Marine Toxins Targeting Kv1 Channels: Pharmacological Tools and Therapeutic Scaffolds

 ,
,  and
and
Abstract
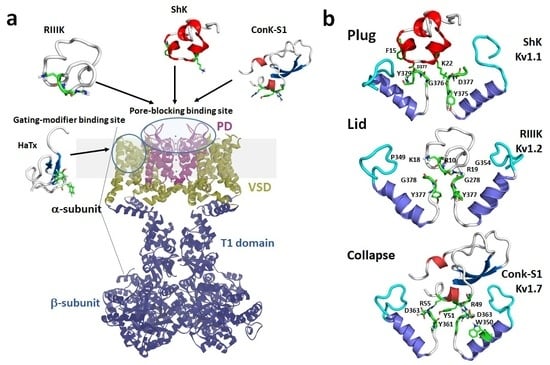
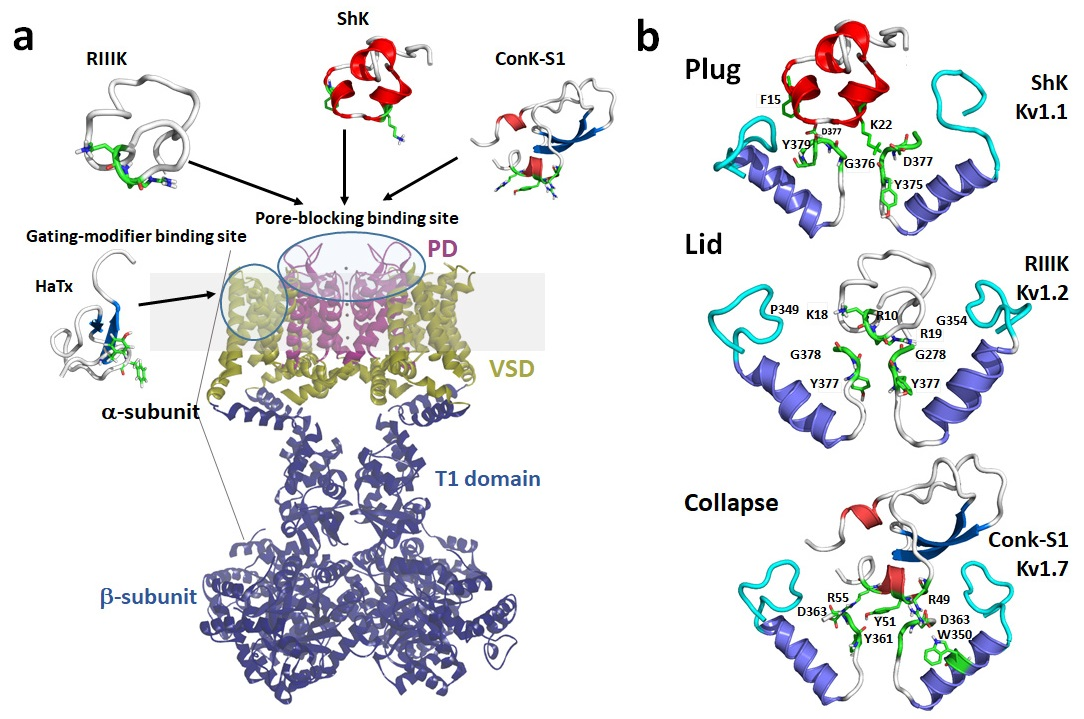
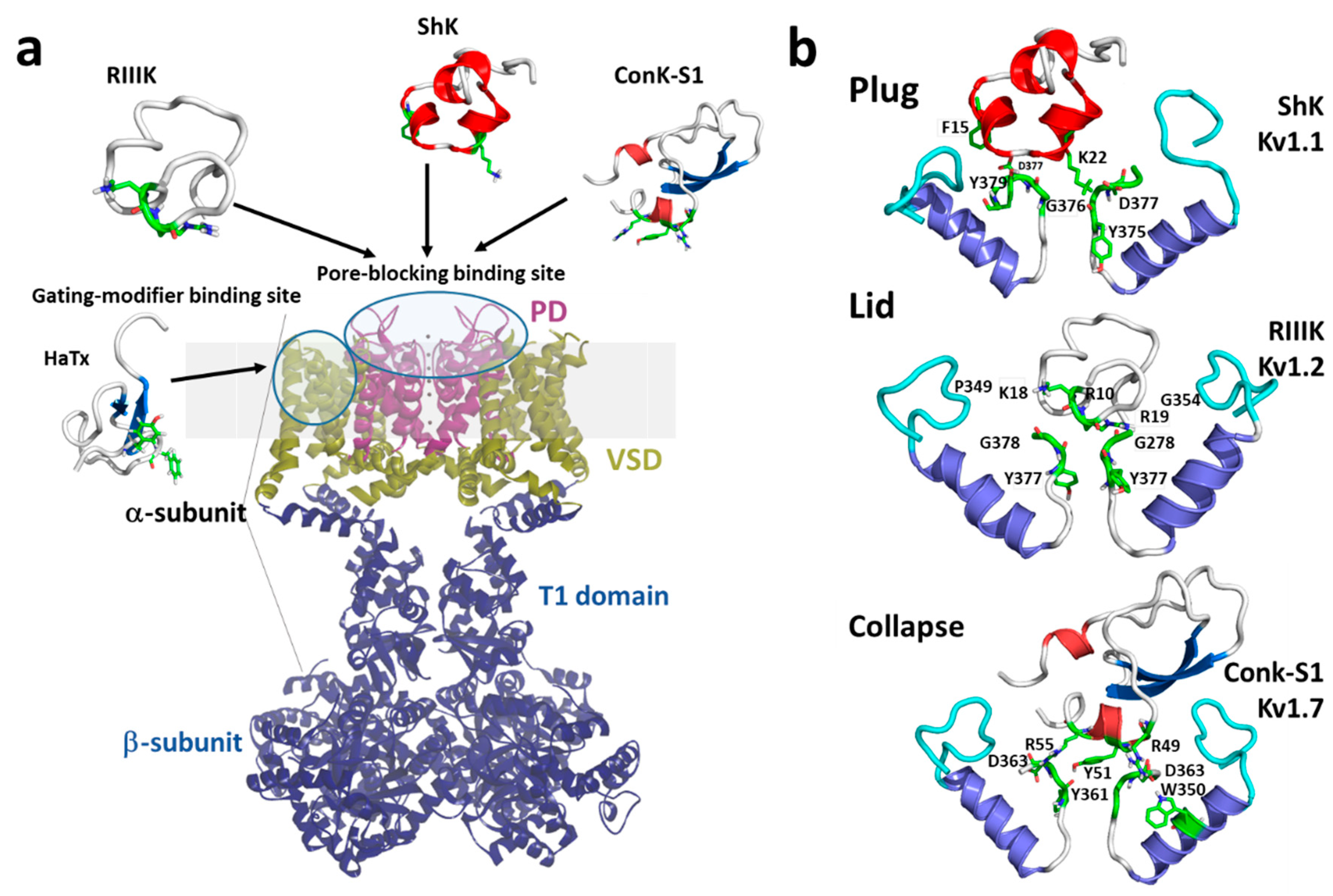
1. Introduction
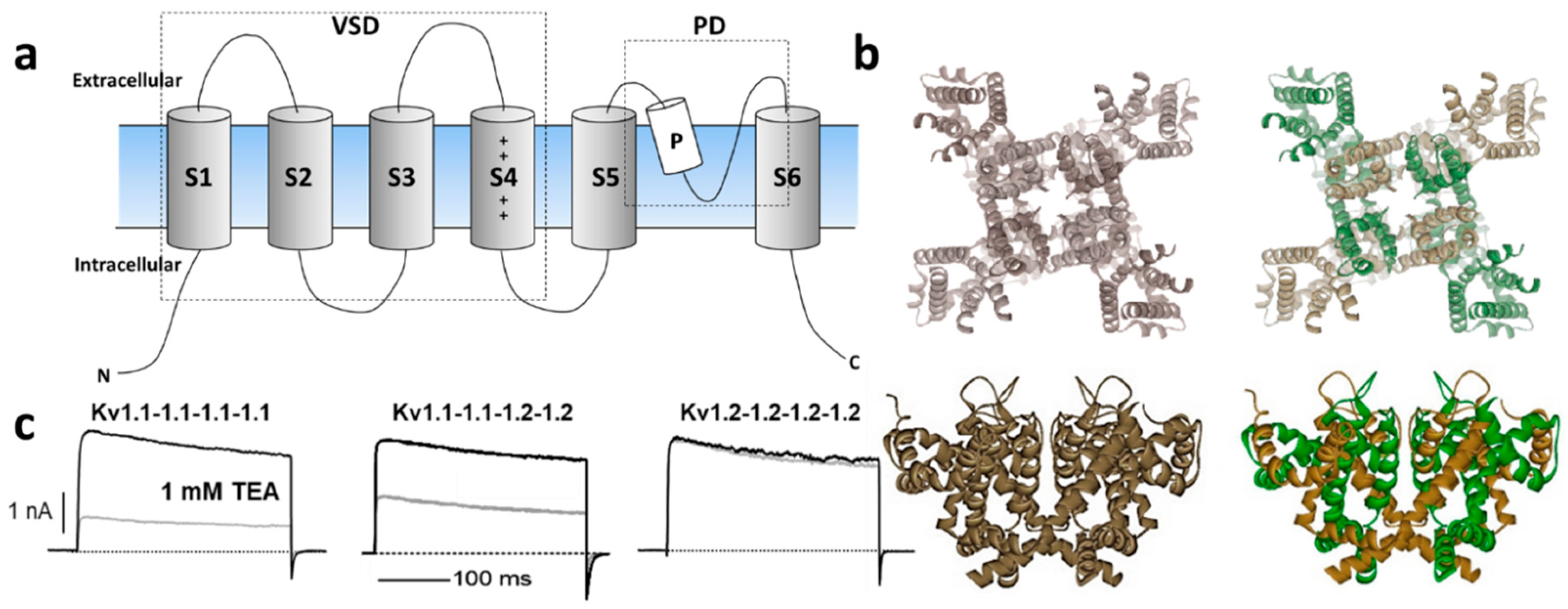
1.1. Kv1 Channels
1.2. Mechanisms of Kv Channel Inhibition by Marine Toxins

2. Molluscan Peptides that Inhibit Kv1 Channels
2.1. κM-RIIIJ

| Conopeptide | Source | Family | Target Channel(s) (IC50) | References |
|---|---|---|---|---|
| CPY-Pl1 | C. planorbis | CPY | Kv1.2 (2 μM); Kv1.6 (170 nM) | [84] |
| CPY-Fe1 | C. ferruginesus | CPY | Kv1.2 (30 μM); Kv1.6 (8.8 μM) | [84] |
| κM-RIIIJ | C. radiatus | M | hKv1.2 (33 nM) | [80] |
| κM-RIIIK | C. radiatus | M | hKv1.2 (300 nM) rKv1.2 (335 nM) | [79] |
| Pl14a (κJ-PlXIVA) | C. planorbis | J | hKv1.6 (1.6 μM) | [76] |
| κ-ViTx | C. vigro | I2 | rKv1.1 (1.6 μM) rKv1.3 | [85] |
| Conkunitzin-S1 | C. Striatus | Conkunitzins | Kv1.7 (< nM) | [12] |
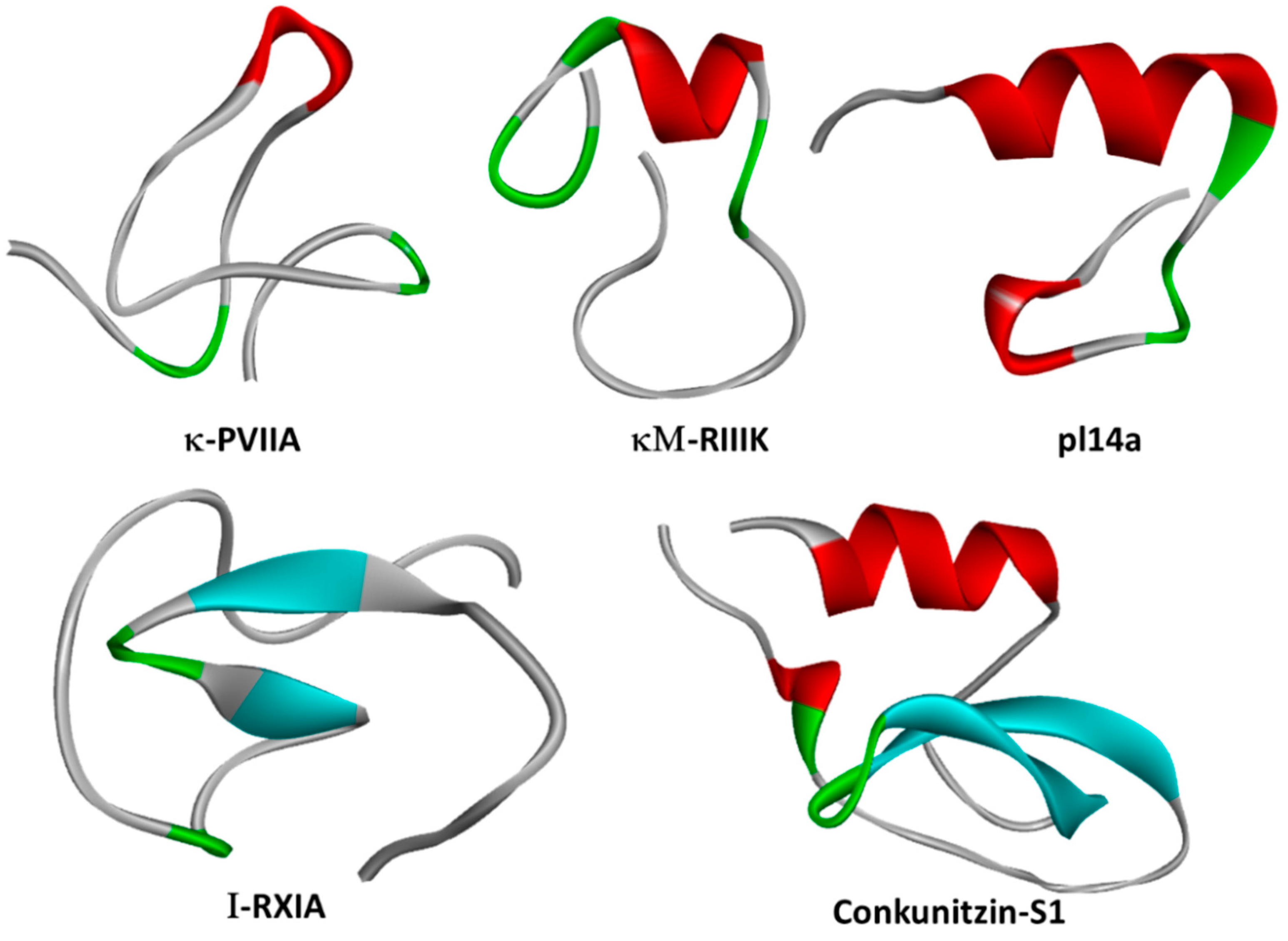
2.2. Conk-S1
2.3. κ-PVIIA
2.4. κ-ViTx
2.5. SrXIa
2.6. Promiscuous Conotoxins Interacting with Kv1 Channels
2.6.1. pl14a
2.6.2. Tyrosine-Rich Conopeptides CPY-Pl1 and CPY-Fe1
2.6.3. µ-PIIIA
2.6.4. κP-Crassipeptides
3. Cnidarian Peptides that Inhibit Kv1 Channels
| Toxin | Source | Inhibited Kv1 Channels | References |
|---|---|---|---|
| Type 1 | |||
| ShK | Stichodactyla helianthus | Kv1.1, Kv1.3, Kv1.4, 1.6 | [106,107] |
| AeK | Actinia equina | 125I α-DTX binding to synaptosomal membranes (IC50 22 nM) | [108] |
| AETX K | Anemonia erythraea | 125I α-dendrotoxinDTX binding to synaptosomal membranes (IC50 91 nM) | [109] |
| AsKS | Anemonia sulcata | Kv1.2 | [110,111] |
| BcsTX1/2 | Bunodosoma caissarum | BcsTx1 Kv1.2, Kv1.6 BcsTx2 Kv1.1, Kv1.2, Kv 1.3, Kv1.6, Shaker IR with nM IC50 | |
| BgK | Bunodosoma granulifera | Kv1.1, Kv1.2, Kv1.3, Kv1.6 | [112,113] |
| HmK | Heteractis (Radianthus) magnifica | Kv1.2, Kv1.3 | [114,115] |
| Type 2 | |||
| AsKC1 | Anemonia sulcata | Kv1.2 | [111] |
| AsKC2 | Anemonia sulcata | Kv1.2 | [116] |
| AsKC3 | Anemonia sulcata | Kv1.2 | [116] |
| APEXTx1 | Anthopleura elegantissima | Kv1.1 | |
| SHTXIII | Stichodactyla haddoni | 125I α-DTXdendrotoxin binding to synaptosomal membranes (IC50 270 nM) | [117] |
| Type 3 | |||
| BDS-I | Anemonia sulcata | Kv1.1–5 < 20% inhibition at 10 µM | [116] |
| APETx1/2/4 | Anthopleura elegantissima | Kv1.1-6 < 30% inhibition at 100 nM | |
| PhcrTx2 | Phymanthus crucifer | Slight inhibition on DRG Kv currents at µM concentrations | [118,119] |
| Type 4 | |||
| SHTX I/II | Stichodactyla haddoni | None | |
| Type 5 | |||
| BcsTx3 | Bunodosoma caissarum | Kv1.1, Kv1.2, Kv 1.3, Kv1.6, Shaker IR | [110] |
| PhcrTx1 | Phymanthus crucifer | Slight inhibition on DRG Kv currents at µM concentrations | [120] |
| Type 6 | |||
| AbeTx1 | Actinia bermudensis | Kv1.1, Kv1.2, Kv1.6, Shaker IR | [121] |
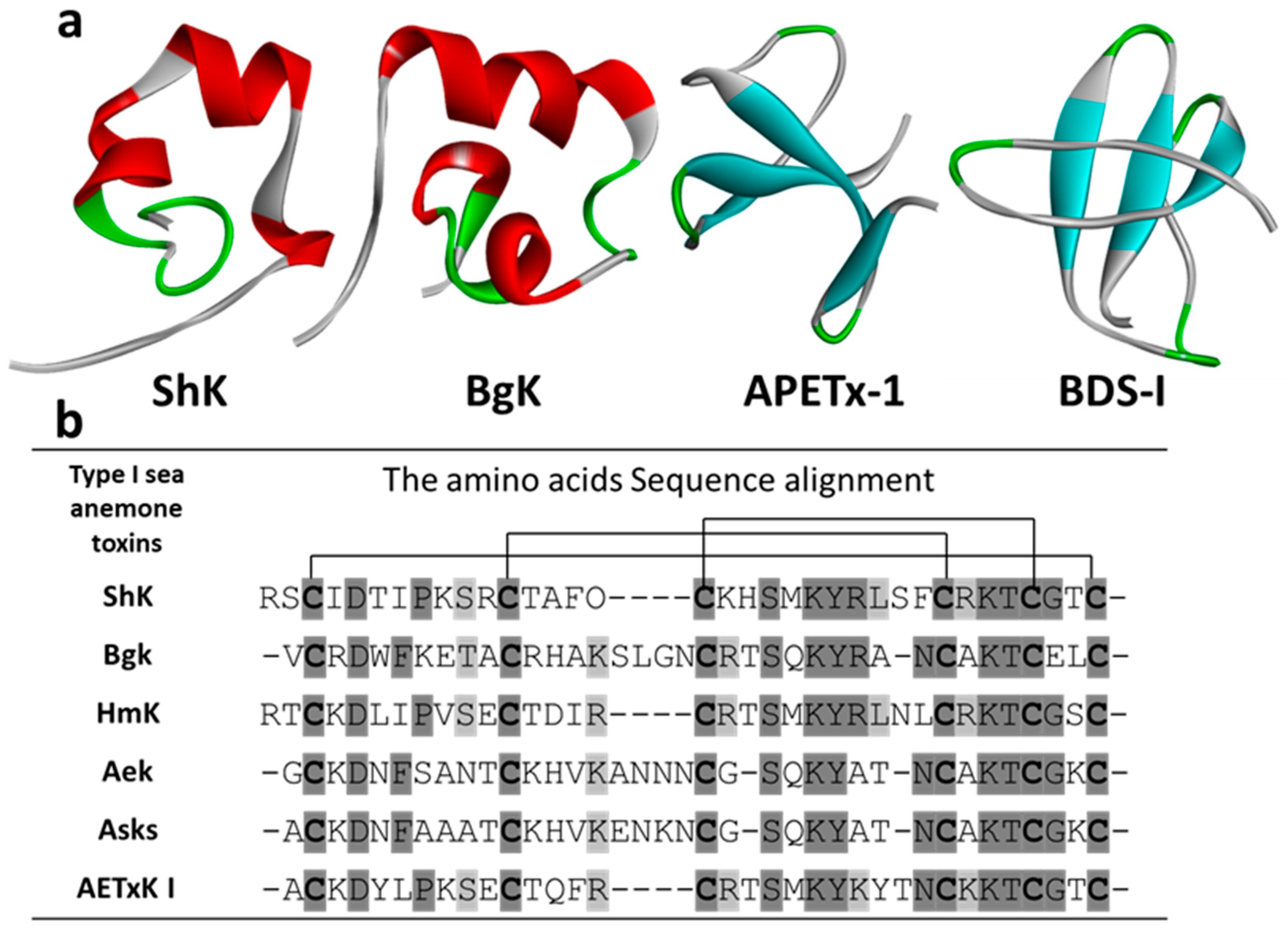
3.1. Kv Type 1 Anemone Toxins
3.1.1. ShK
3.1.2. BgK
3.1.3. BcsTx1/2
3.1.4. Other Kv Type 1 Toxins
3.2. Kv Type 2 Anemone Toxins
3.3. Kv Type 3 Anemone Toxins
3.4. Kv Type 4 Anemone Toxins
3.5. Kv Type 5 Anemone Toxins
3.6. Kv Type 6 Anemone Toxins
4. Non-Peptidyl Kv1 Channel Inhibitors
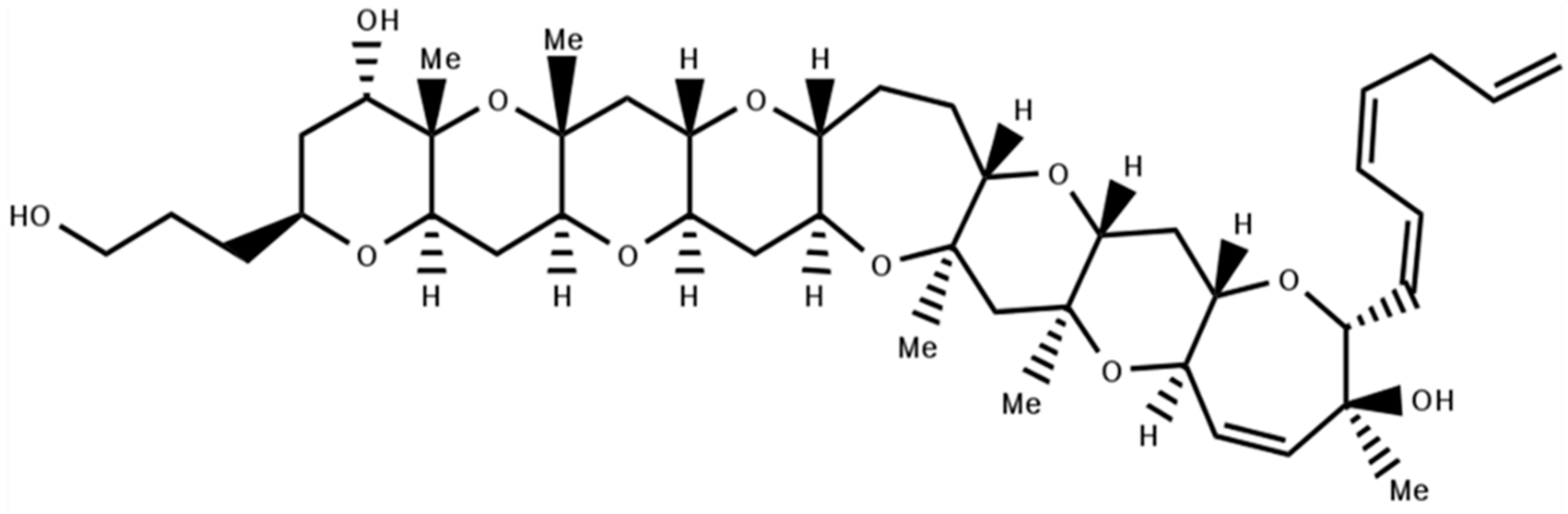
4.1. Gambierol
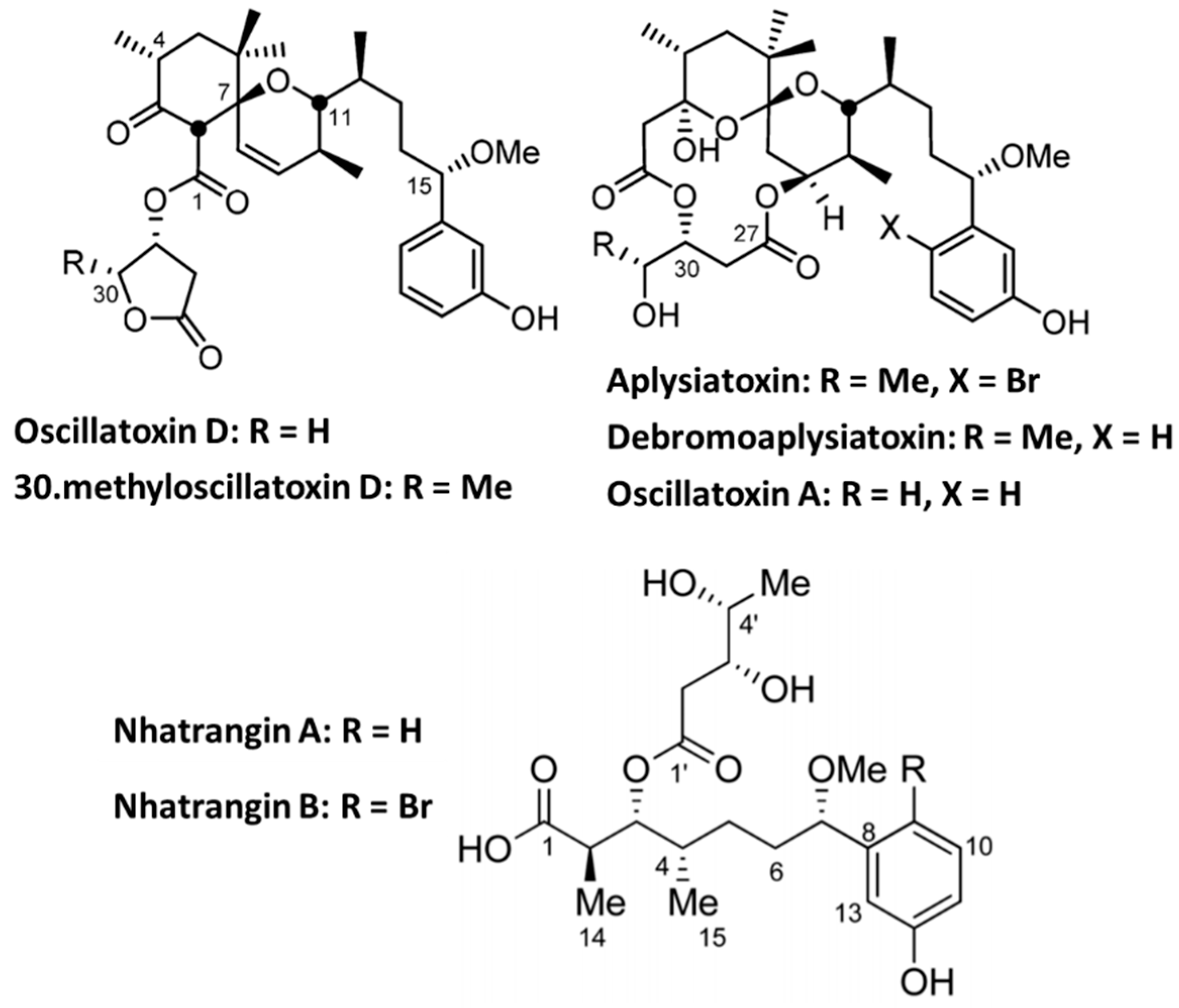
4.2. Aplysiatoxin Derivatives
5. Kv1-Active Toxins in Research and Drug Discovery
6. Challenges and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hille, B. Ion channels of excitable membranes, 3rd ed.; Sunderland, Mass. Sinauer: New York, NY, USA, 2001. [Google Scholar]
- Gutman, G.A.; Chandy, K.G.; Grissmer, S.; Lazdunski, M.; McKinnon, D.; Pardo, L.A.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.C.; Stuhmer, W.; et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 2005, 57, 473–508. [Google Scholar] [CrossRef] [PubMed]
- Rudy, B. Diversity and ubiquity of K channels. Neuroscience 1988, 25, 729–749. [Google Scholar] [CrossRef]
- Vacher, H.; Mohapatra, D.P.; Trimmer, J.S. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol. Rev. 2008, 88, 1407–1447. [Google Scholar] [CrossRef] [PubMed]
- Barry, D.M.; Trimmer, J.S.; Merlie, J.P.; Nerbonne, J.M. Differential expression of voltage-gated K+ channel subunits in adult rat heart. Relation to functional K+ channels? Circ. Res. 1995, 77, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Kalman, K.; Nguyen, A.; Tseng-Crank, J.; Dukes, I.D.; Chandy, G.; Hustad, C.M.; Copeland, N.G.; Jenkins, N.A.; Mohrenweiser, H.; Brandriff, B.; et al. Genomic organization, chromosomal localization, tissue distribution, and biophysical characterization of a novel mammalian Shaker-related voltage-gated potassium channel, Kv1.7. J. Biol. Chem. 1998, 273, 5851–5857. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, H.; Liman, E.R.; Hess, P.; Koren, G. Pretranslational mechanisms determine the type of potassium channels expressed in the rat skeletal and cardiac muscles. J. Biol. Chem. 1991, 266, 13324–13328. [Google Scholar]
- Bose, T.; Cieslar-Pobuda, A.; Wiechec, E. Role of ion channels in regulating Ca2+ homeostasis during the interplay between immune and cancer cells. Cell Death Dis. 2015, 6, e1648. [Google Scholar] [CrossRef]
- Dubois, J.M.; Rouzaire-Dubois, B. The influence of cell volume changes on tumour cell proliferation. Eur. Biophys. J. 2004, 33, 227–232. [Google Scholar] [CrossRef]
- Rouzaire-Dubois, B.; Dubois, J.M. A quantitative analysis of the role of K+ channels in mitogenesis of neuroblastoma cells. Cell. Signal. 1991, 3, 333–339. [Google Scholar] [CrossRef]
- Koo, G.C.; Blake, J.T.; Talento, A.; Nguyen, M.; Lin, S.; Sirotina, A.; Shah, K.; Mulvany, K.; Hora, D., Jr.; Cunningham, P.; et al. Blockade of the voltage-gated potassium channel Kv1.3 inhibits immune responses in vivo. J. Immunol. 1997, 158, 5120–5128. [Google Scholar]
- Finol-Urdaneta, R.K.; Remedi, M.S.; Raasch, W.; Becker, S.; Clark, R.B.; Struver, N.; Pavlov, E.; Nichols, C.G.; French, R.J.; Terlau, H. Block of Kv1.7 potassium currents increases glucose-stimulated insulin secretion. EMBO Mol. Med. 2012, 4, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005, 309, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Kurata, H.T.; Fedida, D. A structural interpretation of voltage-gated potassium channel inactivation. Prog. Biophys. Mol. Biol. 2006, 92, 185–208. [Google Scholar] [CrossRef] [PubMed]
- Bahring, R.; Covarrubias, M. Mechanisms of closed-state inactivation in voltage-gated ion channels. J. Physiol. 2011, 589 (Pt 3), 461–479. [Google Scholar] [CrossRef]
- Aldrich, R.W. Fifty years of inactivation. Nature 2001, 411, 643–644. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Sukomon, N.; Flood, E.; Allen, T.W.; Nimigean, C.M. Ball-and-chain inactivation in a calcium-gated potassium channel. Nature 2020. [Google Scholar]
- Pau, V.; Zhou, Y.; Ramu, Y.; Xu, Y.; Lu, Z. Crystal structure of an inactivated mutant mammalian voltage-gated K+ channel. Nat. Struct. Mol. Biol. 2017, 24, 857–865. [Google Scholar] [CrossRef]
- Hoshi, T.; Armstrong, C.M. C-type inactivation of voltage-gated K+ channels: Pore constriction or dilation? J. Gen. Physiol. 2013, 141, 151–160. [Google Scholar] [CrossRef]
- Valiyaveetil, F.I. A glimpse into the C-type-inactivated state for a Potassium Channel. Nat. Struct. Mol. Biol. 2017, 24, 787–788. [Google Scholar] [CrossRef]
- Sahoo, N.; Hoshi, T.; Heinemann, S.H. Oxidative modulation of voltage-gated potassium channels. Antioxid. Redox Signal. 2014, 21, 933–952. [Google Scholar] [CrossRef]
- Finol-Urdaneta, R.K.; Struver, N.; Terlau, H. Molecular and Functional Differences between Heart mKv1.7 Channel Isoforms. J. Gen. Physiol. 2006, 128, 133–145. [Google Scholar] [CrossRef]
- Al-Sabi, A.; Kaza, S.K.; Dolly, J.O.; Wang, J. Pharmacological characteristics of Kv1.1- and Kv1.2-containing channels are influenced by the stoichiometry and positioning of their alpha subunits. Biochem. J. 2013, 454, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, M.P.; Hurst, R.S.; Yakel, J.; Varnum, M.D.; Adelman, J.P.; North, R.A. Multiple subunits of a voltage-dependent potassium channel contribute to the binding site for tetraethylammonium. Neuron 1992, 8, 493–497. [Google Scholar] [CrossRef]
- Wulff, H.; Castle, N.A.; Pardo, L.A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 2009, 8, 982–1001. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.H.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; et al. The Concise Guide To Pharmacology 2019/20: Introduction and Other Protein Targets. Br. J. Pharmacol. 2019, 176 (Suppl. 1), S1–S20. [Google Scholar] [CrossRef]
- Ovsepian, S.V.; LeBerre, M.; Steuber, V.; O’Leary, V.B.; Leibold, C.; Oliver Dolly, J. Distinctive role of KV1.1 subunit in the biology and functions of low threshold K+ channels with implications for neurological disease. Pharmacol. Ther. 2016, 159, 93–101. [Google Scholar] [CrossRef]
- Parcej, D.N.; Scott, V.E.; Dolly, J.O. Oligomeric properties of alpha-dendrotoxin-sensitive potassium ion channels purified from bovine brain. Biochemistry 1992, 31, 11084–11088. [Google Scholar] [CrossRef]
- Pongs, O.; Schwarz, J.R. Ancillary subunits associated with voltage-dependent K+ channels. Physiol. Rev. 2010, 90, 755–796. [Google Scholar] [CrossRef]
- Coetzee, W.A.; Amarillo, Y.; Chiu, J.; Chow, A.; Lau, D.; McCormack, T.; Moreno, H.; Nadal, M.S.; Ozaita, A.; Pountney, D.; et al. Molecular diversity of K+ channels. Ann. N. Y. Acad. Sci. 1999, 868, 233–285. [Google Scholar] [CrossRef]
- Panyi, G.; Deutsch, C. Assembly and suppression of endogenous Kv1.3 channels in human T cells. J. Gen. Physiol. 1996, 107, 409–420. [Google Scholar] [CrossRef]
- Shen, N.V.; Pfaffinger, P.J. Molecular recognition and assembly sequences involved in the subfamily-specific assembly of voltage-gated K+ channel subunit proteins. Neuron 1995, 14, 625–633. [Google Scholar] [CrossRef]
- Xu, J.; Yu, W.; Jan, Y.N.; Jan, L.Y.; Li, M. Assembly of voltage-gated potassium channels. Conserved hydrophilic motifs determine subfamily-specific interactions between the alpha-subunits. J. Biol. Chem. 1995, 270, 24761–24768. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Deutsch, C. Evidence for dimerization of dimers in K+ channel assembly. Biophys. J. 1999, 76, 2004–2017. [Google Scholar] [CrossRef]
- Stuhmer, W.; Ruppersberg, J.P.; Schroter, K.H.; Sakmann, B.; Stocker, M.; Giese, K.P.; Perschke, A.; Baumann, A.; Pongs, O. Molecular basis of functional diversity of voltage-gated potassium channels in mammalian brain. EMBO J. 1989, 8, 3235–3244. [Google Scholar] [CrossRef] [PubMed]
- Coleman, S.K.; Newcombe, J.; Pryke, J.; Dolly, J.O. Subunit composition of Kv1 channels in human CNS. J. Neurochem. 1999, 73, 849–858. [Google Scholar] [CrossRef]
- Koch, R.O.; Wanner, S.G.; Koschak, A.; Hanner, M.; Schwarzer, C.; Kaczorowski, G.J.; Slaughter, R.S.; Garcia, M.L.; Knaus, H.G. Complex subunit assembly of neuronal voltage-gated K+ channels. Basis for high-affinity toxin interactions and pharmacology. J. Biol. Chem. 1997, 272, 27577–27581. [Google Scholar] [CrossRef]
- Koschak, A.; Bugianesi, R.M.; Mitterdorfer, J.; Kaczorowski, G.J.; Garcia, M.L.; Knaus, H.G. Subunit composition of brain voltage-gated potassium channels determined by hongotoxin-1, a novel peptide derived from Centruroides limbatus venom. J. Biol. Chem. 1998, 273, 2639–2644. [Google Scholar] [CrossRef]
- Ruppersberg, J.P.; Schroter, K.H.; Sakmann, B.; Stocker, M.; Sewing, S.; Pongs, O. Heteromultimeric channels formed by rat brain potassium-channel proteins. Nature 1990, 345, 535–537. [Google Scholar] [CrossRef]
- Shamotienko, O.G.; Parcej, D.N.; Dolly, J.O. Subunit combinations defined for K+ channel Kv1 subtypes in synaptic membranes from bovine brain. Biochemistry 1997, 36, 8195–8201. [Google Scholar] [CrossRef]
- Schendel, V.; Rash, L.D.; Jenner, R.A.; Undheim, E.A.B. The Diversity of Venom: The Importance of Behavior and Venom System Morphology in Understanding Its Ecology and Evolution. Toxins (Basel) 2019, 11, 666. [Google Scholar] [CrossRef]
- Mouhat, S.; Andreotti, N.; Jouirou, B.; Sabatier, J.M. Animal toxins acting on voltage-gated potassium channels. Curr. Pharm. Des. 2008, 14, 2503–2518. [Google Scholar] [CrossRef]
- Eriksson, M.A.; Roux, B. Modeling the structure of agitoxin in complex with the Shaker K+ channel: A computational approach based on experimental distance restraints extracted from thermodynamic mutant cycles. Biophys. J. 2002, 83, 2595–2609. [Google Scholar] [CrossRef]
- Gao, Y.D.; Garcia, M.L. Interaction of agitoxin 2, charybdotoxin, and iberiotoxin with potassium channels: Selectivity between voltage-gated and Maxi-K channels. Proteins 2003, 52, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Miller, C. The charybdotoxin family of K+ channel-blocking peptides. Neuron 1995, 15, 5–10. [Google Scholar] [CrossRef]
- Dauplais, M.; Lecoq, A.; Song, J.; Cotton, J.; Jamin, N.; Gilquin, B.; Roumestand, C.; Vita, C.; de Medeiros, C.L.; Rowan, E.G.; et al. On the convergent evolution of animal toxins. Conservation of a diad of functional residues in potassium channel-blocking toxins with unrelated structures. J. Biol. Chem. 1997, 272, 4302–4309. [Google Scholar] [CrossRef]
- Gilquin, B.; Racape, J.; Wrisch, A.; Visan, V.; Lecoq, A.; Grissmer, S.; Menez, A.; Gasparini, S. Structure of the BgK-Kv1.1 complex based on distance restraints identified by double mutant cycles. Molecular basis for convergent evolution of Kv1 channel blockers. J. Biol. Chem. 2002, 277, 37406–37413. [Google Scholar] [CrossRef]
- Savarin, P.; Guenneugues, M.; Gilquin, B.; Lamthanh, H.; Gasparini, S.; Zinn-Justin, S.; Menez, A. Three-dimensional structure of kappa-conotoxin PVIIA, a novel potassium channel-blocking toxin from cone snails. Biochemistry 1998, 37, 5407–5416. [Google Scholar] [CrossRef]
- Srinivasan, K.N.; Sivaraja, V.; Huys, I.; Sasaki, T.; Cheng, B.; Kumar, T.K.; Sato, K.; Tytgat, J.; Yu, C.; San, B.C.; et al. kappa-Hefutoxin1, a novel toxin from the scorpion Heterometrus fulvipes with unique structure and function. Importance of the functional diad in potassium channel selectivity. J. Biol. Chem. 2002, 277, 30040–30047. [Google Scholar] [CrossRef]
- Jouirou, B.; Mouhat, S.; Andreotti, N.; De Waard, M.; Sabatier, J.M. Toxin determinants required for interaction with voltage-gated K+ channels. Toxicon 2004, 43, 909–914. [Google Scholar] [CrossRef]
- Al-Sabi, A.; Lennartz, D.; Ferber, M.; Gulyas, J.; Rivier, J.E.; Olivera, B.M.; Carlomagno, T.; Terlau, H. KappaM-conotoxin RIIIK, structural and functional novelty in a K+ channel antagonist. Biochemistry 2004, 43, 8625–8635. [Google Scholar] [CrossRef]
- Bayrhuber, M.; Vijayan, V.; Ferber, M.; Graf, R.; Korukottu, J.; Imperial, J.; Garrett, J.E.; Olivera, B.M.; Terlau, H.; Zweckstetter, M.; et al. Conkunitzin-S1 is the first member of a new Kunitz-type neurotoxin family. Structural and functional characterization. J. Biol. Chem. 2005, 280, 23766–23770. [Google Scholar] [CrossRef]
- Tudor, J.E.; Pallaghy, P.K.; Pennington, M.W.; Norton, R.S. Solution structure of ShK toxin, a novel potassium channel inhibitor from a sea anemone. Nat. Struct. Biol. 1996, 3, 317–320. [Google Scholar] [CrossRef]
- Takahashi, H.; Kim, J.I.; Min, H.J.; Sato, K.; Swartz, K.J.; Shimada, I. Solution structure of hanatoxin1, a gating modifier of voltage-dependent K+ channels: Common surface features of gating modifier toxins. J. Mol. Biol. 2000, 297, 771–780. [Google Scholar] [CrossRef] [PubMed]
- de Vries, S.J.; van Dijk, M.; Bonvin, A.M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef] [PubMed]
- van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- DeLano Scientific. The PyMOL Molecular Graphics System; DeLano Scientific: Palo Alto, CA, USA, 2002. [Google Scholar]
- Kalia, J.; Milescu, M.; Salvatierra, J.; Wagner, J.; Klint, J.K.; King, G.F.; Olivera, B.M.; Bosmans, F. From foe to friend: Using animal toxins to investigate ion channel function. J. Mol. Biol. 2015, 427, 158–175. [Google Scholar] [CrossRef]
- Swartz, K.J.; MacKinnon, R. Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron 1997, 18, 665–673. [Google Scholar] [CrossRef]
- Karbat, I.; Altman-Gueta, H.; Fine, S.; Szanto, T.; Hamer-Rogotner, S.; Dym, O.; Frolow, F.; Gordon, D.; Panyi, G.; Gurevitz, M.; et al. Pore-modulating toxins exploit inherent slow inactivation to block K+ channels. Proc. Natl. Acad. Sci. USA 2019, 116, 18700–18709. [Google Scholar] [CrossRef]
- Dave, K.; Lahiry, A. Conotoxins: Review and docking studies to determine potentials of conotoxin as an anticancer drug molecule. Curr. Top. Med. Chem. 2012, 12, 845–851. [Google Scholar] [CrossRef]
- Puillandre, N.; Duda, T.F.; Meyer, C.; Olivera, B.M.; Bouchet, P. One, four or 100 genera? A new classification of the cone snails. J. Molluscan Stud. 2015, 81, 1–23. [Google Scholar] [CrossRef]
- Dutertre, S.; Jin, A.H.; Vetter, I.; Hamilton, B.; Sunagar, K.; Lavergne, V.; Dutertre, V.; Fry, B.G.; Antunes, A.; Venter, D.J.; et al. Evolution of separate predation- and defence-evoked venoms in carnivorous cone snails. Nat. Commun. 2014, 5, 3521. [Google Scholar] [CrossRef]
- Morales Duque, H.; Campos Dias, S.; Franco, O.L. Structural and Functional Analyses of Cone Snail Toxins. Mar. Drugs 2019, 17, 370. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M.; Raghuraman, S.; Schmidt, E.W.; Safavi-Hemami, H. Linking neuroethology to the chemical biology of natural products: Interactions between cone snails and their fish prey, a case study. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 2017, 203, 717–735. [Google Scholar] [CrossRef] [PubMed]
- Teichert, R.W.; Schmidt, E.W.; Olivera, B.M. Constellation pharmacology: A new paradigm for drug discovery. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Han, T.S.; Teichert, R.W.; Olivera, B.M.; Bulaj, G. Conus venoms—A rich source of peptide-based therapeutics. Curr. Pharm. Des. 2008, 14, 2462–2479. [Google Scholar] [CrossRef]
- Jin, A.H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.W.A.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and Biology. Chem. Rev. 2019, 119, 11510–11549. [Google Scholar] [CrossRef]
- Lavergne, V.; Harliwong, I.; Jones, A.; Miller, D.; Taft, R.J.; Alewood, P.F. Optimized deep-targeted proteotranscriptomic profiling reveals unexplored Conus toxin diversity and novel cysteine frameworks. Proc. Natl. Acad. Sci. USA 2015, 112, E3782–E3791. [Google Scholar] [CrossRef]
- Buczek, O.; Bulaj, G.; Olivera, B.M. Conotoxins and the posttranslational modification of secreted gene products. Cell. Mol. Life Sci. 2005, 62, 3067–3079. [Google Scholar] [CrossRef]
- Kaas, Q.; Westermann, J.C.; Halai, R.; Wang, C.K.; Craik, D.J. ConoServer, a database for conopeptide sequences and structures. Bioinformatics 2008, 24, 445–446. [Google Scholar] [CrossRef]
- Terlau, H.; Shon, K.J.; Grilley, M.; Stocker, M.; Stuhmer, W.; Olivera, B.M. Strategy for rapid immobilization of prey by a fish-hunting marine snail. Nature 1996, 381, 148–151. [Google Scholar] [CrossRef]
- Tabakmakher, V.M.; Krylov, N.A.; Kuzmenkov, A.I.; Efremov, R.G.; Vassilevski, A.A. Kalium 2.0, a comprehensive database of polypeptide ligands of potassium channels. Sci. Data 2019, 6, 73. [Google Scholar] [CrossRef]
- Massilia, G.R.; Eliseo, T.; Grolleau, F.; Lapied, B.; Barbier, J.; Bournaud, R.; Molgo, J.; Cicero, D.O.; Paci, M.; Schinina, M.E.; et al. Contryphan-Vn: A modulator of Ca2+-dependent K+ channels. Biochem. Biophys. Res. Commun. 2003, 303, 238–246. [Google Scholar] [CrossRef]
- Scanlon, M.J.; Naranjo, D.; Thomas, L.; Alewood, P.F.; Lewis, R.J.; Craik, D.J. Solution structure and proposed binding mechanism of a novel potassium channel toxin kappa-conotoxin PVIIA. Structure 1997, 5, 1585–1597. [Google Scholar] [CrossRef]
- Imperial, J.S.; Bansal, P.S.; Alewood, P.F.; Daly, N.L.; Craik, D.J.; Sporning, A.; Terlau, H.; Lopez-Vera, E.; Bandyopadhyay, P.K.; Olivera, B.M. A novel conotoxin inhibitor of Kv1.6 channel and nAChR subtypes defines a new superfamily of conotoxins. Biochemistry 2006, 45, 8331–8340. [Google Scholar] [CrossRef] [PubMed]
- Ferber, M.; Al-Sabi, A.; Stocker, M.; Olivera, B.M.; Terlau, H. Identification of a mammalian target of kappaM-conotoxin RIIIK. Toxicon 2004, 43, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Gray, W.R.; Olivera, B.M.; Zeikus, R.D.; Kerr, L.; Yoshikami, D.; Moczydlowski, E. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J. Biol. Chem. 1985, 260, 9280–9288. [Google Scholar]
- Ferber, M.; Sporning, A.; Jeserich, G.; DeLaCruz, R.; Watkins, M.; Olivera, B.M.; Terlau, H. A novel conus peptide ligand for K+ channels. J. Biol. Chem. 2003, 278, 2177–2183. [Google Scholar] [CrossRef]
- Chen, P.; Dendorfer, A.; Finol-Urdaneta, R.K.; Terlau, H.; Olivera, B.M. Biochemical characterization of kappaM-RIIIJ, a Kv1.2 channel blocker: Evaluation of cardioprotective effects of kappaM-conotoxins. J. Biol. Chem. 2010, 285, 14882–14889. [Google Scholar] [CrossRef]
- Verdier, L.; Al-Sabi, A.; Rivier, J.E.; Olivera, B.M.; Terlau, H.; Carlomagno, T. Identification of a novel pharmacophore for peptide toxins interacting with K+ channels. J. Biol. Chem. 2005, 280, 21246–21255. [Google Scholar] [CrossRef]
- Cordeiro, S.; Finol-Urdaneta, R.K.; Kopfer, D.; Markushina, A.; Song, J.; French, R.J.; Kopec, W.; de Groot, B.L.; Giacobassi, M.J.; Leavitt, L.S.; et al. Conotoxin kappaM-RIIIJ, a tool targeting asymmetric heteromeric Kv1 channels. Proc. Natl. Acad. Sci. USA 2019, 116, 1059–1064. [Google Scholar] [CrossRef]
- Giacobassi, M.J.; Leavitt, L.S.; Raghuraman, S.; Alluri, R.; Chase, K.; Finol-Urdaneta, R.K.; Terlau, H.; Teichert, R.W.; Olivera, B.M. An integrative approach to the facile functional classification of dorsal root ganglion neuronal subclasses. Proc. Natl. Acad. Sci. USA 2020. [Google Scholar] [CrossRef]
- Imperial, J.S.; Chen, P.; Sporning, A.; Terlau, H.; Daly, N.L.; Craik, D.J.; Alewood, P.F.; Olivera, B.M. Tyrosine-rich conopeptides affect voltage-gated K+ channels. J. Biol. Chem. 2008, 283, 23026–23032. [Google Scholar] [CrossRef] [PubMed]
- Kauferstein, S.; Huys, I.; Lamthanh, H.; Stocklin, R.; Sotto, F.; Menez, A.; Tytgat, J.; Mebs, D. A novel conotoxin inhibiting vertebrate voltage-sensitive potassium channels. Toxicon 2003, 42, 43–52. [Google Scholar] [CrossRef]
- Dy, C.Y.; Buczek, P.; Imperial, J.S.; Bulaj, G.; Horvath, M.P. Structure of conkunitzin-S1, a neurotoxin and Kunitz-fold disulfide variant from cone snail. Acta Crystallogr. D Biol. Crystallogr. 2006, 62 (Pt 9), 980–990. [Google Scholar] [CrossRef]
- Korukottu, J.; Bayrhuber, M.; Montaville, P.; Vijayan, V.; Jung, Y.S.; Becker, S.; Zweckstetter, M. Fast high-resolution protein structure determination by using unassigned NMR data. Angew. Chem. Int. Ed. Engl. 2007, 46, 1176–1179. [Google Scholar] [CrossRef] [PubMed]
- Cuello, L.G.; Jogini, V.; Cortes, D.M.; Perozo, E. Structural mechanism of C-type inactivation in K+ channels. Nature 2010, 466, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Finol-Urdaneta, R.K. Investigation of the Heterologous Expression of the Voltage Activated Potassium Channel Kv1.7; George-August University: Goettingen, Germany, 2004. [Google Scholar]
- Zhang, S.J.; Yang, X.M.; Liu, G.S.; Cohen, M.V.; Pemberton, K.; Downey, J.M. CGX-1051, a peptide from Conus snail venom, attenuates infarction in rabbit hearts when administered at reperfusion. J. Cardiovasc. Pharmacol. 2003, 42, 764–771. [Google Scholar] [CrossRef]
- Lubbers, N.L.; Campbell, T.J.; Polakowski, J.S.; Bulaj, G.; Layer, R.T.; Moore, J.; Gross, G.J.; Cox, B.F. Postischemic administration of CGX-1051, a peptide from cone snail venom, reduces infarct size in both rat and dog models of myocardial ischemia and reperfusion. J. Cardiovasc. Pharmacol. 2005, 46, 141–146. [Google Scholar] [CrossRef]
- Mahdavi, S.; Kuyucak, S. Why the Drosophila Shaker K+ channel is not a good model for ligand binding to voltage-gated Kv1 channels. Biochemistry 2013, 52, 1631–1640. [Google Scholar] [CrossRef]
- Tanaka, J.; Abe, J.; Futagi, Y. A case of late infantile ceroid lipofuscinosis--an electrophysiological follow-up study. Hattatsu 1987, 19, 415–419. [Google Scholar]
- Aguilar, M.B.; Perez-Reyes, L.I.; Lopez, Z.; de la Cotera, E.P.; Falcon, A.; Ayala, C.; Galvan, M.; Salvador, C.; Escobar, L.I. Peptide sr11a from Conus spurius is a novel peptide blocker for Kv1 potassium channels. Peptides 2010, 31, 1287–1291. [Google Scholar] [CrossRef]
- Mondal, S.; Babu, R.M.; Bhavna, R.; Ramakumar, S. In silico detection of binding mode of J-superfamily conotoxin pl14a with Kv1.6 channel. Silico Biol 2007, 7, 175–186. [Google Scholar]
- Leipold, E.; Ullrich, F.; Thiele, M.; Tietze, A.A.; Terlau, H.; Imhof, D.; Heinemann, S.H. Subtype-specific block of voltage-gated K+ channels by mu-conopeptides. Biochem. Biophys. Res. Commun. 2017, 482, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Finol-Urdaneta, R.K.; McArthur, J.R.; Korkosh, V.S.; Huang, S.; McMaster, D.; Glavica, R.; Tikhonov, D.B.; Zhorov, B.S.; French, R.J. Extremely Potent Block of Bacterial Voltage-Gated Sodium Channels by micro-Conotoxin PIIIA. Mar. Drugs 2019, 17, 510. [Google Scholar] [CrossRef]
- Kaufmann, D.; Tietze, A.A.; Tietze, D. In Silico Analysis of the Subtype Selective Blockage of KCNA Ion Channels through the micro-Conotoxins PIIIA, SIIIA, and GIIIA. Mar. Drugs 2019, 17, 180. [Google Scholar] [CrossRef]
- Imperial, J.S.; Cabang, A.B.; Song, J.; Raghuraman, S.; Gajewiak, J.; Watkins, M.; Showers-Corneli, P.; Fedosov, A.; Concepcion, G.P.; Terlau, H.; et al. A family of excitatory peptide toxins from venomous crassispirine snails: Using Constellation Pharmacology to assess bioactivity. Toxicon 2014, 89, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Honma, T.; Shiomi, K. Peptide toxins in sea anemones: Structural and functional aspects. Mar. Biotechnol. (N. Y.) 2006, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kuyucak, S.; Norton, R.S. Computational approaches for designing potent and selective analogs of peptide toxins as novel therapeutics. Future Med. Chem. 2014, 6, 1645–1658. [Google Scholar] [CrossRef]
- Prentis, P.J.; Pavasovic, A.; Norton, R.S. Sea Anemones: Quiet Achievers in the Field of Peptide Toxins. Toxins (Basel) 2018, 10, 36. [Google Scholar] [CrossRef]
- Madio, B.; King, G.F.; Undheim, E.A.B. Sea Anemone Toxins: A Structural Overview. Mar. Drugs 2019, 17, 325. [Google Scholar] [CrossRef]
- Gasparini, S.; Gilquin, B.; Menez, A. Comparison of sea anemone and scorpion toxins binding to Kv1 channels: An example of convergent evolution. Toxicon 2004, 43, 901–908. [Google Scholar] [CrossRef]
- Mouhat, S.; Jouirou, B.; Mosbah, A.; De Waard, M.; Sabatier, J.M. Diversity of folds in animal toxins acting on ion channels. Biochem. J. 2004, 378 (Pt 3), 717–726. [Google Scholar] [CrossRef]
- Kalman, K.; Pennington, M.W.; Lanigan, M.D.; Nguyen, A.; Rauer, H.; Mahnir, V.; Paschetto, K.; Kem, W.R.; Grissmer, S.; Gutman, G.A.; et al. ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. J. Biol. Chem. 1998, 273, 32697–32707. [Google Scholar] [CrossRef] [PubMed]
- Pennington, M.W.; Mahnir, V.M.; Khaytin, I.; Zaydenberg, I.; Byrnes, M.E.; Kem, W.R. An essential binding surface for ShK toxin interaction with rat brain potassium channels. Biochemistry 1996, 35, 16407–16411. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, S.; Ishida, M.; Nagashima, Y.; Shiomi, K. Primary structure of a potassium channel toxin from the sea anemone Actinia equina. FEBS Lett. 1998, 427, 149–151. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Honma, T.; Nagai, H.; Ishida, M.; Nagashima, Y.; Shiomi, K. Isolation and cDNA cloning of a potassium channel peptide toxin from the sea anemone Anemonia erythraea. Toxicon 2006, 48, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Orts, D.J.; Moran, Y.; Cologna, C.T.; Peigneur, S.; Madio, B.; Praher, D.; Quinton, L.; De Pauw, E.; Bicudo, J.E.; Tytgat, J.; et al. BcsTx3 is a founder of a novel sea anemone toxin family of potassium channel blocker. FEBS J. 2013, 280, 4839–4852. [Google Scholar] [CrossRef] [PubMed]
- Schweitz, H.; Bruhn, T.; Guillemare, E.; Moinier, D.; Lancelin, J.M.; Beress, L.; Lazdunski, M. Kalicludines and kaliseptine. Two different classes of sea anemone toxins for voltage sensitive K+ channels. J. Biol. Chem. 1995, 270, 25121–25126. [Google Scholar] [CrossRef]
- Cotton, J.; Crest, M.; Bouet, F.; Alessandri, N.; Gola, M.; Forest, E.; Karlsson, E.; Castaneda, O.; Harvey, A.L.; Vita, C.; et al. A potassium-channel toxin from the sea anemone Bunodosoma granulifera, an inhibitor for Kv1 channels. Revision of the amino acid sequence, disulfide-bridge assignment, chemical synthesis, and biological activity. Eur. J. Biochem. 1997, 244, 192–202. [Google Scholar] [CrossRef]
- Racape, J.; Lecoq, A.; Romi-Lebrun, R.; Liu, J.; Kohler, M.; Garcia, M.L.; Menez, A.; Gasparini, S. Characterization of a novel radiolabeled peptide selective for a subpopulation of voltage-gated potassium channels in mammalian brain. J. Biol. Chem. 2002, 277, 3886–3893. [Google Scholar] [CrossRef]
- Gendeh, G.S.; Young, L.C.; de Medeiros, C.L.; Jeyaseelan, K.; Harvey, A.L.; Chung, M.C. A new potassium channel toxin from the sea anemone Heteractis magnifica: Isolation, cDNA cloning, and functional expression. Biochemistry 1997, 36, 11461–11471. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, J.; Yuan, X.; Peng, B.; Liu, W.; Han, S.; He, X. Toxins Targeting the Kv1.3 Channel: Potential Immunomodulators for Autoimmune Diseases. Toxins (Basel) 2015, 7, 1749–1764. [Google Scholar] [CrossRef] [PubMed]
- Diochot, S.; Lazdunski, M. Sea anemone toxins affecting potassium channels. Prog. Mol. Subcell Biol. 2009, 46, 99–122. [Google Scholar] [PubMed]
- Honma, T.; Kawahata, S.; Ishida, M.; Nagai, H.; Nagashima, Y.; Shiomi, K. Novel peptide toxins from the sea anemone Stichodactyla haddoni. Peptides 2008, 29, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Diochot, S.; Loret, E.; Bruhn, T.; Beress, L.; Lazdunski, M. APETx1, a new toxin from the sea anemone Anthopleura elegantissima, blocks voltage-gated human ether-a-go-go-related gene potassium channels. Mol. Pharmacol. 2003, 64, 59–69. [Google Scholar] [CrossRef]
- Rodriguez, A.A.; Garateix, A.; Salceda, E.; Peigneur, S.; Zaharenko, A.J.; Pons, T.; Santos, Y.; Arreguin, R.; Standker, L.; Forssmann, W.G.; et al. PhcrTx2, a New Crab-Paralyzing Peptide Toxin from the Sea Anemone Phymanthus crucifer. Toxins (Basel) 2018, 10, 72. [Google Scholar] [CrossRef]
- Rodriguez, A.A.; Salceda, E.; Garateix, A.G.; Zaharenko, A.J.; Peigneur, S.; Lopez, O.; Pons, T.; Richardson, M.; Diaz, M.; Hernandez, Y.; et al. A novel sea anemone peptide that inhibits acid-sensing ion channels. Peptides 2014, 53, 3–12. [Google Scholar] [CrossRef]
- DJ, B.O.; Peigneur, S.; Silva-Goncalves, L.C.; Arcisio-Miranda, M.; Je, P.W.B.; Tytgat, J. AbeTx1 Is a Novel Sea Anemone Toxin with a Dual Mechanism of Action on Shaker-Type K+ Channels Activation. Mar. Drugs 2018, 16. [Google Scholar]
- Chagot, B.; Escoubas, P.; Villegas, E.; Bernard, C.; Ferrat, G.; Corzo, G.; Lazdunski, M.; Darbon, H. Solution structure of Phrixotoxin 1, a specific peptide inhibitor of Kv4 potassium channels from the venom of the theraphosid spider Phrixotrichus auratus. Protein Sci. 2004, 13, 1197–1208. [Google Scholar] [CrossRef]
- Driscoll, P.C.; Gronenborn, A.M.; Beress, L.; Clore, G.M. Determination of the three-dimensional solution structure of the antihypertensive and antiviral protein BDS-I from the sea anemone Anemonia sulcata: A study using nuclear magnetic resonance and hybrid distance geometry-dynamical simulated annealing. Biochemistry 1989, 28, 2188–2198. [Google Scholar] [CrossRef]
- Rauer, H.; Pennington, M.; Cahalan, M.; Chandy, K.G. Structural conservation of the pores of calcium-activated and voltage-gated potassium channels determined by a sea anemone toxin. J. Biol. Chem. 1999, 274, 21885–21892. [Google Scholar] [CrossRef]
- Castaneda, O.; Sotolongo, V.; Amor, A.M.; Stocklin, R.; Anderson, A.J.; Harvey, A.L.; Engstrom, A.; Wernstedt, C.; Karlsson, E. Characterization of a potassium channel toxin from the Caribbean Sea anemone Stichodactyla helianthus. Toxicon 1995, 33, 603–613. [Google Scholar] [CrossRef]
- Pennington, M.W.; Mahnir, V.M.; Krafte, D.S.; Zaydenberg, I.; Byrnes, M.E.; Khaytin, I.; Crowley, K.; Kem, W.R. Identification of three separate binding sites on SHK toxin, a potent inhibitor of voltage-dependent potassium channels in human T-lymphocytes and rat brain. Biochem. Biophys. Res. Commun. 1996, 219, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.C.; Huq, R.; Chhabra, S.; Beeton, C.; Pennington, M.W.; Smith, B.J.; Norton, R.S. N-Terminally extended analogues of the K+ channel toxin from Stichodactyla helianthus as potent and selective blockers of the voltage-gated potassium channel Kv1.3. FEBS J. 2015, 282, 2247–2259. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.K.; Qian, Y.X.; Liu, B.; Elliott, R.; Aral, J.; Park, C.; Zhang, X.; Stenkilsson, M.; Salyers, K.; Rose, M.; et al. Pharmaceutical Optimization of Peptide Toxins for Ion Channel Targets: Potent, Selective, and Long-Lived Antagonists of Kv1.3. J. Med. Chem. 2015, 58, 6784–6802. [Google Scholar] [CrossRef]
- Yan, L.; Herrington, J.; Goldberg, E.; Dulski, P.M.; Bugianesi, R.M.; Slaughter, R.S.; Banerjee, P.; Brochu, R.M.; Priest, B.T.; Kaczorowski, G.J.; et al. Stichodactyla helianthus peptide, a pharmacological tool for studying Kv3.2 channels. Mol. Pharmacol. 2005, 67, 1513–1521. [Google Scholar] [CrossRef]
- Garcia-Fernandez, R.; Peigneur, S.; Pons, T.; Alvarez, C.; Gonzalez, L.; Chavez, M.A.; Tytgat, J. The Kunitz-Type Protein ShPI-1 Inhibits Serine Proteases and Voltage-Gated Potassium Channels. Toxins (Basel) 2016, 8, 110. [Google Scholar] [CrossRef]
- Chi, V.; Pennington, M.W.; Norton, R.S.; Tarcha, E.J.; Londono, L.M.; Sims-Fahey, B.; Upadhyay, S.K.; Lakey, J.T.; Iadonato, S.; Wulff, H.; et al. Development of a sea anemone toxin as an immunomodulator for therapy of autoimmune diseases. Toxicon 2012, 59, 529–546. [Google Scholar] [CrossRef]
- Pennington, M.W.; Harunur Rashid, M.; Tajhya, R.B.; Beeton, C.; Kuyucak, S.; Norton, R.S. A C-terminally amidated analogue of ShK is a potent and selective blocker of the voltage-gated potassium channel Kv1.3. FEBS Lett. 2012, 586, 3996–4001. [Google Scholar] [CrossRef]
- Gilquin, B.; Braud, S.; Eriksson, M.A.; Roux, B.; Bailey, T.D.; Priest, B.T.; Garcia, M.L.; Menez, A.; Gasparini, S. A variable residue in the pore of Kv1 channels is critical for the high affinity of blockers from sea anemones and scorpions. J. Biol. Chem. 2005, 280, 27093–27102. [Google Scholar] [CrossRef]
- Alessandri-Haber, N.; Lecoq, A.; Gasparini, S.; Grangier-Macmath, G.; Jacquet, G.; Harvey, A.L.; de Medeiros, C.; Rowan, E.G.; Gola, M.; Menez, A.; et al. Mapping the functional anatomy of BgK on Kv1.1, Kv1.2, and Kv1.3. Clues to design analogs with enhanced selectivity. J. Biol. Chem. 1999, 274, 35653–35661. [Google Scholar] [CrossRef]
- Beraud, E.; Viola, A.; Regaya, I.; Confort-Gouny, S.; Siaud, P.; Ibarrola, D.; Le Fur, Y.; Barbaria, J.; Pellissier, J.F.; Sabatier, J.M.; et al. Block of neural Kv1.1 potassium channels for neuroinflammatory disease therapy. Ann. Neurol. 2006, 60, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Peigneur, S.; Billen, B.; Derua, R.; Waelkens, E.; Debaveye, S.; Beress, L.; Tytgat, J. A bifunctional sea anemone peptide with Kunitz type protease and potassium channel inhibiting properties. Biochem. Pharmacol. 2011, 82, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Mourao, C.B.; Schwartz, E.F. Protease inhibitors from marine venomous animals and their counterparts in terrestrial venomous animals. Mar. Drugs 2013, 11, 2069–2112. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Defensins: Antimicrobial peptides of vertebrates. Comptes Rendus Biol. 2004, 327, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Chagot, B.; Escoubas, P.; Diochot, S.; Bernard, C.; Lazdunski, M.; Darbon, H. Solution structure of APETx2, a specific peptide inhibitor of ASIC3 proton-gated channels. Protein Sci. 2005, 14, 2003–2010. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Blumenthal, K.M. Site-3 sea anemone toxins: Molecular probes of gating mechanisms in voltage-dependent sodium channels. Toxicon 2007, 49, 159–170. [Google Scholar] [CrossRef]
- Beress, L.D.; Doppelfeld, I.S.; Etschenberg, E.; Graf, E.; Henschen, A.; Zwick, J. Polypeptides, Process for Their Preparation, and Their Use as Hypotensive Active Compounds. Patent No. DE3324689, 17 January 1985. [Google Scholar]
- Liu, P.; Jo, S.; Bean, B.P. Modulation of neuronal sodium channels by the sea anemone peptide BDS-I. J. Neurophysiol. 2012, 107, 3155–3167. [Google Scholar] [CrossRef]
- Moreels, L.; Peigneur, S.; Galan, D.T.; De Pauw, E.; Beress, L.; Waelkens, E.; Pardo, L.A.; Quinton, L.; Tytgat, J. APETx4, a Novel Sea Anemone Toxin and a Modulator of the Cancer-Relevant Potassium Channel Kv10.1. Mar. Drugs 2017, 15, 287. [Google Scholar] [CrossRef]
- Daly, M.; Chaudhuri, A.; Gusmao, L.; Rodriguez, E. Phylogenetic relationships among sea anemones (Cnidaria: Anthozoa: Actiniaria). Mol. Phylogenet. Evol. 2008, 48, 292–301. [Google Scholar] [CrossRef]
- Daly, N.L.; Craik, D.J. Bioactive cystine knot proteins. Curr. Opin. Chem. Biol. 2011, 15, 362–368. [Google Scholar] [CrossRef]
- Konoki, K.; Suga, Y.; Fuwa, H.; Yotsu-Yamashita, M.; Sasaki, M. Evaluation of gambierol and its analogs for their inhibition of human Kv1.2 and cytotoxicity. Bioorg. Med. Chem. Lett. 2015, 25, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J. Ciguatera: Australian perspectives on a global problem. Toxicon 2006, 48, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Cuypers, E.; Abdel-Mottaleb, Y.; Kopljar, I.; Rainier, J.D.; Raes, A.L.; Snyders, D.J.; Tytgat, J. Gambierol, a toxin produced by the dinoflagellate Gambierdiscus toxicus, is a potent blocker of voltage-gated potassium channels. Toxicon 2008, 51, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Kopljar, I.; Labro, A.J.; Cuypers, E.; Johnson, H.W.; Rainier, J.D.; Tytgat, J.; Snyders, D.J. A polyether biotoxin binding site on the lipid-exposed face of the pore domain of Kv channels revealed by the marine toxin gambierol. Proc. Natl. Acad. Sci. USA 2009, 106, 9896–9901. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Ji, X.; Fu, M.; Zhang, W.; Zhang, D.; Xiao, Z. Electrostatic interaction between inactivation ball and T1-S1 linker region of Kv1.4 channel. Biochim. Biophys. Acta 2012, 1818, 55–63. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tang, Y.H.; Liang, T.T.; Fan, T.T.; Keen, L.J.; Zhang, X.D.; Xu, L.; Zhao, Q.; Zeng, R.; Han, B.N. Neo-debromoaplysiatoxin C, with new structural rearrangement, derived from debromoaplysiatoxin. Nat. Prod. Res. 2019, 1–6. [Google Scholar] [CrossRef]
- Nokura, Y.; Araki, Y.; Nakazaki, A.; Nishikawa, T. Synthetic Route to Oscillatoxin D and Its Analogues. Org. Lett. 2017, 19, 5992–5995. [Google Scholar] [CrossRef]
- Dias, L.C.; Polo, E.C. Nhatrangin A: Total Syntheses of the Proposed Structure and Six of Its Diastereoisomers. J. Org. Chem. 2017, 82, 4072–4112. [Google Scholar] [CrossRef]
- Fan, T.T.; Zhang, H.H.; Tang, Y.H.; Zhang, F.Z.; Han, B.N. Two New Neo-debromoaplysiatoxins-A Pair of Stereoisomers Exhibiting Potent Kv1.5 Ion Channel Inhibition Activities. Mar. Drugs 2019, 17, 652. [Google Scholar] [CrossRef]
- Feng, J.; Wang, Z.; Li, G.R.; Nattel, S. Effects of class III antiarrhythmic drugs on transient outward and ultra-rapid delayed rectifier currents in human atrial myocytes. J. Pharmacol. Exp. Ther. 1997, 281, 384–392. [Google Scholar]
- Hurst, R.S.; Kavanaugh, M.P.; Yakel, J.; Adelman, J.P.; North, R.A. Cooperative interactions among subunits of a voltage-dependent potassium channel. Evidence from expression of concatenated cDNAs. J. Biol. Chem. 1992, 267, 23742–23745. [Google Scholar] [PubMed]
- Hurst, R.S.; North, R.A.; Adelman, J.P. Potassium channel assembly from concatenated subunits: Effects of proline substitutions in S4 segments. Recept. Channels 1995, 3, 263–272. [Google Scholar] [PubMed]
- Klein-Schwartz, W.; Stassinos, G.L.; Isbister, G.K. Treatment of sulfonylurea and insulin overdose. Br. J. Clin. Pharmacol. 2016, 81, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Beeton, C.; Pennington, M.W.; Norton, R.S. Analogs of the sea anemone potassium channel blocker ShK for the treatment of autoimmune diseases. Inflamm. Allergy Drug Targets 2011, 10, 313–321. [Google Scholar] [CrossRef]
- Pennington, M.W.; Beeton, C.; Galea, C.A.; Smith, B.J.; Chi, V.; Monaghan, K.P.; Garcia, A.; Rangaraju, S.; Giuffrida, A.; Plank, D.; et al. Engineering a stable and selective peptide blocker of the Kv1.3 channel in T lymphocytes. Mol. Pharmacol. 2009, 75, 762–773. [Google Scholar] [CrossRef]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef]
- Stefan, A.; Ceccarelli, A.; Conte, E.; Monton Silva, A.; Hochkoeppler, A. The multifaceted benefits of protein co-expression in Escherichia coli. J. Vis. Exp. 2015. [Google Scholar] [CrossRef]
- Fahnert, B. Using folding promoting agents in recombinant protein production: A review. Methods Mol. Biol. 2012, 824, 3–36. [Google Scholar]
- Khan, K.H. Gene expression in Mammalian cells and its applications. Adv. Pharm. Bull. 2013, 3, 257–263. [Google Scholar]
- Luna-Ramirez, K.; Csoti, A.; McArthur, J.R.; Chin, Y.K.Y.; Anangi, R.; Najera, R.D.C.; Possani, L.D.; King, G.F.; Panyi, G.; Yu, H.; et al. Structural basis of the potency and selectivity of Urotoxin, a potent Kv1 blocker from scorpion venom. Biochem. Pharmacol. 2020, 174, 113782. [Google Scholar] [CrossRef]
- Baneyx, F.; Mujacic, M. Recombinant protein folding and misfolding in Escherichia coli. Nat. Biotechnol. 2004, 22, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Denks, K.; Vogt, A.; Sachelaru, I.; Petriman, N.A.; Kudva, R.; Koch, H.G. The Sec translocon mediated protein transport in prokaryotes and eukaryotes. Mol. Membr. Biol. 2014, 31, 58–84. [Google Scholar] [CrossRef] [PubMed]
- Lindequist, U. Marine-Derived Pharmaceuticals—Challenges and Opportunities. Biomol. Ther. (Seoul) 2016, 24, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.; Silva, J.; Pinteus, S.; Gaspar, H.; Alpoim, M.C.; Botana, L.M.; Pedrosa, R. From Marine Origin to Therapeutics: The Antitumor Potential of Marine Algae-Derived Compounds. Front. Pharmacol. 2018, 9, 777. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Finol-Urdaneta, R.K.; Belovanovic, A.; Micic-Vicovac, M.; Kinsella, G.K.; McArthur, J.R.; Al-Sabi, A. Marine Toxins Targeting Kv1 Channels: Pharmacological Tools and Therapeutic Scaffolds. Mar. Drugs 2020, 18, 173. https://doi.org/10.3390/md18030173
Finol-Urdaneta RK, Belovanovic A, Micic-Vicovac M, Kinsella GK, McArthur JR, Al-Sabi A. Marine Toxins Targeting Kv1 Channels: Pharmacological Tools and Therapeutic Scaffolds. Marine Drugs. 2020; 18(3):173. https://doi.org/10.3390/md18030173
Chicago/Turabian StyleFinol-Urdaneta, Rocio K., Aleksandra Belovanovic, Milica Micic-Vicovac, Gemma K. Kinsella, Jeffrey R. McArthur, and Ahmed Al-Sabi. 2020. "Marine Toxins Targeting Kv1 Channels: Pharmacological Tools and Therapeutic Scaffolds" Marine Drugs 18, no. 3: 173. https://doi.org/10.3390/md18030173
APA StyleFinol-Urdaneta, R. K., Belovanovic, A., Micic-Vicovac, M., Kinsella, G. K., McArthur, J. R., & Al-Sabi, A. (2020). Marine Toxins Targeting Kv1 Channels: Pharmacological Tools and Therapeutic Scaffolds. Marine Drugs, 18(3), 173. https://doi.org/10.3390/md18030173