small-Molecule Inhibitors Design. bioRxiv 2020. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CL(pro)) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.; Rodriguez, A.D.; Berlinck, R.G.; Fusetani, N. Marine pharmacology in 2007–8: Marine compounds with antibacterial, anticoagulant, antifungal, anti-inflammatory, antimalarial, antiprotozoal, antituberculosis, and antiviral activities; affecting the immune and nervous system, and other miscellaneous mechanisms of action. Comp. Biochem. Physiol. C. Toxicol. Pharmacol. 2011, 153, 191–222. [Google Scholar] [PubMed]
- Sunseri, J.; Koes, D.R. Pharmit: Interactive exploration of chemical space. Nucleic. Acids Res. 2016, 44, W442–W448. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Glombitza, K.W.; Keusgen, M. Fuhalols and Deshydroxyfuhalols from the Brown Alga Sargassum-Spinuligerum. Phytochemistry 1995, 38, 987–995. [Google Scholar] [CrossRef]
- Liu, L.; Heinrich, M.; Myers, S.; Dworjanyn, S.A. Towards a better understanding of medicinal uses of the brown seaweed Sargassum in Traditional Chinese Medicine: a phytochemical and pharmacological review. J. Ethnopharmacol. 2012, 142, 591–619. [Google Scholar] [CrossRef]
- Li, Y.; Fu, X.; Duan, D.; Liu, X.; Xu, J.; Gao, X. Extraction and Identification of Phlorotannins from the Brown Alga, Sargassum fusiforme (Harvey) Setchell. Mar. Drugs 2017, 15, 49. [Google Scholar] [CrossRef] [PubMed]
- Artan, M.; Li, Y.; Karadeniz, F.; Lee, S.H.; Kim, M.M.; Kim, S.K. Anti-HIV-1 activity of phloroglucinol derivative, 6,6’-bieckol, from Ecklonia cava. Bioorg. Med. Chem. 2008, 16, 7921–7926. [Google Scholar] [CrossRef]
- Kong, C.S.; Kim, J.A.; Yoon, N.Y.; Kim, S.K. Induction of apoptosis by phloroglucinol derivative from Ecklonia Cava in MCF-7 human breast cancer cells. Food Chem. Toxicol. 2009, 47, 1653–1658. [Google Scholar] [CrossRef]
- Zhang, R.; Kang, K.A.; Piao, M.J.; Ko, D.O.; Wang, Z.H.; Lee, I.K.; Kim, B.J.; Jeong, I.Y.; Shin, T.; Park, J.W.; et al. Eckol protects V79–4 lung fibroblast cells against gamma-ray radiation-induced apoptosis via the scavenging of reactive oxygen species and inhibiting of the c-Jun NH(2)-terminal kinase pathway. Eur. J. Pharmacol. 2008, 591, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.J.; Ko, S.C.; Cha, S.H.; Kang, D.H.; Park, H.S.; Choi, Y.U.; Kim, D.; Jung, W.K.; Jeon, Y.J. Effect of phlorotannins isolated from Ecklonia cava on melanogenesis and their protective effect against photo-oxidative stress induced by UV-B radiation. Toxicol. In Vitro 2009, 23, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Yoon, N.Y.; Eom, T.K.; Kim, M.M.; Kim, S.K. Inhibitory effect of phlorotannins isolated from Ecklonia cava on mushroom tyrosinase activity and melanin formation in mouse B16F10 melanoma cells. J. Agric. Food Chem. 2009, 57, 4124–4129. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lee, S.H.; Le, Q.T.; Kim, M.M.; Kim, S.K. Anti-allergic effects of phlorotannins on histamine release via binding inhibition between IgE and Fc epsilonRI. J. Agric. Food Chem 2008, 56, 12073–12080. [Google Scholar] [CrossRef] [PubMed]
- Myung, C.S.; Shin, H.C.; Bao, H.Y.; Yeo, S.J.; Lee, B.H.; Kang, J.S. Improvement of memory by dieckol and phlorofucofuroeckol in ethanol-treated mice: possible involvement of the inhibition of acetylcholinesterase. Arch. Pharm. Res. 2005, 28, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, J.H.; Kwon, J.M.; Kwon, H.J.; Jeong, H.J.; Kim, Y.M.; Kim, D.; Lee, W.S.; Ryu, Y.B. Dieckol, a SARS-CoV 3CL(pro) inhibitor, isolated from the edible brown algae Ecklonia cava. Bioorg. Med. Chem. 2013, 21, 3730–3737. [Google Scholar] [CrossRef]
- Nakao, Y.; Masuda, A.; Matsunaga, S.; Fusetani, N. Pseudotheonamides, serine protease inhibitors from the marine sponge Theonella swinhoei. J. Am. Chem. Soc. 1999, 121, 2425–2431. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Xi, K.; Ratia, K.; Santarsiero, B.D.; Fu, W.; Harcourt, B.H.; Rota, P.A.; Baker, S.C.; Johnson, M.E.; Mesecar, A.D. Design and synthesis of peptidomimetic severe acute respiratory syndrome chymotrypsin-like protease inhibitors. J. Med. Chem. 2005, 48, 6767–6771. [Google Scholar] [CrossRef]
- Shie, J.J.; Fang, J.M.; Kuo, T.H.; Kuo, C.J.; Liang, P.H.; Huang, H.J.; Wu, Y.T.; Jan, J.T.; Cheng, Y.S.; Wong, C.H. Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic alpha,beta-unsaturated esters. Bioorg. Med. Chem. 2005, 13, 5240–5252. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Xi, K.; Grum-Tokars, V.; Xu, X.; Ratia, K.; Fu, W.; Houser, K.V.; Baker, S.C.; Johnson, M.E.; Mesecar, A.D. Structure-based design, synthesis, and biological evaluation of peptidomimetic SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 5876–5880. [Google Scholar] [CrossRef]
- Moharram, F.A.; El Dib, R.; Marzouk, M.S.; El-Shenawy, S.M.; Ibrahim, H.A. New Apigenin Glycoside, Polyphenolic Constituents, Anti-inflammatory and Hepatoprotective Activities of Gaillardia grandiflora and Gaillardia pulchella Aerial Parts. Pharmacogn. Mag. 2017, 13, S244–S249. [Google Scholar] [PubMed]
- Meng, S.; Zhu, Y.; Li, J.F.; Wang, X.; Liang, Z.; Li, S.Q.; Xu, X.; Chen, H.; Liu, B.; Zheng, X.Y.; et al. Apigenin inhibits renal cell carcinoma cell proliferation. Oncotarget 2017, 8, 19834–19842. [Google Scholar] [CrossRef] [PubMed]
- Samet, I.; Villareal, M.O.; Motojima, H.; Han, J.; Sayadi, S.; Isoda, H. Olive leaf components apigenin 7-glucoside and luteolin 7-glucoside direct human hematopoietic stem cell differentiation towards erythroid lineage. Differentiation 2015, 89, 146–155. [Google Scholar] [CrossRef]
- Grosser, J.W.; Gmitter, F.G., Jr.; Tusa, N.; Recupero, G.R.; Cucinotta, P. Further evidence of a cybridization requirement for plant regeneration from citrus leaf protoplasts following somatic fusion. Plant. Cell. Rep. 1996, 15, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Siracusa, L.; Ruberto, G.; Carrubba, A.; Lazzara, S.; Speciale, A.; Cimino, F.; Saija, A.; Cristani, M. Phytochemical profiles, phototoxic and antioxidant properties of eleven Hypericum species - A comparative study. Phytochemistry 2018, 152, 162–173. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Luo, C.; Liu, H.; Xu, W.; Chen, G.; Liew, O.W.; Zhu, W.; Puah, C.M.; Shen, X.; et al. Binding interaction of quercetin-3-beta-galactoside and its synthetic derivatives with SARS-CoV 3CL(pro): Structure-activity relationship studies reveal salient pharmacophore features. Bioorg. Med. Chem. 2006, 14, 8295–8306. [Google Scholar] [CrossRef]
- Barf, T.; Lehmann, F.; Hammer, K.; Haile, S.; Axen, E.; Medina, C.; Uppenberg, J.; Svensson, S.; Rondahl, L.; Lundback, T. N-Benzyl-indolo carboxylic acids: Design and synthesis of potent and selective adipocyte fatty-acid binding protein (A-FABP) inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 1745–1748. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 2004, 10, 155–164. [Google Scholar] [CrossRef]
- Alemán, C.; Luque, F.J.; Orozco, M. Suitability of the PM3-derived molecular electrostatic potentials. J. Comput. Chem. 1993, 14, 799–808. [Google Scholar] [CrossRef]
- Qiao, F.; Luo, L.; Peng, H.; Luo, S.; Huang, W.; Cui, J.; Li, X.; Kong, L.; Jiang, D.; Chitwood, D.J.; et al. Characterization of Three Novel Fatty Acid- and Retinoid-Binding Protein Genes (Ha-far-1, Ha-far-2 and Hf-far-1) from the Cereal Cyst Nematodes Heterodera avenae and H. filipjevi. PLoS ONE 2016, 11, e0160003. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. YASARA View - molecular graphics for all devices - from smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Floresta, G.; Amata, E.; Barbaraci, C.; Gentile, D.; Turnaturi, R.; Marrazzo, A.; Rescifina, A. A Structure- and Ligand-Based Virtual Screening of a Database of "Small" Marine Natural Products for the Identification of "Blue" Sigma-2 Receptor Ligands. Mar. Drugs 2018, 16, E384. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Dunbrack, R.L., Jr.; Hooft, R.W.; Krieger, B. Assignment of protonation states in proteins and ligands: combining pKa prediction with hydrogen bonding network optimization. Methods Mol. Biol 2012, 819, 405–421. [Google Scholar]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New Ways to Boost Molecular Dynamics Simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Galimberti, M.; Barbera, V.; Guerra, S.; Bernardi, A. Facile Functionalization of Sp(2) Carbon Allotropes with a Biobased Janus Molecule. Rubber Chem. Technol. 2017, 90, 285–307. [Google Scholar] [CrossRef]
- Di Lodovico, S.; Napoli, E.; Di Campli, E.; Di Fermo, P.; Gentile, D.; Ruberto, G.; Nostro, A.; Marini, E.; Cellini, L.; Di Giulio, M. Pistacia vera L. oleoresin and levofloxacin is a synergistic combination against resistant Helicobacter pylori strains. Sci. Rep. 2019, 9, 4646. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Gentile, D.; Perrini, G.; Patamia, V.; Rescifina, A. Computational Tools in the Discovery of FABP4 Ligands: A Statistical and Molecular Modeling Approach. Mar. Drugs 2019, 17, 624. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Amata, E.; Gentile, D.; Romeo, G.; Marrazzo, A.; Pittalà, V.; Salerno, L.; Rescifina, A. Fourfold Filtered Statistical/Computational Approach for the Identification of Imidazole Compounds as HO-1 Inhibitors from Natural Products. Mar. Drugs 2019, 17, 113. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound/Name | MNP ID | Structure | ΔGB Vina | ΔGB Autodock4 | Average ΔGB | ΔGB Re-docking Vina |
|---|---|---|---|---|---|---|
| 1 Heptafuhalol A | 76265-30-0 |  | −15.4 | −13.8 | −14.6 | −18.0 |
| 2 Phlorethopentafuhalol B | 138551-15-2 |  | −14.6 | −13.9 | −14.2 | −13.8 |
| 3 Pseudopentafuhalol C | 202211-82-3 |  | −14.5 | −13.8 | −14.2 | −15.8 |
| 4 Phlorethopentafuhalol A | 138529-06-3 |  | −14.0 | −14.3 | −14.1 | −12.3 |
| 5 Hydroxypentafuhalol A | 137809-92-8 |  | −14.6 | −11.7 | −13.1 | −13.7 |
| 6 Pentaphlorethol B |  | −13.9 | −12.2 | −13.1 | −13.9 | |
| 7 8,8’-Bieckol | 89445-12-5 |  | −13.7 | −12.1 | −12.9 | −13.5 |
| 8 Apigenin-7-O-neohesperidoside | 36790-49-5 |  | −12.4 | −12.3 | −12.4 | −12.8 |
| 9 Luteolin-7-rutinoside | 20633-84-5 |  | −12.1 | −12.3 | −12.2 | −12.2 |
| 10 6,6’-Bieckol | 88095-81-2 |  | −12.2 | −12.0 | −12.1 | −15.8 |
| 11 Dieckol | 88095-77-6 |  | −12.0 | −12.1 | −12.1 | −12.9 |
| 12 Pseudotheonamide D | 224577-36-0 |  | −12.2 | −10.9 | −11.6 | −12.2 (−14.9) a |
| 13 Aeruginosin 98B | 167228-01-5 |  | −12.1 | −10.4 | −11.3 | −12.5 |
| 14 Resinoside B | 144027-79-2 |  | −12.2 | −10.2 | −11.2 | −11.4 |
| 15 Pentaphlorethol A | 164176-23-2 |  | −12.8 | −9.4 | −11.1 | −14.5 |
| 16 Tunichrome An2 | 115982-31-5 |  | −11.5 | −10.5 | −11.0 | −13.5 |
| 17 Pseudotheonamide C | 224577-35-9 |  | −10.5 | −10.9 | −10.7 | −11.0 (−14.4) a |
| 18 N-[(5-methylisoxazol-3-yl)carbonyl]alanyl-l-valyl-N1-((1R,2Z)-4-(benzyloxy)-4-oxo-1-{[(3R)-2-oxopyrrolidin-3-yl]methyl}but-2-enyl)-l-leucinamide | PRD_002214 |  | −11.9 (−15.0) a | −11.0 (−15.3) a | −11.4 (−15.1) a | −14.5 (−15.1) a |
| 19 Lopinavir | 92727 |  | −10.3 | −10.3 | −10.3 | −12.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gentile, D.; Patamia, V.; Scala, A.; Sciortino, M.T.; Piperno, A.; Rescifina, A. Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study. Mar. Drugs 2020, 18, 225. https://doi.org/10.3390/md18040225
Gentile D, Patamia V, Scala A, Sciortino MT, Piperno A, Rescifina A. Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study. Marine Drugs. 2020; 18(4):225. https://doi.org/10.3390/md18040225
Chicago/Turabian StyleGentile, Davide, Vincenzo Patamia, Angela Scala, Maria Teresa Sciortino, Anna Piperno, and Antonio Rescifina. 2020. "Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study" Marine Drugs 18, no. 4: 225. https://doi.org/10.3390/md18040225
APA StyleGentile, D., Patamia, V., Scala, A., Sciortino, M. T., Piperno, A., & Rescifina, A. (2020). Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study. Marine Drugs, 18(4), 225. https://doi.org/10.3390/md18040225