Identification and Characterization of a New Type III Polyketide Synthase from a Marine Yeast, Naganishia uzbekistanensis

 , ,
, ,  ,
,  ,
, 
Abstract
1. Introduction
2. Results
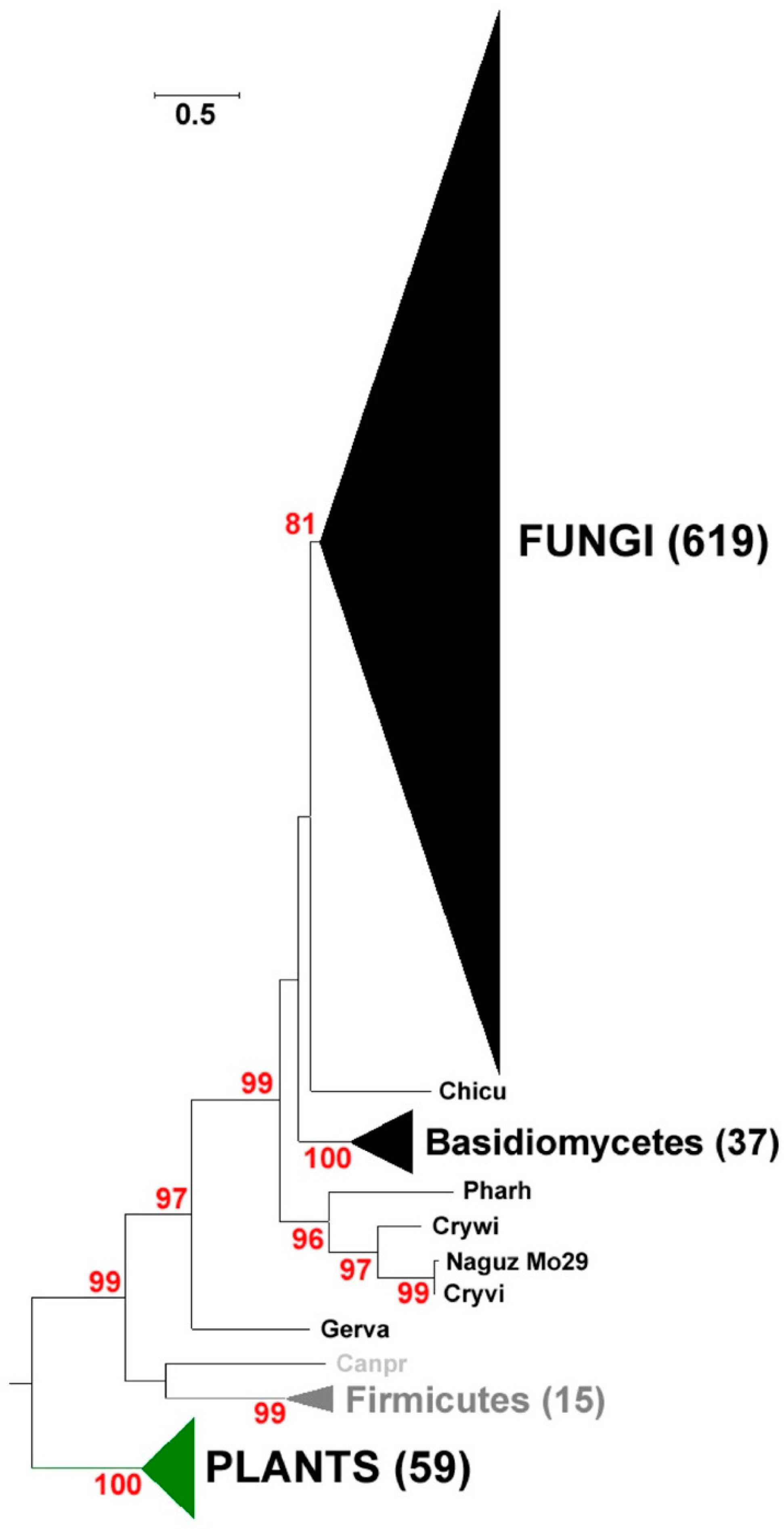
2.1. Identification and Phylogenetic Analysis of Pksiii Gene from Marine Yeast
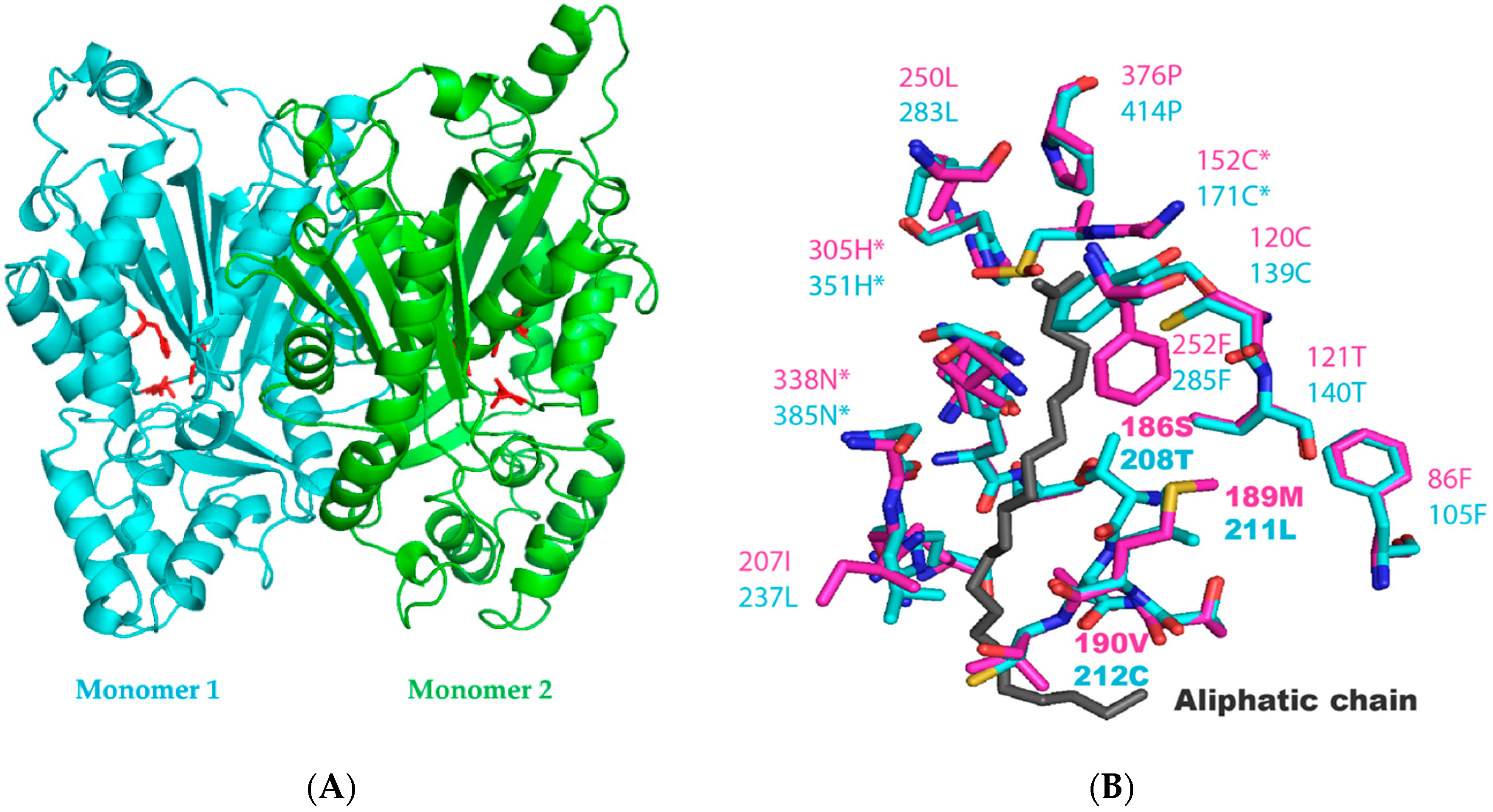
2.2. Identification of the Key Molecular Features by Structural Modelling of N. Uzbekistanensis PKSIII Protein
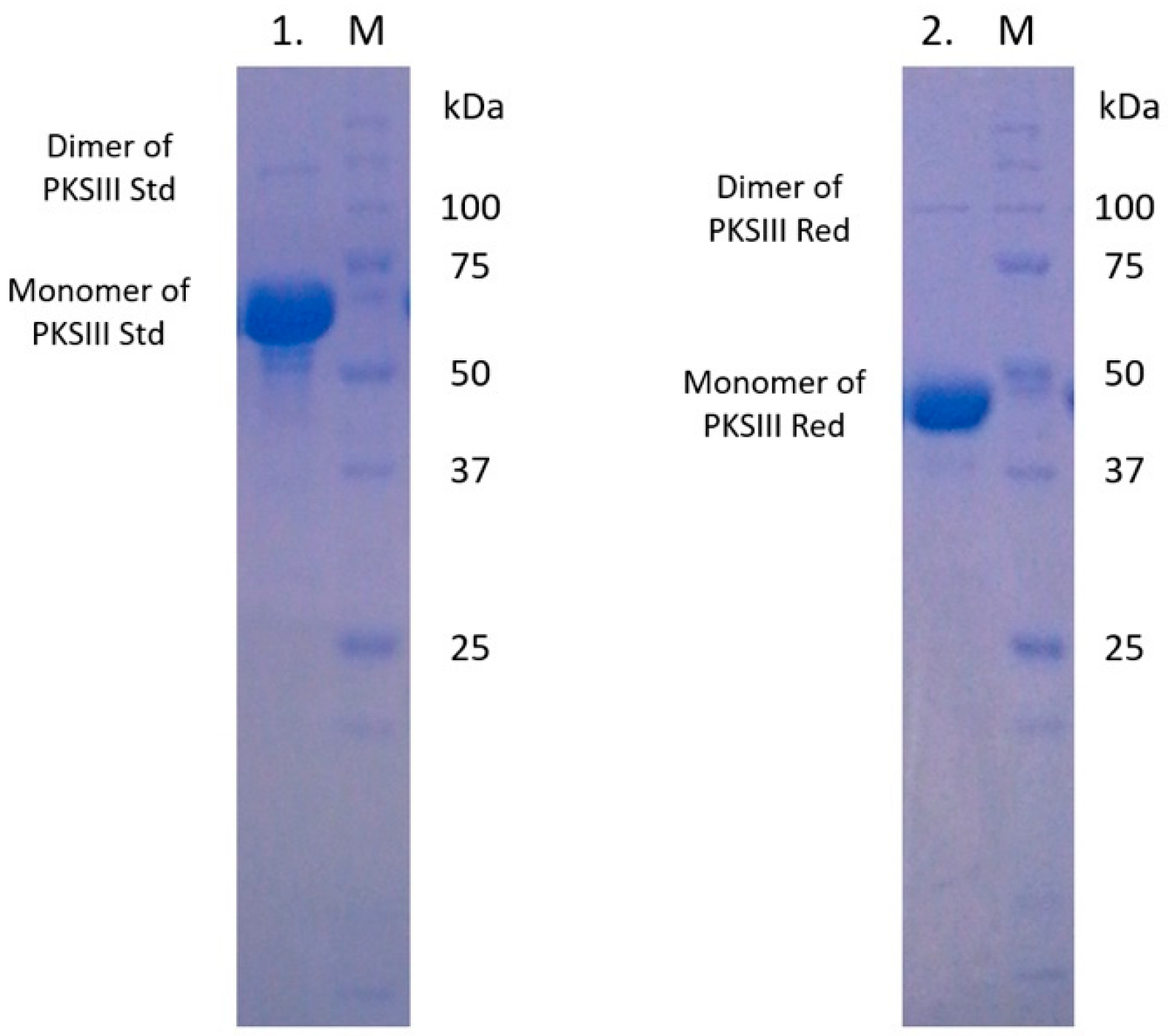
2.3. Expression and Enzymatic Activities of Recombinant PKSIII Mo29 (Full Length and Without C-Terminus Extension)
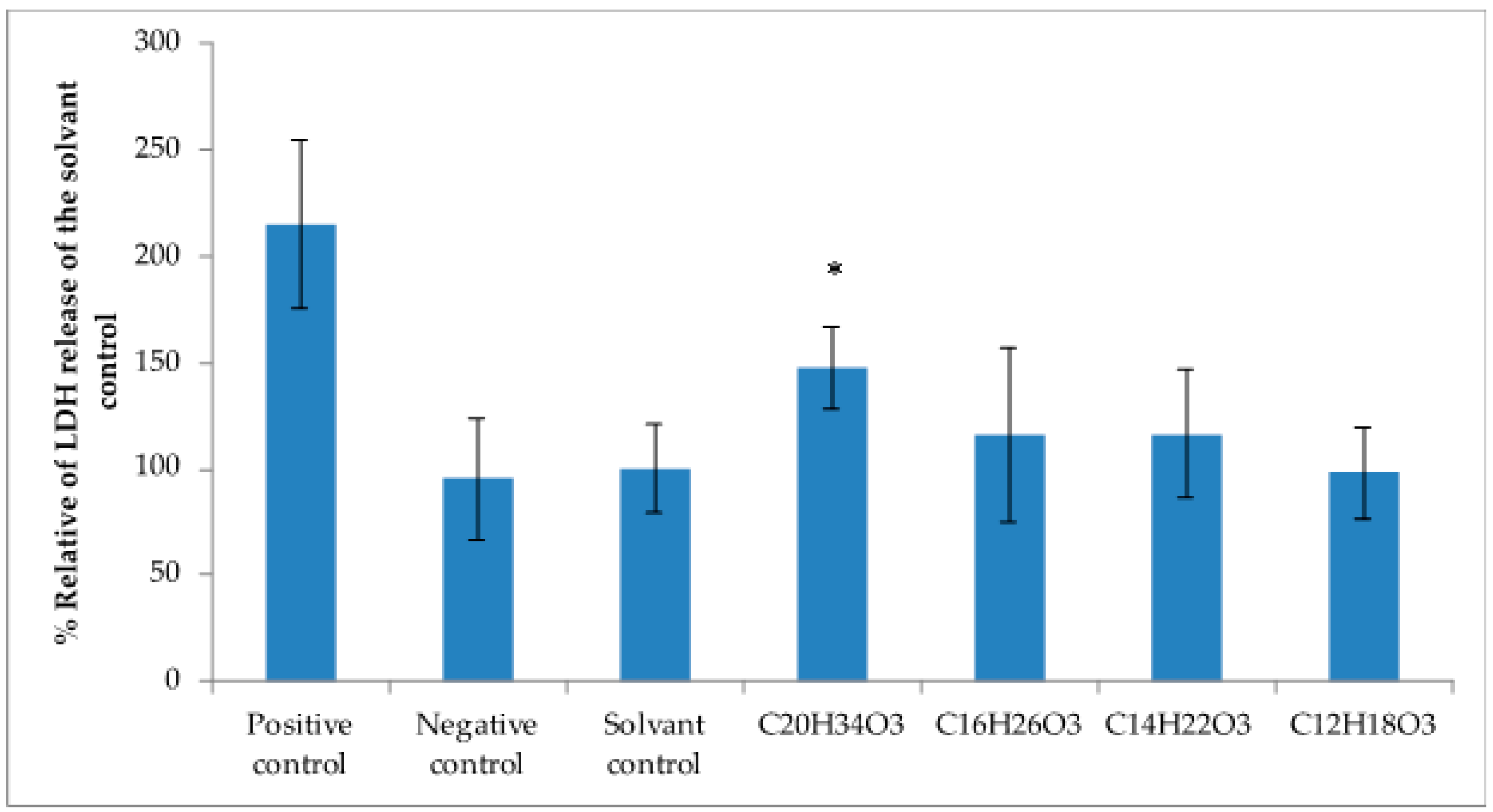
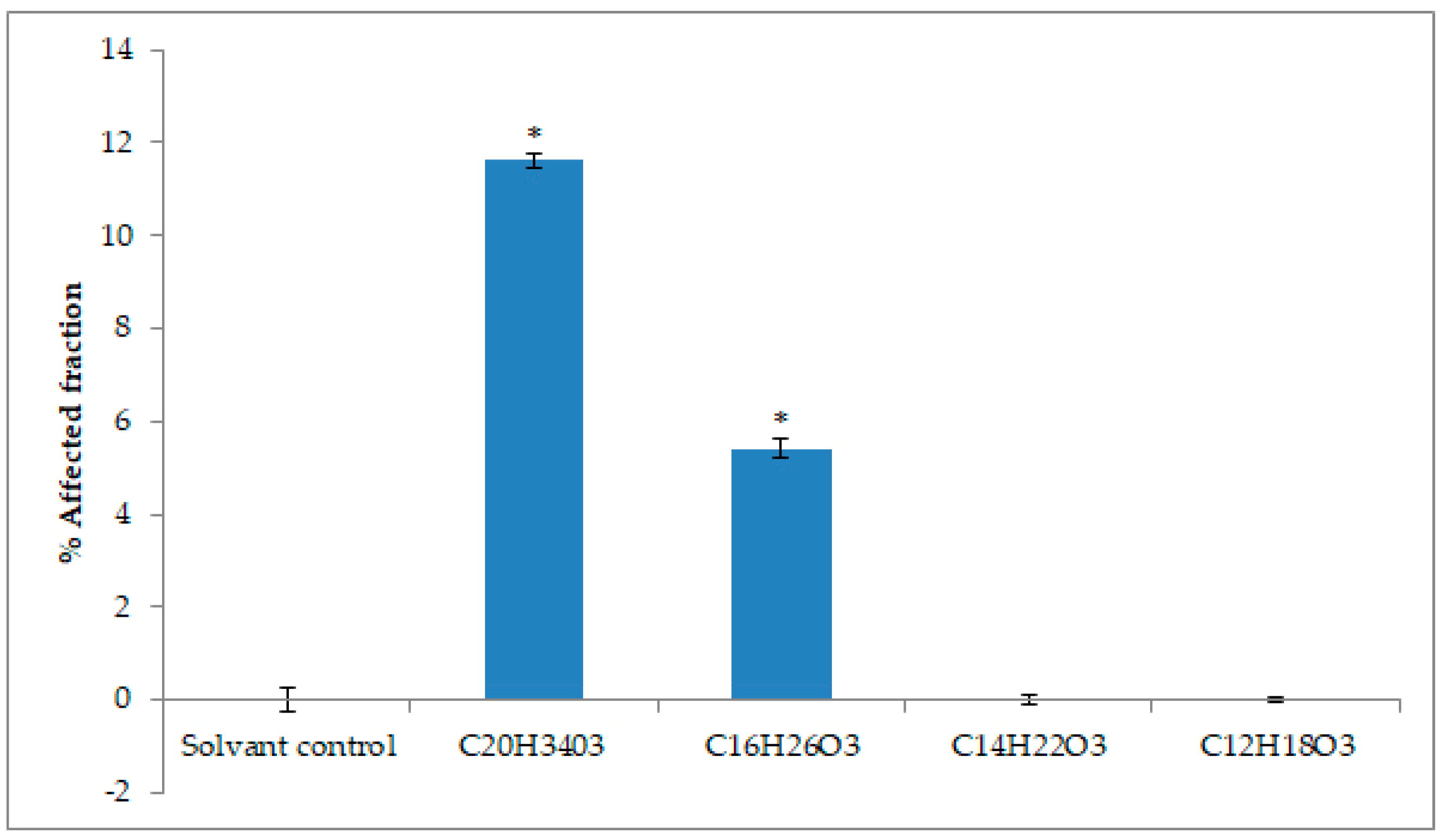
2.4. Cytotoxicity Activity against Tumoral Cell Lines
3. Discussion
4. Materials and Methods
4.1. Fungal Strains
4.2. Bacterial Strains
4.3. Phylogenetic Analysis
4.4. Cloning of Polyketide Synthase and Sequence Validation
4.5. Expression and Purification of Recombinant Proteins
4.6. Enzyme Assays
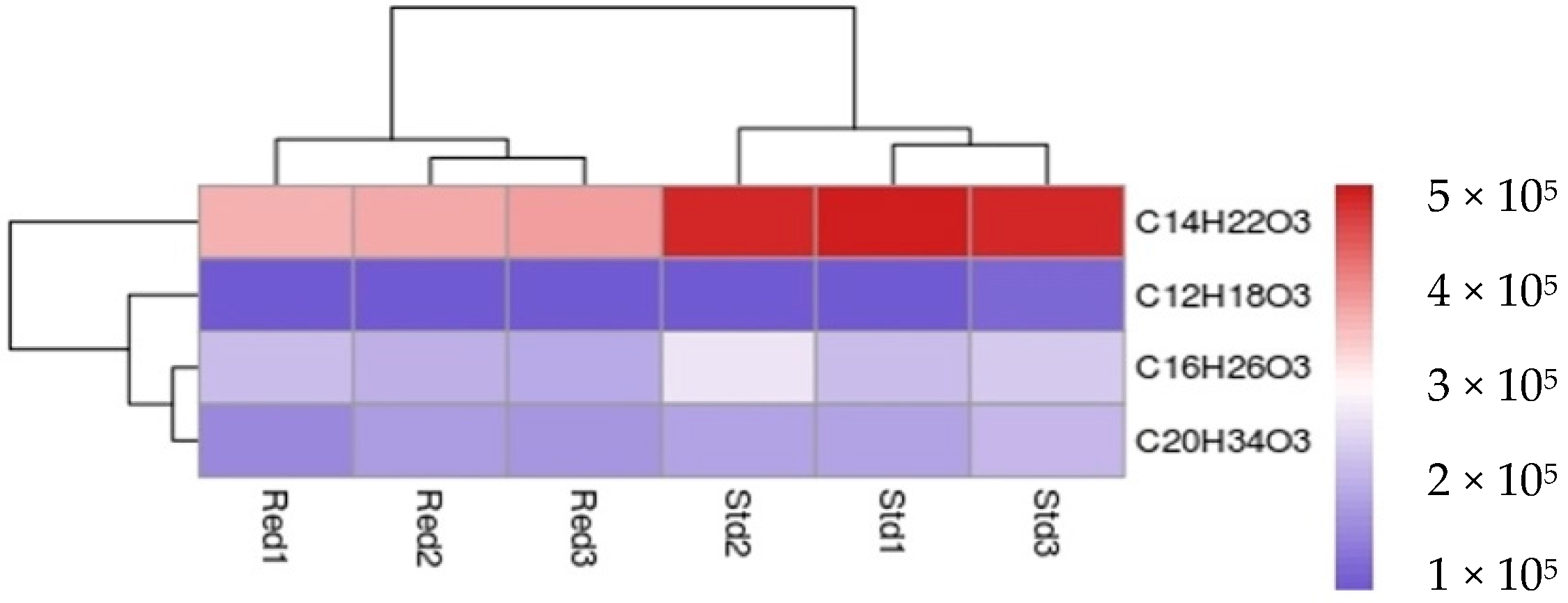
4.7. LC-MS
4.8. Cell Culture Conditions
4.9. Lactate Dehydrogenase Assays
4.10. Evaluation of Cytotoxicity by Mitochondrial Activity
4.11. Statistical Analysis for Cell Viability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berdy, J. Thoughts and facts about antibiotics: Where we are now and where we are heading. J. Antibiot. 2012, 65, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. Br. J. Exp. Pathol. 1929, 10, 226. [Google Scholar] [CrossRef]
- Alberts, A.W.; Chen, J.; Kuron, G.; Hunt, V.; Huff, J.; Hoffman, C.; Rothrock, J.; Lopez, M.; Joshua, H.; Harris, E.; et al. Mevinolin: A highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc. Natl. Acad. Sci. USA 1980, 77, 3957–3961. [Google Scholar] [CrossRef] [PubMed]
- Barrios-Gonzalez, J.; Miranda, R.U. Biotechnological production and applications of statins. Appl. Microbiol. Biotechnol. 2010, 85, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Fliri, H.; Baumann, G.; Enz, A.; Kallen, J.; Luyten, M.; Mikol, V.; Movva, R.; Quesniaux, V.; Schreier, M.; Walkinshaw, M.; et al. Cyclosporins. Structure-activity relationships. Ann. N. Y. Acad. Sci. 1993, 696, 47–53. [Google Scholar] [CrossRef]
- Demain, A.L.; Vaishnav, P. Natural products for cancer chemotherapy. Microb. Biotechnol. 2011, 4, 687–699. [Google Scholar] [CrossRef]
- Stierle, A.; Strobel, G.; Stierle, D. Taxol and taxane production by Taxomyces andreanae, an endophytic fungus of Pacific yew. Science 1993, 260, 214–216. [Google Scholar] [CrossRef]
- Barghoorn, E.S. Marine fungi: Their taxonomy and biology. Farlowia 1944, 1, 395–467. [Google Scholar]
- Kohlmeyer, J.; Kohlmeyer, E. Marine Mycology: The Higher Fungi; Press, N.Y.A., Ed.; Elsevier: Amsterdam, The Netherlands, 1979; p. 690. [Google Scholar]
- Jones, E.B.G.; Suetrong, S.; Sakayaroj, J.; Bahkali, A.; Abdel-Wahab, M.; Boekhout, T.; Pang, K.-L. Classification of marine Ascomycota, Basidiomycota, Blastocladiomycota and Chytridiomycota. Fungal Divers. 2015, 73, 1–72. [Google Scholar] [CrossRef]
- Ciobanu, M.-C.; Burgaud, G.; Dufresne, A.; Breuker, A.; Rédou, V.; Ben Maamar, S.; Gaboyer, F.; Vandenabeele-Trambouze, O.; Lipp, J.S.; Schippers, A.; et al. Microorganisms persist at record depths in the subseafloor of the Canterbury Basin. ISME J. 2014, 8, 1370–1380. [Google Scholar] [CrossRef]
- Orsi, W.; Biddle, J.F.; Edgcomb, V. Deep Sequencing of Subseafloor Eukaryotic rRNA Reveals Active Fungi across Marine Subsurface Provinces. PLoS ONE 2013, 8, e56335. [Google Scholar] [CrossRef] [PubMed]
- Gadanho, M.; Sampaio, J.P. Occurrence and diversity of yeasts in the mid-atlantic ridge hydrothermal fields near the Azores Archipelago. Microb. Ecol. 2005, 50, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Burgaud, G.; Arzur, D.; Durand, L.; Cambon-Bonavita, M.-A.; Barbier, G. Marine culturable yeasts in deep-sea hydrothermal vents: Species richness and association with fauna. FEMS Microbiol. Ecol. 2010, 73, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Burgaud, G.; Le Calvez, T.; Arzur, D.; Vandenkoornhuyse, P.; Barbier, G. Diversity of culturable marine filamentous fungi from deep-sea hydrothermal vents. Environ. Microbiol. 2009, 11, 1588–1600. [Google Scholar] [CrossRef] [PubMed]
- Pachiadaki, M.G.; Rédou, V.; Beaudoin, D.J.; Burgaud, G.; Edgcomb, V.P. Fungal and Prokaryotic Activities in the Marine Subsurface Biosphere at Peru Margin and Canterbury Basin Inferred from RNA-Based Analyses and Microscopy. Front. Microbiol. 2016, 7, 846. [Google Scholar] [CrossRef] [PubMed]
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef] [PubMed]
- Ebada, S.S.; Proksch, P. Marine-derived fungal metabolites. In Springer Handbook of Marine Biotechnology; Springer: Berlin/Heidelberg, Germany, 2015; pp. 759–788. [Google Scholar]
- Rédou, V.; Navarri, M.; Meslet-Cladière, L.; Barbier, G.; Burgaud, G. Species Richness and Adaptation of Marine Fungi from Deep-Subseafloor Sediments. Appl. Environ. Microbiol. 2015, 81, 3571–3583. [Google Scholar] [CrossRef]
- Silber, J.; Kramer, A.; Labes, A.; Tasdemir, D. From discovery to production: Biotechnology of marine fungi for the production of new antibiotics. Mar. Drugs 2016, 14, 137. [Google Scholar] [CrossRef]
- Navarri, M.; Jegou, C.; Meslet-Cladiere, L.; Brillet, B.; Barbier, G.; Burgaud, G.; Fleury, Y. Deep Subseafloor Fungi as an Untapped Reservoir of Amphipathic Antimicrobial Compounds. Mar. Drugs 2016, 14, 50. [Google Scholar] [CrossRef]
- Cheng, M.-M.; Tang, X.-L.; Sun, Y.-T.; Song, D.-Y.; Cheng, Y.-J.; Liu, H.; Li, P.-L.; Li, G.-Q. Biological and Chemical Diversity of Marine Sponge-Derived Microorganisms over the Last Two Decades from 1998 to 2017. Molecules 2020, 25, 853. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Z.; Dong, X.; Feng, Y.; Liu, X.; Gao, B.; Wang, J.; Zhang, L.; Wang, J.; Shi, S.; et al. Identification and functional characterization of three type III polyketide synthases from Aquilaria sinensis calli. Biochem. Biophys. Res. Commun. 2017, 486, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Abe, I. Novel applications of plant polyketide synthases. Curr. Opin. Chem. Biol. 2012, 16, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Ogata, H.; Goto, S. TypeIII Polyketide Synthases: Functional Classification and Phylogenomics. ChemBioChem 2017, 18, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Staunton, J.; Weissman, K.J. Polyketide biosynthesis: A millennium review. Nat. Prod. Rep. 2001, 18, 380–416. [Google Scholar] [CrossRef] [PubMed]
- Shen, B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr. Opin. Chem. Biol. 2003, 7, 285–295. [Google Scholar] [CrossRef]
- Lim, Y.P.; Go, M.K.; Yew, W.S. Exploiting the Biosynthetic Potential of Type III Polyketide Synthases. Molecules 2016, 21, 806. [Google Scholar] [CrossRef]
- Lee, J.S. Recent advances in the synthesis of 2-pyrones. Mar. Drugs 2015, 13, 1581–1620. [Google Scholar] [CrossRef]
- Austin, M.B.; Noel, J.P. The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep. 2003, 20, 79–110. [Google Scholar] [CrossRef]
- Ferrer, J.L.; Jez, J.M.; Bowman, M.E.; Dixon, R.A.; Noel, J.P. Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nat. Struct. Biol. 1999, 6, 775–784. [Google Scholar]
- Achkar, J.; Xian, M.; Zhao, H.; Frost, J. Biosynthesis of phloroglucinol. J. Am. Chem. Soc. 2005, 127, 5332–5333. [Google Scholar] [CrossRef]
- Funa, N.; Ohnishi, Y.; Fujii, I.; Shibuya, M.; Ebizuka, Y.; Horinouchi, S. A new pathway for polyketide synthesis in microorganisms. Nature 1999, 400, 897–899. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, Y.; Ohnishi, Y. Chapter Sixteen—Type III Polyketide Synthases in Microorganisms. In Methods in Enzymology; Hopwood, D.A., Ed.; Academic Press: Cambridge, MA, USA, 2012; Volume 515, pp. 359–377. [Google Scholar]
- Meslet-Cladière, L.; Delage, L.; Leroux, C.J.-J.; Goulitquer, S.; Leblanc, C.; Creis, E.; Gall, E.A.; Stiger-Pouvreau, V.; Czjzek, M.; Potin, P. Structure/Function Analysis of a Type III Polyketide Synthase in the Brown Alga Ectocarpus siliculosus Reveals a Biochemical Pathway in Phlorotannin Monomer Biosynthesis. Plant Cell 2013, 25, 3089–3103. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Xu, F.; Zeng, J.; Zhan, J. Type III polyketide synthases in natural product biosynthesis. IUBMB Life 2012, 64, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Funa, N.; Awakawa, T.; Horinouchi, S. Pentaketide Resorcylic Acid Synthesis by Type III Polyketide Synthase from Neurospora crassa. J. Biol. Chem. 2007, 282, 14476–14481. [Google Scholar] [CrossRef] [PubMed]
- Seshime, Y.; Juvvadi, P.R.; Kitamoto, K.; Ebizuka, Y.; Fujii, I. Identification of csypyrone B1 as the novel product of Aspergillus oryzae type III polyketide synthase CsyB. Bioorg. Med. Chem. 2010, 18, 4542–4546. [Google Scholar] [CrossRef] [PubMed]
- Seshime, Y.; Juvvadi, P.R.; Kitamoto, K.; Ebizuka, Y.; Nonaka, T.; Fujii, I. Aspergillus oryzae type III polyketide synthase CsyA is involved in the biosynthesis of 3,5-dihydroxybenzoic acid. Bioorg. Med. Chem. Lett. 2010, 20, 4785–4788. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Zeng, J.; Chen, D.; Zhan, J. Characterization and reconstitution of a new fungal type III polyketide synthase from Aspergillus oryzae. Enzym. Microb. Technol. 2010, 46, 575–580. [Google Scholar] [CrossRef]
- Li, J.; Luo, Y.; Lee, J.-K.; Zhao, H. Cloning and characterization of a type III polyketide synthase from Aspergillus niger. Bioorg. Med. Chem. Lett. 2011, 21, 6085–6089. [Google Scholar] [CrossRef]
- Sun, L.; Wang, S.; Zhang, S.; Yu, D.; Qin, Y.; Huang, H.; Wang, W.; Zhan, J. Identification of a type III polyketide synthase involved in the biosynthesis of spirolaxine. Appl. Microbiol. Biotechnol. 2016, 100, 7103–7113. [Google Scholar] [CrossRef]
- Navarro-Muñoz, J.C.; Collemare, J. Evolutionary Histories of Type III Polyketide Synthases in Fungi. Front. Microbiol. 2020, 10, 3018. [Google Scholar] [CrossRef]
- Liu, X.Z.; Wang, Q.M.; Göker, M.; Groenewald, M.; Kachalkin, A.V.; Lumbsch, H.T.; Millanes, A.M.; Wedin, M.; Yurkov, A.M.; Boekhout, T.; et al. Towards an integrated phylogenetic classification of the Tremellomycetes. Stud. Mycol. 2015, 81, 85–147. [Google Scholar] [CrossRef] [PubMed]
- Rédou, V.; Kumar, A.; Hainaut, M.; Henrissat, B.; Record, E.; Barbier, G.; Burgaud, G. Draft Genome Sequence of the Deep-Sea Basidiomycetous Yeast Cryptococcus sp. Strain Mo29 Reveals Its Biotechnological Potential. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Saxena, P.; Rahman, A.; Singh, P.K.; Kasbekar, D.P.; Gokhale, R.S.; Sankaranarayanan, R. Structural insights into biosynthesis of resorcinolic lipids by a type III polyketide synthase in Neurospora crassa. J. Struct. Biol. 2008, 162, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Parvez, A.; Giri, S.; Bisht, R.; Saxena, P. New Insights on Cyclization Specificity of Fungal Type III Polyketide Synthase, PKSIIINc in Neurospora crassa. Indian J. Microbiol. 2018, 58, 268–277. [Google Scholar] [CrossRef]
- Saxena, P.; Yadav, G.; Mohanty, D.; Gokhale, R.S. A new family of type III polyketide synthases in Mycobacterium tuberculosis. J. Biol. Chem. 2003, 278, 44780–44790. [Google Scholar] [CrossRef]
- Seshime, Y.; Juvvadi, P.R.; Fujii, I.; Kitamoto, K. Discovery of a novel superfamily of type III polyketide synthases in Aspergillus oryzae. Biochem. Biophys. Res. Commun. 2005, 331, 253–260. [Google Scholar] [CrossRef]
- Jeya, M.; Kim, T.-S.; Tiwari, M.K.; Li, J.; Zhao, H.; Lee, J.-K. The Botrytis cinerea type III polyketide synthase shows unprecedented high catalytic efficiency toward long chain acyl-CoAs. Mol. Biosyst. 2012, 8, 2864–2867. [Google Scholar] [CrossRef]
- Austin, M.B.; Bowman, M.E.; Ferrer, J.L.; Schroder, J.; Noel, J.P. An aldol switch discovered in stilbene synthases mediates cyclization specificity of type III polyketide synthases. Chem. Biol. 2004, 11, 1179–1194. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, D.; Tiwari, M.K.; Manoharan, G.; Sairam, T.; Thangamani, R.; Lee, J.-K.; Marimuthu, J. Molecular characterization of two alkylresorcylic acid synthases from Sordariomycetes fungi. Enzym. Microb. Technol. 2018, 115, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Rubin-Pitel, S.B.; Zhang, H.; Vu, T.; Brunzelle, J.S.; Zhao, H.; Nair, S.K. Distinct structural elements dictate the specificity of the type III pentaketide synthase from Neurospora crassa. Chem. Biol. 2008, 15, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Kirimura, K.; Watanabe, S.; Kobayashi, K. Heterologous gene expression and functional analysis of a type III polyketide synthase from Aspergillus niger NRRL 328. Biochem. Biophys. Res. Commun. 2016, 473, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chen, C.; Mo, S.; Liu, J.; Wang, W.; Zang, Y.; Li, H.; Chai, C.; Zhu, H.; Hu, Z.; et al. Fusaresters A-E, new γ-pyrone-containing polyketides from fungus Fusarium sp. Hungcl and structure revision of fusariumin D. Org. Biomol. Chem. 2019, 17, 5526–5532. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Cai, J.; Zhong, W.; Xu, G.; Wang, F.; Tian, X.; Zhou, X.; Liu, Q.; Liu, Y.; Wang, J. Protein tyrosine phosphatase 1B (PTP1B) inhibitorsfrom the deep-sea fungus Penicillium chrysogenum SCSIO 07007. Bioorg. Chem. 2020, 96, 103646. [Google Scholar] [CrossRef]
- Keisham, S.S. Pyrone-derived Marine Natural Products: A Review on Isolation, Bio-activities and Synthesis. Curr. Org. Chem. 2020, 24, 354–401. [Google Scholar]
- Zhu, H.; Li, D.; Yan, Q.; An, Y.; Huo, X.; Zhang, T.; Zhang, M.; Wang, C.; Xia, M.; Ma, X.; et al. α-Pyrones, secondary metabolites from fungus Cephalotrichum microsporum and their bioactivities. Bioorg. Chem. 2019, 83, 129–134. [Google Scholar] [CrossRef]
- Rédou, V.; Vallet, M.; Meslet-Cladière, L.; Kumar, A.; Pang, K.-L.; Pouchus, Y.-F.; Barbier, G.; Grovel, O.; Bertrand, S.; Prado, S.; et al. Marine Fungi. In The Marine Microbiome: An Untapped Source of Biodiversity and Biotechnological Potential; Stal, L.J., Cretoiu, M.S., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 99–153. [Google Scholar]
- Pang, K.-L.; Overy, D.P.; Jones, E.B.G.; da Luz Calado, M.; Burgaud, G.; Walker, A.K.; Johnson, J.A.; Kerr, R.G.; Cha, H.-J.; Bills, G.F. ‘Marine fungi’ and ‘marine-derived fungi’ in natural product chemistry research: Toward a new consensual definition. Fungal Biol. Rev. 2016, 30, 163–175. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Residues Near the Active Site (aa Involved in the Tunnel and Binding with Stearoyl-CoA) | |||||||||||||||||||||
| PKSIIINc | 86F | 120C | 121T | 125N | 186S | 189M | 190V | 206G | 207I | 210F | 211S | 250L | 252F | 261V | 306P | 307G | 308G | 309A | 310T | 311I | 312L | 313S |
| PKS18 | 109F | 143S | 144T | 148A | 205C | 210V | 211F | 220I | 221H | 224F | 225G | 264I | 266L | 275C | 314P | 315G | 316G | 317P | 318K | 319I | 320I | 321E |
| AoCsyA | 101F | 135C | 136T | 140N | 201C | 204F | 205F | 221A | 222M | 225F | 226G | 266I | 268F | 277P | 323P | 324G | 325G | 326Y | 327S | 328I | 329A | 330V |
| AoCsyB | 89F | 123C | 124T | 128H | 189P | 192F | 193A | 209A | 210M | 213F | 214G | 254A | 256F | 265A | 311P | 312G | 313G | 314Y | 315A | 316V | 317L | 318V |
| BcPKS | 99F | 133C | 134T | 138N | 199S | 202L | 203V | 219G | 220V | 223F | 224S | 263L | 265F | 274V | 318P | 319G | 320G | 321A | 322T | 323I | 324L | 325T |
| AnPKS | 101F | 135V | 136T | 140A | 201C | 204H | 205L | 221A | 222P | 225F | 226S | 265M | 267Y | 276A | 317P | 318G | 319G | 320R | 321A | 322V | 323I | 324Q |
| PKSIII Mo29 | 105F | 139C | 140T | 144Y | 208T | 211L | 212C | 236S | 237L | 240F | 241S | 283L | 285F | 294A | 352P | 353G | 354G | 355S | 356L | 357I | 358I | 359S |
| Protein | Residues of active site | |||||||||||||||||||||
| PKSIIINc | 152C | 305H | 338N | |||||||||||||||||||
| PKS18 | 175C | 313H | 346N | |||||||||||||||||||
| AoCsyA | 167C | 322H | 355N | |||||||||||||||||||
| AoCsyB | 155C | 310H | 343N | |||||||||||||||||||
| BcPKS | 165C | 317H | 350N | |||||||||||||||||||
| AnPKS | 167C | 316H | 349N | |||||||||||||||||||
| PKSIII Mo29 | 171C | 351H | 385N | |||||||||||||||||||
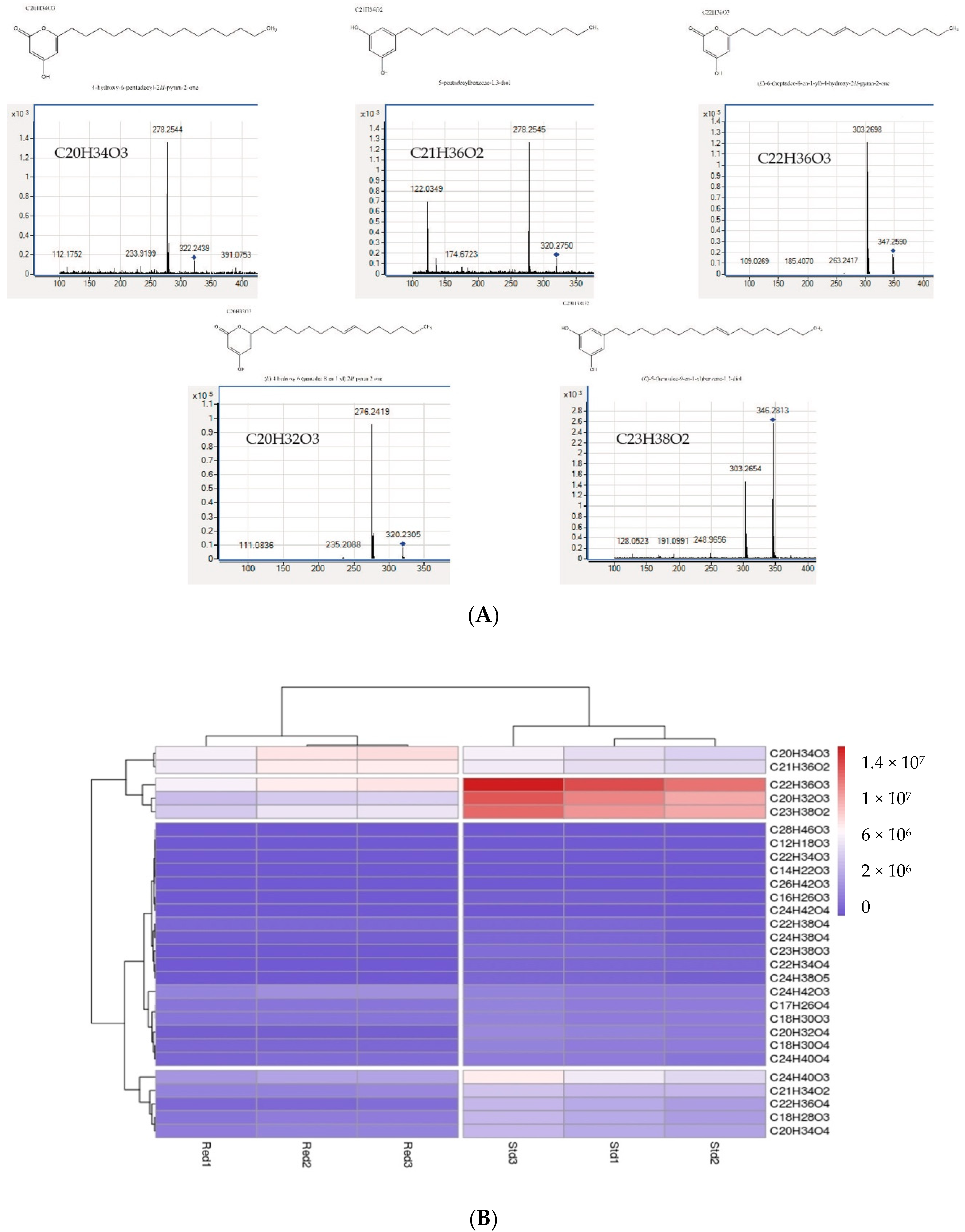
| Starter Substrat | Products | Rt LC/min | Parent Ion m/z | Fragment Ion m/z |
|---|---|---|---|---|
| Acetyl-CoA | ND | / | / | |
| Malonyl-CoA | ND | / | / | |
| Hexanoyl-CoA | ND | / | / | |
| Octanoyl-CoA | C12H18O3 | 17.50 | 209.1300 | ND |
| Decanoyl-CoA | C14H22O3 | 19.80 | 238.1567 | 194.1599 |
| Lauroyl-CoA | C16H26O3 | 22.05 | 266.1884 | 222.1952 |
| Palmytoyl-CoA | C20H34O3 | 26.03 | 322.2507 | 278.2554 |
| Products | Name of Products | RT (min) | Parent Ion m/z (ESI(-)) | Fragment Ion m/z |
|---|---|---|---|---|
| C12H18O3 | Pyrone | 17.5 | 209.1300 | ND |
| C14H22O3 | Pyrone | 19.735 | 238.1567 | 194.1599 |
| C16H26O3 | Pyrone | 21.973 | 266.1884 | 222.1952 |
| C17H26O4 | Pyrone | 24.11 | 293.1800 | ND |
| C18H28O3 | Pyrone | 22.48 | 292.2038 | 248.2109 |
| C18H30O3 | Pyrone | 24.125 | 294.2192 | 250.2236 |
| C18H30O4 | Pyrone | 20.656 | 310.2143 | 125.0231 |
| C18H32O3 | Pyrone | 24.6 | 295.2200 | ND |
| C20H32O3 | Pyrone | 24.421 | 320.2349 | 276.2421 |
| C20H32O4 | Pyrone | 21.27 | 336.2296 | 125.0225 |
| C20H34O3 | Pyrone | 26.10 | 322.2507 | 278.2554 |
| C20H34O4 | Pyrone | 22.8 | 338.2450 | 125.0221 |
| C21H34O2 | Resorcinol | 25.037 | 318.2555 | 122.0376 276.2415 |
| C21H36O2 | Resorcinol | 26.57 | 320.2714 | 122.0349 278.2545 |
| C22H34O4 | Pyrone | 26.5 (27.2) | 361.2400 | ND |
| C22H36O3 | Pyrone | 26.22 | 348.2663 | 303.2700 |
| C22H36O4 | Pyrone | 23.21 | 364.2620 | 125.0223 |
| C22H38O3 | Pyrone | 27.97 | 350.2832 | 305.2855 |
| C22H38O4 | Pyrone | 26.572 | 366.2766 | 319.2635 |
| C23H38O2 | Resorcinol | 26.65 | 346.2869 | 122.0338 304.2729 |
| C23H38O3 | Resorcinol | 23.455 (27.24) | 362.2814 | 123.0448 |
| C24H38O4 | Pyrone | 25.49 | 389.2700 | ND |
| C24H38O5 | Pyrone | 22.98 | 406.2716 | 125.0232 |
| C24H40O3 | Pyrone | 27.95 | 376.2975 | 331.2974 |
| C24H40O4 | Pyrone | 26.657 | 392.2923 | 345.2814 |
| C24H42O3 | Pyrone | 30.315 | 378.3131 | 333.3145 |
| C24H42O4 | Pyrone | 28.308 | 394.3075 | 347.2919 |
| C26H42O3 | Pyrone | 27.279 | 402.3130 | 123.0795 357.3123 |
| C28H46O3 | Pyrone | 28.918 | 430.3442 | 385.3474 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinelli, L.; Redou, V.; Cochereau, B.; Delage, L.; Hymery, N.; Poirier, E.; Le Meur, C.; Le Foch, G.; Cladiere, L.; Mehiri, M.; et al. Identification and Characterization of a New Type III Polyketide Synthase from a Marine Yeast, Naganishia uzbekistanensis. Mar. Drugs 2020, 18, 637. https://doi.org/10.3390/md18120637
Martinelli L, Redou V, Cochereau B, Delage L, Hymery N, Poirier E, Le Meur C, Le Foch G, Cladiere L, Mehiri M, et al. Identification and Characterization of a New Type III Polyketide Synthase from a Marine Yeast, Naganishia uzbekistanensis. Marine Drugs. 2020; 18(12):637. https://doi.org/10.3390/md18120637
Chicago/Turabian StyleMartinelli, Laure, Vanessa Redou, Bastien Cochereau, Ludovic Delage, Nolwenn Hymery, Elisabeth Poirier, Christophe Le Meur, Gaetan Le Foch, Lionel Cladiere, Mohamed Mehiri, and et al. 2020. "Identification and Characterization of a New Type III Polyketide Synthase from a Marine Yeast, Naganishia uzbekistanensis" Marine Drugs 18, no. 12: 637. https://doi.org/10.3390/md18120637
APA StyleMartinelli, L., Redou, V., Cochereau, B., Delage, L., Hymery, N., Poirier, E., Le Meur, C., Le Foch, G., Cladiere, L., Mehiri, M., Demont-Caulet, N., & Meslet-Cladiere, L. (2020). Identification and Characterization of a New Type III Polyketide Synthase from a Marine Yeast, Naganishia uzbekistanensis. Marine Drugs, 18(12), 637. https://doi.org/10.3390/md18120637